

Abstract
Disulfides are conventionally viewed as structurally stabilizing elements in proteins but emerging evidence suggests two disulfide subproteomes exist. One group mediates the well known role of structural stabilization. A second redox-active group are best known for their catalytic functions but are increasingly being recognized for their roles in regulation of protein function. Redox-active disulfides are, by their very nature, more susceptible to reduction than structural disulfides; and conversely, the Cys pairs that form them are more susceptible to oxidation. In this study, we searched for potentially redox-active Cys Pairs by scanning the Protein Data Bank for structures of proteins in alternate redox states. The PDB contains over 1134 unique redox pairs of proteins, many of which exhibit conformational differences between alternate redox states. Several classes of structural changes were observed, proteins that exhibit: disulfide oxidation following expulsion of metals such as zinc; major reorganisation of the polypeptide backbone in association with disulfide redox-activity; order/disorder transitions; and changes in quaternary structure. Based on evidence gathered supporting disulfide redox activity, we propose disulfides present in alternate redox states are likely to have physiologically relevant redox activity.
Keywords: disulfide redox activity, thiol-based redox signaling, OxyR, oxidative stress, CLIC1, zinc signaling, disordered proteins, molten globule
Introduction
Emerging evidence supports the concept of two distinct types of disulfides in protein structures which have different functional roles.1 The best known group act as structural stabilizers in proteins, significantly lowering the Gibb's free energy of the protein chain by crosslinking it, resulting in proteins that are more resistant to denaturation. The second group are redox-active and best known for their catalytic role in proteins such as thiol-disulfide oxidoreductases. An increasing role for the redox-active group in protein redox regulation is emerging.1,2 These disulfides act as redox-sensitive switches and are best viewed as post-translational modifications akin to phosphorylation. It has now been established that oxidative regulation: important in processes such as the cell cycle, apoptosis, and response to oxidative stress; is an important control mechanism in the cytosol. Transient oxidation mediates control of enzymes, signaling pathways, as well as transcriptional and translational control. However, these characterized cytosolic proteins likely only represent the tip of the iceberg. Increasingly, reduction of disulfides is also emerging as a key regulator of protein function in oxidizing environments.3–5 Importantly, ageing and several major diseases including neurodegenerative diseases such as Alzheimer's disease, type II diabetes, cancer, and cardiovascular disease have been associated with abnormal redox conditions.
Structural and redox-active disulfides can be distinguished by their redox potentials. Disulfide redox potentials measured in thiol-disulfide oxidoreductases range from −120 to −270 mV.6–9 For disulfides serving structural purposes, the redox potential can be as low as −470 mV.10 Computational methods for distinguishing the two groups would be a boon. Reactivity of a redox-active Cys is modulated by its immediate protein environment. In addition to the effects of the electrostatic environment, physical stress applied to a disulfide bond both through stretching and twisting enhances its reactivity. These forces can be applied to a protein dynamically11 or may be captured during the protein folding process, rendering the disulfide poised to act upon exposure to variations in redox conditions.12 Because of their enhanced reactivity, redox-active disulfides are also more susceptible to cleavage/oxidation under nonphysiologic conditions. For instance, redox-active disulfides are prone to cleavage by synchrotron radiation during the process of X-ray structure determination.13–15 Other factors such as the pH of crystallization or the presence of mutations that change the electrostatic environment near a disulfide may also influence reactivity. Here, we mined the Protein Data Bank (PDB)16 for highly similar proteins that have been solved in multiple redox states, that is, disulfide-bonded in one structure and reduced in another. We present evidence that most of the disulfides present in our Redox Pairs dataset are likely to be physiologically redox-active. Many of the proteins have known thiol-based redox activity, while others have a high likelihood of thiol-based redox activity based on their involvement with redox processes. We also observed and classified types of conformational changes which occur during redox transitions.
Results
Redox processes are enriched in the redox pair dataset
The Redox Pairs dataset contains 18,003 pairs of protein structures consisting of 4333 protein chains, of which 1238 (28.57%) are high-resolution structures (<2.2 Å). Many structures are of the same protein solved in different redox environments. A filtered dataset consists of 322 unique protein groups clustered at 95% sequence identity.
Further evidence of disulfide redox activity was sought based on experimental data in the literature, association of protein function with known redox activity, and implication of the disulfide in redox activity by independent computational prediction methods. For 41 proteins, direct experimental evidence of redox activity for the particular disulfide was available (designated by “K” in Table S1, Supporting Information). The proteins belong to several major groups known to have redox activity. 11 unique Redox Pairs are associated with aerobic metabolism in eukaryotes or carbon fixation in plants (in groups D and E of Table S1); a further five are associated with maturation of proteins of these pathways (in groups E and G). Four Redox Pairs are associated with ion channel activity (group A): two of these, the chloride channel CLIC1 and the detoxification enzyme arsenate reductase, have known redox activity17,18; while a third, aquaporin, was recently implicated as a pore for the reactive oxygen species hydrogen peroxide, as part of the redox signaling process.19 Seven Redox Pairs are associated with the cell cycle, a process known to be redox regulated20 (Group B). Formation of disulfides involving the catalytic Cys in the phosphatases PR-1 and Cdc25B regulate their resident protein's function.21,22 41 Redox Pairs (group d) are involved in transcriptional control, replication, and DNA synthesis including the oxidative stress response transcription factor OxyR23 and ribonucleotide reductases from several species in multiple redox states.24 12 Redox Pairs generated or destroyed reactive oxygen species (Groups F and Y). The latter comprised three different peroxiredoxins25 as well as superoxide dismutases from several species.26 Four different amine oxidases generate peroxide while inducible nitric oxide synthase generates NO. Other large groups consist of proteins devoted to cell entry27 (Group b, 15 examples), cell adhesion (group c, nine examples), immunity (group f, 43 examples), apoptosis and protein fate (groups Z and a, 10 examples), and redox signaling (group X, 13 examples). A schematic of supergroups is shown in Figure 1.
Figure 1.
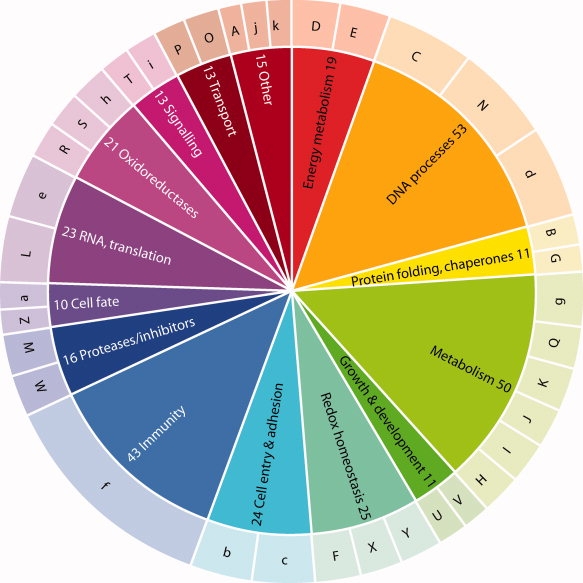
Functional supergroups in the Redox Pairs dataset (inner circle). The outer circle corresponds to groups in Supporting Information Table 1. Key to Groups: A, ion channel, ion pump modifier; B, protein folding, isomerization; C, cell cycle; D, Calvin cycle; E, photosynthesis, respiration, transport, maturation of proteins involved in these processes; F, destruction of oxygen radicals; G, redox metal homeostasis; H, amino acid metabolism; I, sulfur metabolism; J, nucleotide metabolism/homeostasis; K, isoprenoid biosynthesis; L, RNA-interacting; M, proteases; N, DNA repair; O, actin-related; P, carrier proteins; Q, other metabolism; R, NADP+-dependent oxidoreductase; S, other oxidoreductase; T, signaling; U, development; V, cell growth pathways; W, enzyme inhibitors; X, redox homeostasis; Y, ROS generating; Z, apoptosis; a, protein fate; b, cell entry; c, cell adhesion; d, DNA-interacting proteins; e, translation; f, immunity; g, carbohydrate metabolism; h, NAD+-dependent oxidoreductases; i, Ca handling; j, methyltransferase; k, other.
To further assess the likelihood that the presence of proteins in the PDB with disulfides in both redox states is an indicator of physiological disulfide redox activity, we looked for Gene Ontology (GO) terms overrepresented in the Redox Pairs dataset compared to the entire PDB. GO is a dynamic controlled vocabulary of over 16,000 terms used to describe molecular function, process and location of action of a protein in a generic cell.28 The analysis revealed many GO terms consistent with redox function, including cell redox homeostasis and multiple pathways of cysteine biosynthesis (Supporting Information Table 2). Interestingly, several of the enriched GO terms are linked to proteins mediating cell entry, including bacterial toxins and viral entry proteins. Reduction of disulfides, often accompanied by irreversible conformational changes, have previously been implicated as a general mechanism in cell entry proteins of diverse types.27 Experimental evidence directly supporting such a general process exists for HIV gp120, Newcastle disease virus, SARS coronavirus and Chlamydia.29–32 Thus the GO terms support redox roles for proteins in the Redox Pair dataset.
Disulfides were tested computationally for likely redox activity by screening the dataset for oxidized structures where the disulfide had particular properties previously associated with redox activity. Flagged disulfides were those (a) with high torsional energies; (b) found in known redox-active sequence motifs; or (c) which disobeyed known rules of protein stereochemistry.12 Studies of disulfide-bonds in model proteins have shown that those bonds with high torsional energies are more easily cleaved than disulfide-bonds with lower stored energy.33–36 148 disulfides in 139 proteins have torsional energies >15 kJ mol−1: a conservative threshold associated with redox activity.27 These are designated by “H” in Supporting Information Table 1. Redox-pair disulfides were also found in sequence motifs previously shown to be associated with redox activity. 67 proteins, designated by “M” in Table S1, contain known redox-active motifs such as the CXXC motif: a redox-active disulfide motif commonly found in the thioredoxin fold, but which has also arisen convergently in other folds. A further 65 proteins, designated by “F” in Table S1, contain disulfides in protein structures that disobey known rules of protein stereochemistry.37,38 These disulfides, which we refer to as “forbidden” disulfides, conform to canonical structural motifs. Deformation of the resident structure enhances disulfide reactivity by inducing stress in the protein chain.37,38 Forbidden disulfides are proposed to have a functional role through redox activity.12 In summary, a total of 226 proteins in the Redox Pair dataset contain disulfides predicted to be redox-active by a second independent bioinformatic analysis method.
Nonetheless, it is possible that some proteins with alternate Cys-Pair redox states are non-native, that is, not encountered under normal, or abnormal, physiological conditions. We reasoned that proteins demonstrated selective reduction of disulfides were more likely to represent physiological states. Accordingly, for proteins with more than one disulfide, the dataset was partitioned into pairs where all the disulfides were reduced and those where some disulfides were selectively reduced while others remained intact, to further test the likelihood of involvement in physiological redox processes. The enriched GO terms for both the partially and fully reduced sets contained terms previously associated with thiol-based redox regulation indicating both the fully and partially reduced sets have likely redox activity (Supporting Information Table 2).
Conformational changes associated with redox activity
Having established the Redox-Pair dataset is enriched in proteins involved in redox-regulated processes, we studied conformational changes between redox pairs of proteins. For many proteins in the data set there were negligible conformational differences between the SG atoms of each Cys residue of the reduced and oxidized states (see Fig. 2). For this group, other factors may be required for, or preclude, conformational changes in these proteins. Crystal packing forces, for example, may prevent a subsequent conformational change after cleavage of the disulfide by synchrotron radiation. For another group, the small distance changes of 2–4 Å observed are most likely due to rotation of one or both Cys sidechains toward each other to form the S—S bond. Some of the proteins exhibiting these small conformational changes are oxidoreductases with catalytic thiols such as thioredoxin, thioredoxin reductase, and protein disulfide isomerase.
Figure 2.

Change in disulfide separation in protein Redox Pairs. Excluding interchain disulfides, around 21% of redox structure pairs have a change of over 1 Å in the sulfur atom separation between the oxidized and reduced structures. Maximal intrachain differences of up to 18.8 Å were apparent in 1-deoxy-d-xylulose-5-phosphate reductoisomerase (DXP reductoisomerase) and the RNA sulfuration enzyme TrmU.39
For proteins which exhibit major conformational changes, four classes were identified based on the nature of the change. These groups were: proteins that oxidize disulfides following expulsion of metals such as Zn; proteins that exhibited major reorganization or “morphing” of portions of the polypeptide backbone in association with disulfide redox-activity; proteins that exhibited order/disorder transitions; and proteins that exhibited changes in quaternary structure.
Redox sites associated with zinc and other metals
Redox regulation of proteins that form disulfides following expulsion of Zn is an emerging area of signaling.40 For example, reversible redox regulation of the Zn-binding site has recently been proposed for small Tim proteins of the mitochondrial intermembrane space (IMS).41 Small Tim proteins have an essential role in the transport of proteins into the IMS. These proteins can exist in both a reduced Zn-bound state and an oxidized disulfide-bridged state. Both states likely represent physiological forms of the proteins which are adopted in response to changing redox conditions in the IMS.41 In addition, expelled Zn acts as a second messenger to effect further cellular responses.
Proteins that form disulfides following expulsion of metals such as Zn were identified by searching for ligation of metals by Cys in reduced structures. A total of 73 redox pairs in 53 proteins were found (Table I, z in S2). Proteins identified include transcriptional regulators such as SMAD and anti-TRAP; transcriptional silencers such as SIRT2; and the enzymes of sulfur metabolism, BHMT, MetE, and MetH. Also notable were a number of RNA-interacting proteins including amino-acid-tRNA synthetases, RNA/DNA polymerases, ribonucleotide reductase and the ribosome. In addition to Zn, a few other bound metal atoms were represented including Fe, Ni, Cu. There were several examples of inorganic iron-sulfur (Fe-S) clusters associated with redox-active disulfides including: xanthine dehydrogenase, a major source of ROS in the failing heart; and components of respiratory complexes (Table I). Other proteins containing Fe-S clusters contained other redox-active disulfides that were not associated directly with the Fe-S cluster. All proteins containing Fe-S clusters (designated with “C” for cofactor) are listed in Table I regardless of whether the Fe-S cluster is directly associated with a labile dithiol pair or not.
Table I.
Proteins with Likely Redox-Active Metal Sites
| Group | Proteins | Metal site | Sitea | ox | re | M | Ref. |
|---|---|---|---|---|---|---|---|
| Sulfur metabolism | Hs/RnBHMT | 217,299,300 | ZnS3 | 1lt7 | 1lt8 | Y | 42 |
| TmMetH | 207,272,273? | ZnS3 | 1q7m | 1q8j | Y | 43 | |
| TmMetE | H618,620,704 | ZnS2N | 1t7l | 1xdj | Y | ||
| Cell and protein fate | HsNedd8 | 199,202,343,346 | ZnS4 | 1r4nB | 1tt5 | Y | |
| Hssurvivin | 57,60,H77,84 | ZnS3N | 1f3h | 1e31 | N | ||
| HsBirc8 | 300,303,H320,327 | ZnS3N | 1xb1 | 1xb0 | Y | ||
| Nucleotide metabolism | BsG-Deaminase | H53,83,86 | ZnS2N | 1tiy | 1wkq | N | |
| EcATCase | 109,114,138,141 | ZnS4 | 1tugB | 1tugD | P | ||
| EcGTPhI | 110,H113,181 | ZnS2N | 1gtp | 1fbx | Y | 44,45 | |
| Acthymidine kinase | 145,148,183,186 | ZnS4 | 1xx6B | 1xx6A | N | ||
| NAD+-dependent oxidoreductases | TmSir2 | 124,127,148,151 | ZnS4 | 2h59 | 1yc5 | N | |
| HsSIRT2 | 195,200,221,224 | ZnS4 | 1j8fC | 1j8fA | N | ||
| MmADH4 | 97,100,103,111 | ZnS4 | 1e3l | 1e3e | N | ||
| EqADH | 97,100,103,111 | ZnS4 | 1axg | 3bto | N | ||
| Transcriptional regulators | HsSMAD | 64,109,121, H169 | ZnS3N | 1mhd | 1ozj | Y | |
| BsAnti-TRAP | 12,15,26,29 | ZnS4 | 2bx9L | 2bx9A | N | ||
| SARSnsp10 | 117,129,128 | ZnS3? | 2g9t | 2fyg | N | ||
| SARSnsp10 | 74,77,H83,90 | ZnS3N | 2ga6 | 2fyg | N | ||
| HsEstrogen receptor | 7,10,24,27 | ZnS4 | 1hcqB | 1hcqA | N | 46 | |
| DNA repair | DrRecR | 57,60,69,72 | ZnS4 | 1vddC | 1vddA | N | |
| Metallo-chaperones | ScAtxI | T14,15,18 | CuS2O | 1cc7 | 1cc8 | Y | 47 |
| MjHypB | 95,127,95,b127b | ZnS4 | 2hf9 | 2hf8 | Y | ||
| Other viral proteins | HepCNs3Ib | 97,99,145 | ZnS3? | 1w3c | 1dxp | Y | |
| norovirus3c-like protease | 77,83,154 | ZnS3? | 1wqsB | 1wqsA | N | 48 | |
| Oxidoreductases | EcRibD | H49,74,83 | ZnS2N | 2d5n | 2b3z | N | |
| MtCODH | 595,597,B596,B597 | Binuclear Ni/ ZnS2N2,ZnS3 | 1mjg | 1oao | N | ||
| PtNHase | 108,111,B112,113 | CoS3N/S2N2 | 1ugq | 1ugp | Y | 49 | |
| RrpNHase | 110,113,B114,115 | FeS3N | 1ahj | 2cyz | N | ||
| DvNiFe hydrogenase | 81,84,546,549 | NiFeS4 | 1ubk | 1h2r | N | ||
| DsNiFe hydrogenase | 65,68,530,533 | NiFeS4 | 2frv | 1frv | N | ||
| PafdhE | 225,228,256,259 | FeS4 | 2fiyB | 2fiyA | N | ||
| HsNOS-3 | 110,115,110,115 | ZnS4 | 2nsi | 1nsi | Y | 50 | |
| aa-tRNA synthetases | TtVal-tRNAs | 417,420,438,441 | ZnS4 | 1gax | 1ivs | N | 51,52 |
| TtLeu-tRNAs | 439,442,484,487 | ZnS4 | 1obh | 1h3n | Y | ||
| TtIle-tRNAs | 181,184,389,392 | ZnS4 | 1ile | 1jzq | N | ||
| TtIle-tRNAs | 461,464,502,504 | ZnS4 | 1ile | 1jzq | N | ||
| RNA/DNA polymerases | ScRNApolIIB1 | 67,70,77,H80 | ZnS3N | 1r9t | 1i6h | N | 53–55 |
| ScRNA polIIB1 | 107,110,148,167 | ZnS4 | 1r9t | 1i6h | N | 53–55 | |
| ScRNA polIIB2 | 1163,1166,1182,1185 | ZnS4 | 1twh | 1i3q | N | 53–55 | |
| ScRNApolIIB3 | 86,88,92,95 | ZnS4 | 1twh | 1i3q | N | 53–55 | |
| ScRNApolIIB9 | 7,10,29,32 | ZnS4 | 1i6h | 1y1v | N | 53–55 | |
| ScRNApolIIB9 | 75,78,103,106 | ZnS4 | 1i6h | 1i3q | N | 53–55 | |
| ScRNApolIIABC4 | 31,34,48,51 | ZnS4 | 1twh | 1i3q | N | 53–55 | |
| ScRNApolIIABC5 | 7,10,45,46 | ZnS4 | 1r9t | 1i3q | N | 53–55 | |
| TtRNApolβ | 1112,1194,1201,1204 | ZnS4 | 1zyr | 1smy | N | 53–55 | |
| Ribosomal proteins | Tt30SRibosomeS4 | 9,12,26,31 | ZnS4 | 2j02 | 1hr0 | N | |
| Tt30SRibosomeS14 | 24,27,40,43 | ZnS4 | 1n33 | 1fjg | N | ||
| EcrrmA | 5,8,21,H25 | ZnS3N | 1p91B | 1p91A | N | ||
| Replication, DNA synthesis | B4Ribonucleotide reductase III | 543,546,561,564 | ZnS4 | 1h7a | 1hk8 | N | |
| EcDNA pol III τ | 64,73,76,79 | ZnS4 | 1xxh | 1njf | N | ||
| SV40LT-antigen | 302,305,H313,H317 | ZnS2N2 | 1svo | 1svm | N | ||
| Fe-S containing | Rcxanthine dehydrogenase | 103,106,134,136 | [2Fe-2S]S4 | 1jrp | 1jro | N | |
| Ggsuccinate dehydrogenase L | 65,70,73,85 | [Fe-S]S4 | 2h89 | 1yq3 | N | ||
| Ssmitochondrial respiratory complex II, lp subunit | 158,161,164,225 | [4Fe-4S]S4 | 1zp0 | 1zoy | N | ||
| Other | Hsmnk2 | 299,303,311,314 | ZnS4 | 2hw7 | 2ac3 | N |
Ligands in metal sites are Cys unless indicated by a one-letter-code amino acid prefix, B indicates ligation by the backbone of the indicated residue.
ox, oxidized structure; re, reduced structure; M, metal expelled; Y, metal absent from oxidized structure; N, metal present in oxidized structure; P, metal present in oxidized structure with partial occupancy; ADH, alcohol dehydrogenase; Anti-TRAP, tryptophan RNA-binding attenuator protein-inhibitory protein; ATCase, aspartate transcarbamylase; AtxI, metal homeostasis factor ATX1; BHMT, betaine-homocysteine s-methyltransferase; Birc8, baculoviral iap repeat-containing protein 8; CODH, bifunctional carbon monoxide dehydrogenase/acetyl-coA synthase; fdhE-formate dehydrogenase; G-Deaminase, guanine deaminase; GTPhI, GTP hydrolase I; Ile-tRNAs, isoleucyl-tRNA synthetase; HypB, hydrogenase nickel incorporation protein; Leu-tRNAs, Leucyl-tRNA synthetase; LT-antigen, large T-antigen; MetE-B12, independent methionine synthase; MetH-B12, dependent methionine synthase; mnk2, map kinase-interacting serine/threonine-protein kinase 2; Nedd8, E3 ubiquitin-protein ligase Nedd8; NHase, nitrile hydratase; NOS-3, inducible nitric oxide synthase; Ns3, Ns3 Protease; RecR, recombinational repair protein; RibD, riboflavin biosynthesis protein ribD; RNA pol II–RNA polymerase II subunit; rrmA, ribosomal RNA large subunit methyltransferase A; Sir2, sirtuin homolog; SIRT2, sirtuin homolog 2; SMAD, mothers against decapentaplegic homolog 3; Val-tRNAs, valyl-tRNA synthetase; B4, Bacteriophage t4; Bs, Bacillus subtilis; Ds, Desulfovibrio s; Dv, Desulfovibrio vulgaris; Ec, Escherichia coli; Gg, Gallus gallus; HepC, Hepatitis C; Hs, Homo sapiens; Mt, Moorella thermoacetica; Pa, Pseudomonas aeruginosa; Pt, Pseudonocardia thermophila; Rc, Rhodobacter capsulatus; Rrp, Rhodococcus sp R312 plastid; SARS, severe acute respiratory syndrome coronavirus; Sc, Saccharomyces cerevisiae; Ss, Sus scrofa; SV40, Simian Virus 40; Tt, Thermus thermophilus.
Zn sites in italics are catalytic.
Cys residue is in another chain (interchain disulfide).
Crystallographers typically differentiate two types of Zn sites: catalytic Zn sites, which in their simplest, mononuclear form have three amino-acid sidechains and one water molecule as ligands; and structural Zn sites, which have four amino acid ligands. From a redox point of view, a second variable - lability, is important. Some structural Zn sites expel Zn upon variation of physiological redox conditions, whereas others bind Zn tightly, and would not be expected to release Zn under physiological conditions. We refer to this first redox-regulated group as “labile” Zn sites; and the second, purely structural group, as “inert.”
We wished to determine whether Zn sites associated with Redox Pair disulfides detected in the study were labile or inert. The number of Zn ligands and their identity are key indicators of Zn site lability. For catalytic mononuclear Zn sites, three amino acids, which are often His, ligate the Zn. In binuclear Zn sites there are usually six ligands, and the carboxylated residues Asp and Glu predominate over His.56,57 Cys is more commonly found as a ligand in structural Zn sites which generally have four ligands. On consideration of ligand type alone, S4 sites - those with four Cys ligands, are the most labile type of structural Zn site, and are almost as reactive as a free thiol.58 Most of the structural Zn sites identified in this study were of the S4 type (Table I), suggesting they are labile. Four catalytic Zn sites were also found in the dataset. These catalytic Zn sites are highly unusual because of their utilization of Cys as a ligand instead of the more usual His, Glu, or Asp. Two of the catalytic sites are within BHMT and MetH: enzymes of sulfur metabolism, which is redox-regulated in plants.2 Selection of sulfur ligands for Zn in these enzymes may confer an additional level of redox control on sulfur metabolism.
As an additional control, we checked whether metal atoms were expelled from oxidized structures. A metal atom is not present in over a third of the oxidized structures. For another structure, aspartate transcarbamylase (1tug), the metal site was only partially occupied in the oxidized chain. For the remaining structures where the metal atom is retained in the oxidized structure, the shortened SG-SG distance may be caused by a heterogeneous mix of oxidized and reduced, metal-bound structures in the ensemble. We reasoned that if the metal was expelled from some members of the ensemble, or partially expelled (still in the site but held, for example, by only two of four potential ligating Cys), then the B-factor should be higher than for chains where no shortened SG-SG distances are observed. Although B-factors cannot be compared between different structures, for some of the redox pairs, different chains of the same structure in different disulfide redox states could be compared. Our analysis confirmed that the B-factor of the metal was higher in the oxidized chains for 80% of structures (see Fig. 3). A similar result was obtained for proteins with Fe-S clusters. There were no instances of expulsion of the Fe-S cluster from the oxidized structure but oxidation of Fe-ligating Cys residues to form disulfides was generally associated with a higher B-factor of the relevant Fe atom than Fe atoms where the ligating Cys remained reduced. In contrast, in reduced structures all Fe atoms of the Fe-S cluster had similar B-factors.
Figure 3.
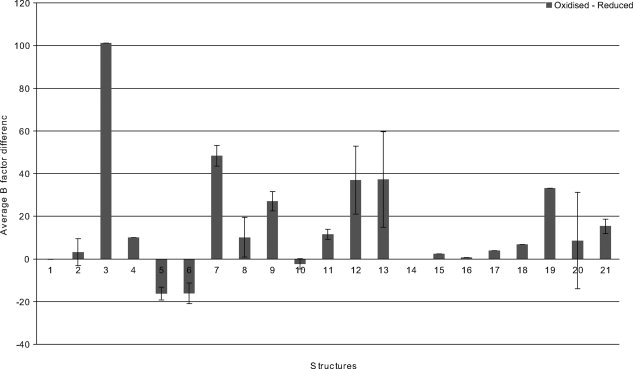
Comparison of B-factors between multiple chains of structures where oxidation of the metal site is heterogeneous Bars represent the difference of the average B-factor of the metal between oxidized and reduced chains. In structures where there is partial oxidation of the metal site, the B-factor of the metal should be higher because it has fewer ligands and hence more conformational freedom. This is true for most structures (bars above the axis). Standard errors are depicted where they could be calculated. Key to structures: Clostridium acetobutylicum thymidine kinase 1-1xx6; E. coli aspartate transcarbamylase 2-1r0b, 3-1tug; Thermotoga maritima Sir2 4-2h59; Homo Sapiens Sir2 5-1j8f; Equus caballus alcohol dehydrogenase 6-1axg; Bacillus subtilis Tryptophan RNA-binding attenuator protein- inhibitory protein (antiTRAP) 7-2bx9; severe acute respiratory syndrome coronavirus nonstructural protein 10 8-2g9t, 9-2ga6; Homo sapiens estrogen receptor 10-1hcq; Deinococcus radiodurans recombinational repair protein 11-1vdd; norovirus 3c-like protease 12-1wqs; Moorella thermoacetica bifunctional carbon monoxide dehydrogenase/acetyl-coa synthase 13-1mjg; Rhodococcus sp R312 nitrile hydratase 14-1ahj; Desulfovibrio s. NiFe hydrogenase 15-1frv; Pseudomonas aeruginosa formate dehydrogenase (fdhE) 16-2fiy; Thermus thermophilus valyl-tRNA synthetase 17-1gax; Thermus thermophilus RNA polymerase 18-2cw0; E. coli ribosomal RNA large subunit methyltransferase A (rrmA) 19-1p91; E. coli DNA polymerase III τ 20-1xxh; Rhodobacter capsulatus xanthine dehydrogenase 21-1jrp. Metal in all structures is Zn unless specified otherwise. Metal atoms in structure 21 are Fe atoms in Fe-S cluster.
Further inspection of proteins with likely redox-active Zn sites reveals that two forbidden disulfide motifs, the Jump Strand Disulfide (JSD) and a type of β-diagonal disulfide (BDD),12 are repetitively associated with redox-active Zn-binding sites in the dataset. Oxidized structures that contain a BDD include isoleucyl tRNA synthetase (1ile 180–184, 502–504), the apoptosis-related proteins birc8 and survivin (1xb1, 300–303;1f3h, 57–60), aspartate transcarbamylase (1r0b, 138–141) and the DNA repair enzyme RecR (1vdd, 57–60). Oxidized structures that contain a JSD include the amino-acyl tRNA synthetases for Leu and Ile (1obh, 439–484; 1jzs, 461–502), the enzymes of sulfur metabolism MetH and BHMT (1q7m, 207–273; 1lt7, 217–300) and GTP hydrolase GTPhI (1gtp, 110–181).
Morphing transitions in the dataset
A large group of proteins in the dataset were observed to undergo plastic deformations involving large scale rearrangements of the polypeptide backbone. We refer to these conformational changes as morphing transitions. These transitions, which typically involve unraveling portions of secondary structure, are illustrated in Figure 4 and in animations in the Supporting Information. The well known redox-sensitive protein OxyR belongs to this group. In OxyR, a protein of 305 residues, significant refolding of the C-terminus occurs involving residues 196–221.23 In the reduced structure, residues 200–204 form a short 310 helix flanked by regions of coil. Upon oxidation, residues 196–221 adopt a different fold with residues 189–193 and 213–218 forming two parallel β-strands joined by a loop with a disulfide-linkage between Cys199 and Cys208. In the reduced OxyR structure, the two Cys that form the disulfide are separated by almost 13 Å. Cys 199 in OxyR is known to undergo alternate modifications such as glutathionylation.60 Because of the spectacular nature of the transition between the reduced and oxidized structures, and the existence of an alternate mode of redox regulation, the physiological relevance of the disulfide-linked structure has been called into question.60
Figure 4.
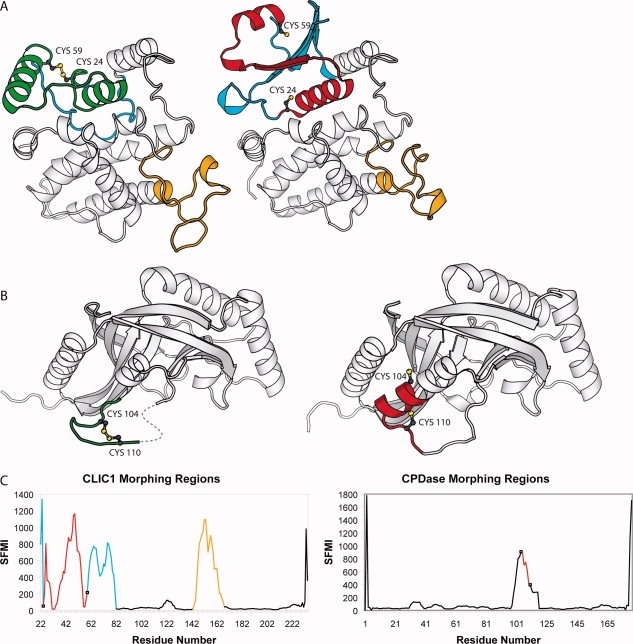
Morphing transitions in protein Redox Pairs. A: A large morphing transition in the chloride channel CLIC1: a protein that forms ion channels by inserting itself into membranes in response to oxidation. B: A smaller morphing transition in cyclic phosphodiesterase (CPDase) a protein involved in the tRNA splicing pathway of yeast, plants and vertebrates. Redox activity of the disulfide modulates access to the active site.59 Oxidized structures are shown on the left. Reduced structures are on the right. The region of the polypeptide chain between the two redox-active Cys is shown in green in the oxidized structure and red in the reduced structure. Other morphing regions are shown in blue. C: Flocco-Mowbray diagram for CLIC1 (left) and CPDase (right). Y-axis SFMI-Smoothed Flocco-Mowbray Index. The polypeptide chain is coloured according to the corresponding oxidized structure. Redox-active thiols are indicated with open squares. Animations of the conformational changes in CLIC1 and CPDase are included in the Supporting Information.
To determine whether the oxidized structure of OxyR is indeed an oddity, or if other proteins exhibit similar conformational changes, we systematically searched for morphing proteins in the dataset by scanning for protein structure pairs that exhibited large differences of the backbone torsional angles over at least seven contiguous residues. Changes in pseudo-dihedral angles calculated using four consecutive Cα atoms exceeding a summed threshold of 1000 degrees over seven consecutive residues were used to detect changes in secondary structure. Twenty-two parents were found in the fully reduced set and four in the partially reduced set (designated m in Supporting Information Table 1). Proteins showing the largest conformational changes are in Table II. These proteins include the Cl− channel CLIC1: a protein that forms ion channels by inserting itself into membranes in response to oxidation; the detoxification enzyme arsenate reductase, which undergoes multiple conformational changes associated with a disulfide relay as part of its reaction cycle17,61 and cyclic phosphodiesterase, a tRNA-splicing enzyme where the disulfide modulates access to the active site.59 Like OxyR, several of these proteins exhibit large changes in Cα separations of the two Cys between alternate redox states (Table II). Thus large physical separation of the reduced Cys pair seems to be a common feature of many of these proteins and does not preclude disulfide formation.
Table II.
Proteins with Morphing Regions
| Structures |
||||||
|---|---|---|---|---|---|---|
| Name | Disulfide | ox | re | Morphing region | ΔSG Å | Transition Reduced → Oxidized |
| EcOxyR | 199–208 | 1i6a | 1i69 | 281–298 | 15.9 | Helix, coil to sheet |
| HsCLIC1 | 24–59 | 1rk4 | 1k0m | 27–56 | 13.5 | Sheet to helix, coil |
| VpChpP | 424–493 | 1v7v | 1v7w | 483–499 | 10.8 | Strand to coil |
| SaArsC | 82–89 | 1lju | 1jf8 | 81–95 | 10.7 | Helix to coil |
| AtCPDase | 104–110 | 1fsi | 1jh6 | 99–119 | 9.0 | C-term of helix to coil |
| Aebcp | 49–54 | 2cx3 | 2cx4 | 44–60 | 7.6 | Helix to β-hairpin |
| Hs/RnPhe hydroxylase | 203–334 | 1tdw | 1phz | 131–149 | 7.2 | Helix to coil |
| HsAKT | 60–77 | 1unr | 1unq | 39–53 | 6.8 | Helix to coil |
| SaArsC | 10–82 | 1lk0 | 1jf8 | 80–94 | 5.7 | Helix to coil |
| HsSSAT | 120–122 | 2b5g | 2g3t | 145–147,155–170 | 5.2 | Coil to strand transition |
| HsIL2 | 58–105 | 1m48 | 1m4a | 29–36, 71–87 | 5.1 | Coil to N-term of helix |
| TfIMPDH | 26–459 | 1meh | 1pvn | 317–327,412–435 | 5.0 | Helical phase shift, helix to coil |
| RnPR-1 Ppase | 49–104 | 1x24 | 12cl | 20–30,46–56,103–110 | 4.9 | R1 helix to coil, R3 coil to helix |
| EcMurD | 208–227 | 1uag | 1eeh | 345–354 | 4.6 | Helix to coil transition |
| HepCRNA polymerase 1a | 303–311 | 1nhu | 1c2p | 303–318 | 3.8 | β-hairpin straddled by disulfide (motif C) curls in oxidized structure |
| TtLeu-tRNAs | 439–484 | 1obh | 1h3n | 150–193, 436–447, 539–546 | 3.8 | Sheet melts, helical phase shift |
| EcATCase | 114–141 | 1tug | 1d09 | 45–58 | 3.7 | Coil to helix transition |
| HepCRNA polymerase 2b | 316–366 | 1yvx | 1yuy | 19–38, 445–451, 539–546 | 3.6 | Helix to coil |
| HsTSP1 | 153–214 | 2es3 | 1za4 | 17–29 | 3.1 | Coil to helix transition |
| MmActivin receptor | 86–91 | 1bte | 2goo | 58–73, 87–94 | 3.0 | R1 helix to coil, R2 coil to helix |
| Schem13 | 193–193a | 1txn | 1tkl | 40–60, 187–202 | NA | R1 coil to strand, R2 coil to helix/strand |
| HsDNA pol III λ | 543–543a | 1xsl | 1xsn | 460–474 | NA | Strand to coil |
| HsActivin | 80–80a | 2arv | 2arp | 20–27, 65–79 | NA | R1 helix to coil transition |
| R2 coil to extension of C-term helix | ||||||
| HsRXR | 269–269a | 1g5y | 1fm9 | 242–264,433–451 | NA | Coil to helix R2 |
Transitions are expressed from reduced to oxidized but are likely to be reversible. Structures where change in SG separation is caused by quaternary structure changes are indicated as NA.
ox, oxidized structure; re, reduced structure; AKT, Rac-alpha serine/threonine kinase; ArsC, arsenate reductase; ATCase, aspartate carbamoyltransferase; bcp, bacterioferritin comigratory protein; ChpP, chitobiose phosphorylase; CLIC1, chloride intracellular channel protein 1; CPDase, cyclic nucleotide phosphodiesterase; DNA pol III λ, DNA polymerase III λ; hem13, coproporphyrinogen III oxidase; IL2, interleukin 2; IMPDH, inosine monophosphate dehydrogenase; Leu-tRNAs, leucyl-tRNA synthetase; MurD, UDP-N-acetylmuramoyl-L-alanine:D-glutamate ligase; OxyR, hydrogen peroxide-inducible genes activator; PR-1 Ppase, protein tyrosine phosphatase 4a1; RXR, retinoic acid receptor; SSAT, spermidine/spermine N1-acetyltransferase; TSP1, thrombospondin-1; Ae, Aeropyrum pernix, At, Arabidopsis thaliana; Ec, E. coli; HepC, Hepatitis C; Hs, Homo sapiens; Mm, Mus musculus; Rn, Rattus norvegus; Sa, Staphylococcus aureus; Sc, Saccharomyces cerevisiae; Tf, Tritrichomonas foetus; Tt, Thermus thermophilus; Vp, Vibrio proteolyticus.
2nd Cys residue is in another chain (interchain disulfide).
Order/disorder transitions
Another recognizable group of conformational changes involves order/disorder transitions. In these transitions, alternate redox states of the protein correlated with differing amounts of disorder in the protein structure, as evidenced by missing electron density. A total of 282 redox pair structures in 27 parents exhibited order/disorder transitions correlated with the redox state of the disulfide (Table III, Fig. 5). Proteins were classified into four groups based on the location of disulfide-forming Cys residues with respect to the disordered region in the protein chain. Proteins in Group A had a single Cys residue in a disordered region, with the other Cys residing on a more stable region of structure. In Group B, both Cys residues reside on a single disordered region. In Group C, both residues lie on disordered regions which are not contiguous in the sequence. In Group D, disulfide reduction is associated with multiple regions of disorder in the reduced protein chain.
Table III.
Order/Disorder Transitions Associated with Disulfide Redox State
| Structures |
|||||||
|---|---|---|---|---|---|---|---|
| Name | ox | re | Disulfide | Disordered Region | Function | Ref. | |
| A | EfAPH | 1j7l | 2bkk | 19–156a | 150a –165a | Protein kinase | |
| PsFBPase | 1d9q | 1dbz | 153–173 | 151–163 | Calvin cycle, chloroplast, Trxf substrate | 62 | |
| HsG6PDH | 1qkiC | 1qkiA | 13–446 | 15–26 | Pentose phosphate pathway, glutathione metabolism | 63 | |
| BcSMase | 2ddrD | 2ddrA | 123–159 | 156–162 | Differentiation, development, aging, and apoptosis | ||
| EcTrmU | 2derB | 2derA | 102–199 | 189–204 | tRNA processing, catalytic disulfide | 39 | |
| RnTRPV2 | 2etc | 2eta | 195–206 | 201–106 | Ion channel | 64 | |
| Hsp53BP1 | 1kzy | 1gzh | 1796–1802 | 1793–1797 | Cell cycle, DNA binding, p53 binding, nucleocytoplasmic | ||
| BpPRD1p2 | 1n7u | 1n7v | 254–277 | 248–269 | Cell entry | 12,27 | |
| TeTAXI1 | 1t6e | 1t6g | 50–71 | 69–78 | Xylanase inhibitor, plant development, plant defense | ||
| B | EcSPase I | 1kn9 | 1t7d | 170–176 | 171–178 | Signal peptidase | |
| AcgdhB | 1c9u | 1qbi | 338–345 | 335–344 | Oxidoreductase, PQQ, pentose phosphate pathway | 65 | |
| HsAKT2 | 1mry | 1mrv | 297–311 | 295–313 | Redox signaling | 65,66 | |
| SaHis-tRNAs | 1qe0A | 1qe0B | 191–194 | 172–230 | Binds tRNA, translation, CXXC | ||
| SpSPE-H | 1eu4 | 1et9 | 77–79 | 76–82 | Immune, toxin, bacterial superantigen | ||
| OcDNAse I | 2a3z | 2a42 | 101–104 | 99–104 | Apoptosis, nuclear envelope, i,i+3 | 68 | |
| HsVit D BP | 1j78 | 1lot | 80–96, 95–106 | 91–111 | Actin scavenging system, carbonylated in Alzheimer's disease | 69 | |
| AtCPDase | 1fsi | 1jh6 | 104–110 | 103–113 | tRNA splicing | 59 | |
| HsDefensin-5 | 1zmpA | 1zmpD | 10–30 | 10–16 | Small toxin/inhibitor fold, immunity | ||
| PvAMA1 | 1w8k | 1w81 | 208–220 | 205–218 | Cell entry | ||
| HsGAS6 | 1h30 | 2c5d | 262–277 | 261–278 | Cell adhesion | ||
| C | Mmnidogen | 1gl4 | 1h4u | 360–373 | 359–366,374–381 | Cell adhesion | |
| TtLeu-tRNAs | 1obh | 1h3n | 439–484 | 440–433, 486–491 | Binds tRNA, translation, reduced more ordered | 51 | |
| D | HsTCRα 3A6 | 1zgl | 1zgl | 139–189 | Multiple | Binds protein, autoimmunity | |
| HsTCRβ | 1ktkE | 1ktkF | 147–212 | Multiple | Binds protein, immunity | ||
| TmPsi55s | 1ze1 | 1ze2 | 92–184 | Multiple | Binds RNA, tRNA processing | ||
| HsHGF | 1gmo | 1gmn | 74–84, 70–96 | Multiple | Binds protein, angiogenesis | ||
| Rn/HsActRIIA | 1nys | 1nyu | 84–103 | Multiple | Binds protein, TGFβ-like receptor | ||
Group A: one Cys lies within the disordered region with the other residing on a more stable region of structure Group B: Both Cys are found in or near a single disordered region; Group C: Both Cys lie within isolated disordered regions; Group D: Massive loss of secondary structure in one monomer of a multimer associated with binding to a ligand. May correspond to Molten Globule state.
ox, oxidized structure; re, reduced structure; ref, evidence for redox activity; ActRIIA, activin receptor type II; AKT2, Rac-beta serine/threonine-protein kinase; AMA1, apical membrane antigen 1; APH, aminoglycoside phosphotransferase; CPDase, cyclic nucleotide phosphodiesterase; DNAse I, deoxyribonuclease-1; FBPase, fructose-1,6-bisphosphatase; G6PDH, glucose-6-phosphate 1-dehydrogenase; GAS6, growth-arrest-specific protein 6; gdhB, glucose dehydrogenase; HGF, hepatocyte growth factor; His-tRNAs, histidyl-tRNA synthetase; Leu-tRNAs, leucyl-tRNA synthetase; p53BP1, tumor suppressor p53-binding protein 1; PRD1p2, adsorption protein p2; Psi55s, pseudouridine 55 synthase; SMase, sphingomyelin phosphodiesterase; SPase I, signal peptidase I; SPE-H, superantigen spe-h; TAXI1, Triticum aestivum xylanase inhibitor I; TCRα 3A6, T-cell receptor α 3A6; TCRβ, T-cell receptor; TrmU, tRNA methyltransferase; TRPV2, transient receptor potential cation channel subfamily V member 2; Vit D; BP, vitamin D binding protein; Ac, Clostridium acetobutylicum; At, Arabidopsis thaliana, Bc, Bacillus cereus; Bp, Bacteriophage, Bs, Bacillus subtilis; Ec, E. coli; Ef, Enterococcus faecalis; Hs, Homo sapiens; Mm, Mus musculus; Oc, Oryctolagus cuniculus; Ps, Pisum sativum; Pv, Plasmodium vivax; Rn, Rattus norvegus; Sa, Staphylococcus aureus; Sp, Streptococcus pyogenes; Te, Triticum aestivum; Tm, Thermotoga maritima; Tt, Thermus thermophilus.
Residue is in another monomer (interchain).
Figure 5.
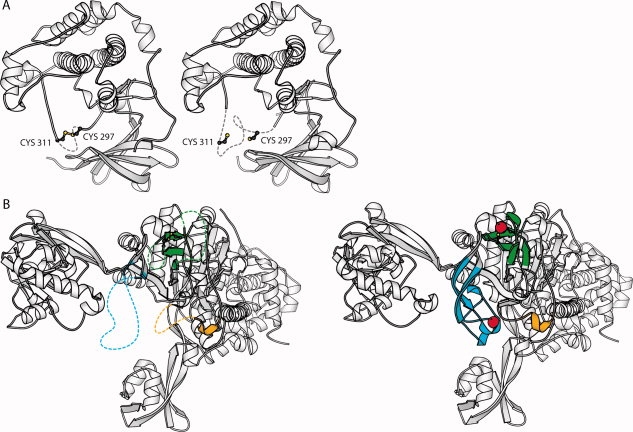
Order/disorder transitions in protein Redox Pairs. A: A small order/disorder transition in the T-loop of the signaling protein Rac-beta serine/threonine-protein kinase (AKT2).67 In the more ordered oxidized structure: a disulfide is formed between Cys 297 and Cys 311 which straddles a key phosphorylation site at Thr 309. Formation of the disulfide is proposed to inhibit kinase activity by recruiting the cognate phosphatase.70 The T-loop was recently identified as a target for selective cancer drugs.66 B: A larger order/disorder transition associated with expulsion of Zn from the oxidized structure of leucyl-tRNA synthetase.71,72 In the more ordered reduced structure, several new secondary structures condense enabling rigid co-ordination of the Zn. Zn is represented by red spheres in the reduced structure. Oxidized structures are shown on the left, reduced structures on the right.
Disorder-to-order transitions have previously been observed upon binding of ligands.73 Acquisition of order upon binding of ligands concomitant with disulfide formation was apparent for the group B oxidoreductase gdhB, where the disulfide straddled part of the PQQ binding site,74 and the group A RNA sulfuration enzyme EcTrmU, where the disulfide straddles the tRNA-binding site. However, the reverse was true for the Thermotoga maritima tRNA-processing enzyme, Psi55s, where significant disorder of the protein chain concomitant with disulfide reduction occurred upon binding of the tRNA substrate fragment.75 The introduction of disorder in this case may facilitate further co-operative binding of the RNA and protein after the initial docking step.75
Analyses of the composition of disordered sequences have detected amino acid biases from global frequencies. In particular, aromatic amino acids and Cys are depleted in disordered sequences, whereas Lys is enriched.76,77 The disordered sequences in the Redox Pair dataset are also depleted in aromatics, in agreement with more general studies of disordered sequences, but are not depleted in Cys, or enriched in Lys; and thus do not conform entirely to previously detected sequence patterns. Redox Pair disordered sequences are instead enriched in oxygen-bearing sidechains such as Asp, Glu, Ser and Thr.
Some changes in secondary structure are also apparent during the order/disorder transitions. In the oxidized state, both Cys residues generally reside on coil in their proteins, but for a small number of pairs the oxidized form has a secondary structure that “melts” upon reduction of the disulfide. Examples include vitamin D-binding protein78,79 and the E. coli oxidoreductase ghdB,74,80 where helices are lost upon reduction; and the bacteriophage cell entry protein PRD181 where a small β-sheet disappears upon reduction.
The amount of disorder in the chains is also of interest. For most reduced structures, the disorder introduced upon reduction of the disulfide affects less than 10% of the polypeptide chain. However, three of the four group D proteins exhibited large regions of disorder in excess of 20% of the involved protein chain. Interestingly, all were associated with a second more-ordered monomer that was oxidized. For example, the Thermotoga maritima tRNA-processing enzyme, Psi55s undergoes a spectacular loss of secondary structure in one monomer upon binding of RNA.75 It is possible that the fully reduced state of these protein dimers is uncrystallisable and that only partially and fully oxidized oligomers are sufficiently ordered to enable structure solution.
Changes in quaternary structure
We also investigated whether formation of interchain disulfide bonds altered the quaternary state of proteins. Different redox states of the interchain disulfides are associated with two general types of quaternary interaction: those where the number of oligomers differs between the reduced and oxidized state; and those where the number of subunits involved in the oligomer stays constant, but the oligomerization interface changes. Some Redox Pairs exhibit a mixture of the two types of quaternary change. For example, disulfide-bonded dimers of the macrophage sialoadhesin form an interface distinct from that found in trimers formed upon binding of sialosides,82 and may negatively regulate ligand binding.
Twenty-nine Redox Pair protein clusters with intermolecular disulfide bonds exhibit changes in quaternary structure upon oxidation/reduction. Redox regulation of the quaternary state has previously been associated with cooperative behavior in proteins. For example, the HoxB5 protein binds DNA in vitro when either oxidized or reduced but formation of an intramolecular disulfide between HoxB5 monomers is necessary for co-operative binding.83 Other well-known examples are OxyR23 and p53.84 Several other proteins in the dataset exhibit co-operative behavior. Cytoglobin is a member of the vertebrate haemoglobin family residing in the nucleus and cytosol that is upregulated under conditions of hypoxia and certain types of stress (see Fig. 6). It has recently been implicated as a tumor suppressor.88 Intermolecular disulfide bonds in cytoglobin modulate the quaternary structure and may regulate the ligand-binding properties89 (Table IV). The homologous lamphrey haemoglobin is in an equilibrium state between a low O2 affinity oligomer and a high-affinity monomer which contributes to its co-operativity.
Figure 6.
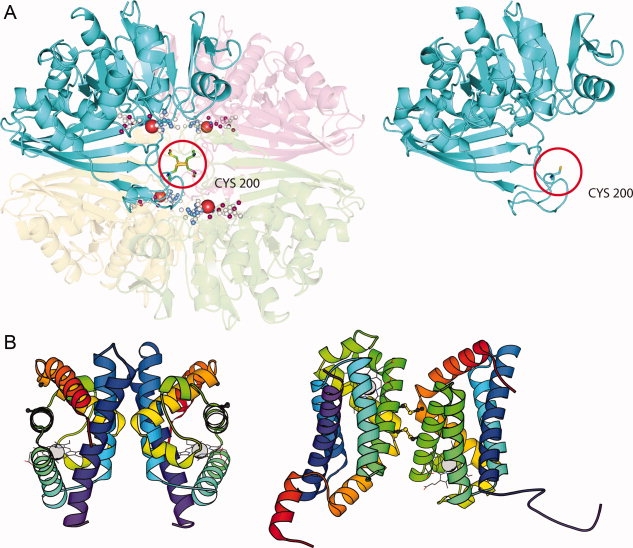
Quaternary changes between different redox states. A: Changes in the number of subunits in the multimer between different redox states of chloroplast GAPDH. The A subunit of spinach chloroplast GAPDH in a reduced, monomeric form (PDB 2hki) is shown on the right and the oxidized homotetramer in complex with NAD+ (PDB 1nbo) is shown on the left. In higher plants, photosynthetic GAPDH exists in vivo mainly as heteromeric isoforms, A2B2 and A8B8 being the most abundant, interconvertible conformations. The B subunit is homologous to the A subunit but has an additional nonhomologous C-terminal extension. Stable homotetramers of A subunits (A4-GAPDH) are found at low levels in chloroplast preparations and may be the only form of photosynthetic GAPDH present in unicellular green algae and cyanobacteria. Calvin cycle enzymes are known, as a group, to be redox regulated.85 The activity of the AB isoform of chloroplast GAPDH is redox regulated by formation of a disulfide between C-terminal thiols in the nonhomologous part of the B chain in light, and its reduction in the dark.86 The interchain disulfide between A subunits may be important for redox control of the A4 isoform, which is known to form complexes with the small chloroplast protein CP12 and phosphoribulosekinase in the dark.87 The disulfide-forming Cys 200 (1nbo numbering) is not present in the B subunit of chloroplast GAPDH which is transcribed from a different gene, or in cytosolic GAPDHs. B: Changes in the oligomeric interface in cytoglobin, a member of the vertebrate haemoglobin family residing in the nucleus and cytosol that is upregulated under conditions of hypoxia and certain types of stress. Intermolecular disulfide bonds in cytoglobin modulate the quaternary structure and may regulate the ligand-binding properties.89 Both the reduced and oxidized states are dimers.
Table IV.
Quaternary Structure Changes Associated with Interchain Disulfide Redox Status
| Protein | Disulfide | Reduced | Oxidized | Function |
|---|---|---|---|---|
| Number of subunits differs between redox states | ||||
| CrRuBisCoL | 247–247a | 8ruc octamer | 1uzd 16mer | CO2 fixation in plants90 |
| NtRuBisCoL | 247–247a | 1ej7 monomer | 4rub tetramer | CO2 fixation in plants90 |
| Spchloroplast GAPDH | 200–200a | 2hki monomer | 1nbo tetramer with NAD | CO2 fixation in plants91 |
| HsGrx | 90–90a | 1bwc, monomer | 2gh5 dimer | Redox homeostasis |
| Aebcp | 80–80a | 2cx3 tetramer | 2cx4 octamer | Redox homeostasis |
| HsPNMT | 48–139a | 1yz3 monomer | 2an5 dimer | Catecholamine biosynthesis, dimer is active |
| MeCbiT | 23–27a | 1kxz octamer | 1l3i hexamer | B12 synthesis, SAM-binding |
| AfDsrC | 114–114a | 1sau monomer | 2a5w trimer | sulfite reductase, [4Fe-4S] |
| HsVEGF | 51–60a | 1bj1 dimer | 1mkg tetramer | Angiogenesis |
| Rnactivin | 90–90a | 2arp monomer | 1nys dimer | TGFβ ligand |
| HsBMP2 | 78–78a | 2goo ternary complex/hexamer | 1rew binary complex/tetramer | TGFβ ligand |
| Hsartemin | 69–69a | 2gyz monomer | 2gyr hexamer | TGFβ ligand92 |
| BPV1-E2 | 340–340a | 2bop dimmer | 1jjh trimer | Viral transcription factor |
| HPV6a-E2 | 295–298a | 2ayb_immer, complex with DNA | 1r8h hexamer | Viral transcription factor |
| FMDVleader protease | 133–133a | 1qmy trimer | 1qol octamer | Viral protease |
| HsAPE-1 | 138–138a | 1de8 dimer, complex with DNA | 1e9n dimer | Repairs oxidative DNA damage |
| MmMacrophage sialoadhesin | 17–17a | 1qfo trimer, complex with sialoadhesin | 2bve dimer | Cell attachment, lectin |
| HsGGA1 | 78–78a | 1py1 tetramer | 1jwg dimer | Sorting and trafficking |
| PsAO | 647–647a | 1ksi dimer | 1w2z tetramer | Oxidoreductase, Pro cis/trans isomerization |
| Spaquaporin | 69–69a | 1z98 dimer/closed | 2b5f tetramer/open | Membrane water pore |
| HsDNA pol III λ | 543–543a | 1xsn monomer | 1xsl tetramer | DNA repair93 |
| Mpmethenyltetrahydrofolate synthetase | 42–42a | 1u3g monomer | 1sbq dimer | Folate biosynthesis |
| Hsgranzyme A | 93–93a | 1orf monomer | 1op8 hexamer | Immunity, apoptosis111 |
| TtEF-Ts | 190–190a | 1tfe monomer | 1aip tetramer | Translation, GEF for EF-Tu, dimer is active112 |
| Dmph sam domain | 32–32a | 1kw4 monomer | 1pk1 dimer | Chromatin protein, development |
| MmGrp1 | 342–342a | 1fgy monomer | 1fhw dimer | Signaling, binds phosphoinositides |
| Oligomerization interface changes between redox states | ||||
| Tdhemerythrin | 9–9a | 1hmd tetramer 1 | 1hmz tetramer 2 | Oxygen transport |
| HsPCBP2 | 24–24a | 2axy tetramer 1 | 1ztg tetramer 2 | Translation regulation |
| Hscytoglobin | 38–83a | 1umo dimer 1 | 2dc3 dimer 2 | Oxygen carrier, upregulated in response to hypoxia |
Italicized prefixes are species abbreviations. Protein names in roman font.
AO, aminooxidase; bcp, bacterial comigratory protein peroxiredoxin; BMP2, bone morphogenetic protein 2; DNA pol III λ, DNA polymerase III λ; EF-Ts, elongation factor Ts; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GEF, guanine nucleotide exchange factor; GGA1, golgi-localized γ adaptin; Grp1, guanine nucleotide exchange factor and integrin binding protein homolog grp1; Grx, glutathione reductase; PCBP2, poly(C)-binding protein-2; pHsam domain, polyhomeotic-proximal chromatin protein sterile alpha motif domain; PNMT, phenylethanolamine N-methyltransferase; RuBisCoL, ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit; SAM-S-adenosyl-l-methionine; VEGF, vascular endothelial growth factor; Ae, Aeropyrum pernix; Af, Archaeoglobus fulgidus; BPV, bovine papilloma virus; Cr, Crithidia fasciculata, Dm, Drosophila melangaster; FMDV, foot and mouth disease virus; HPV, human papilloma virus; Hs, Homo sapiens; Me, Methanobacterium thermoautotrophicum; Mm, Mus musculus; Mp, Mycoplasma pneumoniae; Nt, Nicotiana tabacum; Ps, Pisum sativum (Pea); Rn, Rattus norvegus; Sp, Spinacia oleracea; Td, Themiste dyscritum; Tt, Thermus thermophilus.
2nd Cys in different monomer.
There was also a small group of transmembrane receptors which form covalent dimers in a neck region close to the membrane. These include the low-density oxidized LDL receptor, and the asialoglycoprotein receptor. Covalent dimer formation may promote further quaternary changes such as higher oligomer formation in response to bound ligands as has been recently demonstrated for the metabotropic glutamate receptors.96
Relevance to disease
Importantly, ageing and several major diseases including neurodegenerative diseases such as Alzheimer's disease, type II diabetes, cancer and cardiovascular disease have been associated with abnormal redox conditions. Inadvertent triggering of redox-active disulfide switches is a likely contributor to these phenotypes. This view is supported by the association of several Redox Pairs in the dataset with disease. SOD1 (also known as CuZn-SOD) is an enzyme mutated in familial lateral sclerosis (FALs), an age-dependent degenerative disorder of motor neurons in the spinal cord, brain stem and brain. SOD1 is largely localized to the cytosol but is also found in the intermembrane space of mitochondria. SOD1 is activated by formation of the disulfide and insertion of Cu by the copper carrier CCS. Both of these proteins are represented in the Redox Pair dataset (Tables I and S2). The conserved disulfide in SOD1 is essential for activity and unusual because it remains oxidized in the cytosol. Although the disulfide is intrachain, it is thought to play a critical role in stabilizing the SOD1 homodimer. Formation of the disulfide is part of the SOD1 maturation process which is regulated by physiological oxidative stress. The three reduced proteins in the dataset are three distinct mutants associated with familial lateral sclerosis (FALs): G37R, H46R, and I113T. The exclusive association of the reduced proteins with the disease mutants suggests redox imbalance of this disulfide is implicated in the disease. This hypothesis is supported by the finding that 14 FALs mutants have the common property of being more susceptible to disulfide reduction than wild-type protein.97 Toxicity associated with disulfide-linked multimerization of SOD1 mutants has recently been implicated in the disease mechanism.98
Discussion
Several types of conformational change were observed in the Redox Pair dataset suggesting there may be common structural modes of redox regulation. Proteins exhibited disulfide oxidation following expulsion of metals such as Zn, morphing, order/disorder transitions, and changes in quaternary structure. In an individual protein more than one mode may be employed. For example in Leu-tRNA synthetase, part of the protein chain becomes disordered upon Zn expulsion but this is not the case for all proteins with labile Zn.
Redox regulation of proteins that form disulfides following expulsion of Zn is an emerging area of signaling. Well known and characterized examples including Hsp33 which is activated by Zn expulsion as part of the oxidative stress response99; and the small Tim proteins which are kept in a transport-competent state in the cytosol by conjugation with Zn.41 Upon delivery to mitochondria, Zn is expelled from small Tim proteins which are activated by disulfide formation. Zn is not expelled from all protein Zn-binding sites with equal facility, and is probably not expelled from some sites at all under physiologic conditions. For structural Zn sites an analogous situation may exist to the bipartite distribution of redox-active and structural disulfides in proteins, with labile Zn sites being redox-active and inert Zn sites corresponding to the purely structural group. Key to Zn expulsion under oxidizing conditions is the presence of oxidation-prone Cys as a Zn ligand. Sites with more Cys are likely to be more facile. Based on the number and nature of ligands, the Zn sites detected in the Redox Pair dataset likely belong to the redox-active group.
The repeated association of forbidden disulfides of the BDD type with labile Zn binding sites is particularly interesting because of the recent characterization of the oxidative process that leads to activation of the bacterial chaperone Hsp33.99 The presence of the Zn center keeps Hsp33 monomeric and functionally inactive. Upon exposure to elevated temperatures, the C-terminal domain swings away from the protein's center of mass, exposing the Zn centre. The N-terminal thiols in the Zn site, Cys 232 and Cys 234, are preferentially oxidatively modified. The remaining two Cys, 269 and 272, form a disulfide of the BDD type, releasing Zn and unfolding the C-terminal domain. Unfolding the C-terminus removes a steric barrier to dimerization, activating the protein's chaperone function. The function of disulfide formation between Cys 269 and 272 in the Zn binding site has not been fully delineated. Potential benefits are protection from other oxidation modifications and restriction of the conformational freedom of the protein chain allowing easier refolding upon return of the protein's milieu to normal conditions. However, these benefits apply to any disulfide and the repeated appearance of the BDD in labile Zn sites of proteins of the Redox Pair dataset suggests a more specific function, possibly recognition by a foldase or metallochaperone. We hypothesize the forbidden disulfide would become stressed upon condensation of the β-sheet in its vicinity,12 possibly allowing easier re-insertion of Zn. Without any detailed experiments as a guide, the significance of the other repeated forbidden disulfide motif, the JSD, is not clear, but the same arguments relating to its forbidden disulfide nature would apply. The JSD-containing proteins MetH and BHMT are known to be reversibly inactivated by disulfide formation.100 In vitro, BHMT can be reactivated by dialysis against DTT and Zn,101 but the specific method of reactivation in vivo has not been characterized.
For most structures exhibiting order/disorder transitions in the dataset, the reduced structure is associated with more disorder than the oxidized structure. Energetically, formation of a disulfide bond between Cys residues would have a favorable entropic contribution to the stability of the chain. A notable exception is Leu-tRNA synthetase where the more ordered reduced structure binds Zn (Fig. 5(B)). This may be generally true for order/disorder transitions associated with labile Zn sites: the Zn-bound state is entropically favored over a single disulfide because the polypeptide chain is constrained at three or four points instead of two.40
A subset of Redox Pair proteins exhibiting order/disorder transitions correlated with disulfide redox status contain regions of disorder in excess of 20% of the protein chain. All the proteins in this group exist as dimers with the other monomer being more ordered. The disordered monomers in these class D structures may correspond to the Molten Globule state. For these proteins, the Molten Globule state adopted may be physiologically relevant to their function. Molten Globules are collapsed forms of the protein chain which have some native-like secondary structure but a dynamic tertiary structure as seen by far and near circular dichroism spectroscopy, respectively. The Radius of Gyration of proteins in the Molten Globule state is ∼10% larger than the native state but precise tertiary structures at atomic resolution have not yet been obtained for commonly studied Molten Globule proteins such as lactoglobulin A. However, one of the proteins in group D: Platelet Factor 4, which like hepatocyte growth factor, binds heparin and regulates angiogenesis, has been shown to adopt the Molten Globule state upon reduction of its disulfides.102 It seems likely proteins in group D adopt a Molten Globule state in vivo but can also adopt more ordered states upon binding of appropriate ligands. Several of the proteins bind large polymers such as RNA and heparin and the order/disorder transitions may mediate co-operative behavior between monomers.
General studies of disordered sequences suggest there may be several different types of disordered sequence, three of which have been previously detected using an automatic classifier.103 The oxygen-rich sequences of the disordered regions of proteins of the Redox Pair dataset seem to represent a novel type of disordered sequence not previously recognized. Oxygen-rich sequences may confer bistable switch-like properties on the sequence: an oxygen-rich environment stabilizes the disulfide in the oxidized state by increasing its pKa104; while the subset of negatively charged Asp and Glu residues in oxygen-rich sequences may contribute to disorder in the reduced state via like-charge repulsion. Disordered regions are known to undergo post-translational modifications including acetylation, glycosylation, methylation, and phosphorylation which may regulate the order/disorder transition.73 Redox-activity of resident Cys residues should be added to this list of post-translational modifications.
Perhaps the most astonishing conformational changes associated with disulfide reduction are the morphing transitions. The current paradigm, due to Anfinsen, is that the primary sequence of a protein encodes a unique three-dimensional structure. Various discoveries that have challenged this notion include the spectacular pH-induced conformational change in influenza A haemagglutinin (HA),105 molecular chaperones, and the previously discussed disordered sequences. All of these discoveries have been difficult to reconcile with the concept that protein sequences adopt a unique minimally frustrated fold. HA is believed to be kinetically trapped in a higher energy fold by an N-terminal domain which is subsequently cleaved, allowing it to cascade into a minimally frustrated fold. The conformations of disordered sequences appear to be principally governed by entropic factors: weak sequence preferences for a variety of conformational substates or modes appear to be co-selected by ligands. Morphing structures appear to be a new type of challenge to the concept of a unique minimally frustrated fold. For morphing sequences, the structure appears to be dependent on the environment. Unlike influenza A HA, where kinetic trapping during the folding process clearly plays a part in the structure adopting two different folds, for morphing sequences the two folds are energetic minima for the sequence in a particular environment. As such structural changes are reversible and dependent on environment.
We were previously aware of two instances where subdomain morphing of proteins has been associated with reversible disulfide reduction: a redox-controlled structural reorganization of the ion channel CLIC1 proposed to regulate its insertion into membranes,18 and sequential oxidation of the transcription factor OxyR in response to oxidative stress which modulates its quaternary structure and DNA-binding properties.23 Here we showed that morphing is a common type of major structural change associated with disulfide reduction. The most spectacular morphing transition is that of CLIC1, where an entire subdomain of the protein rearranges in response to different redox conditions. Several Redox Pairs were found exhibiting large morphing conformational transitions of similar magnitude to OxyR, supporting the notion that these types of transitions may be reasonably common. Large separations of the Cys residues in the reduced state are a typical feature of these proteins, and do not preclude disulfide linkages forming. Possibly attack by a helper molecule, such as glutathione, may be required as an intermediate in disulfide formation to bridge the intervening distance. Large physical separation of partner Cys residues may represent an example of negative design against inadvertent disulfide formation under mild oxidizing conditions.
Morphing transitions are not limited to changes in disulfide redox status. Other morphing transitions not associated with disulfide redox state have been noted in Mad2 and lymphotactin.106–108 pH and redox are probably only two of multiple environmental changes associated with structural morphing. The relative importance of these and other environmental changes in protein morphing remain to be determined.
Morphing transitions may also have some relevance to evolutionary bridging states. In 1995, based on structures of Arc repressor, Sauer proposed the existence of evolutionary bridging states. A substructure in wild-type Arc repressor adopts a β-strand conformation which forms a two-stranded β-sheet with the adjacent monomer. In the N11L mutant of Arc repressor, the substructure was shown to fluctuate between the native state and a helical state on a millisecond time scale. A double mutant known as “switch Arc” (N11L, L12N) was shown to be stabilized in the alternate helical conformation. Sauer proposed the fluctuating N11L mutant represented an evolutionary bridge between two stable low energy folds. Proteins that can adopt two different folds from the same sequence could evolve two different functions upon gene duplication and mutation hence acting as an evolutionary bridge between two different folds.109 It is clear morphing proteins could also act as bridge states: deselection of one state over the other by point mutation may lead to loss of the ability to morph in the daughters resulting in proteins with different but related folds.
Although we have concentrated solely on disulfide reduction in this study, several anecdotal examples of disulfide isomerization should be mentioned in the context of the aforementioned discussion. A second example of an evolutionary bridging state was proposed based on a study of cnidarian collagen. The N-terminal domain of minicollagen 1 has 44% sequence identity with the C-terminal domain, but the structures show a different disulfide bonding pattern and fold.110 At this stage it is not clear if the two forms are interconvertible or represent true one-state endpoints evolved from an evolutionary bridging state. A different example where major structural organization is regulated by disulfide isomerization is the activation of P1 lysozyme.111 This conformational change is irreversible and associated with release of a hydrophobic transmembrane sequence by membrane depolarization, suggesting the latent state may be a kinetically trapped state rather than a true morphing protein.
Conclusions
Our study mined pairs of protein structures which contain disulfides in both reduced and oxidized states and represents the first systematic compilation of such a dataset. Quaternary changes, morphing and expulsion of transition metals, particularly Zn, have previously been associated with redox-related changes in proteins and here we showed these conformational changes are more common than anecdotal examples suggest. To our knowledge order/disorder transitions have not previously been linked with redox changes but also seem to be a common type of conformational change. Like morphing transitions, changing redox conditions appear to be only one of many mechanisms regulating order/disorder transitions. The oxygen-rich sequences of the disordered regions of proteins of the Redox Pair dataset seem to be a novel type of disordered sequence not previously recognized. Finally, two forbidden disulfide motifs are associated with redox-regulated Zn binding here for the first time.
The physiological significance of redox-controlled structural changes is only beginning to be appreciated. Previously, all oxidative changes were thought to be irreversible and damaging. It is now known that redox signaling pathways can modulate homeostatic and adaptive responses. However, continued stimulation of adaptive responses may lead to maladaptive responses. The Redox Pair dataset is a valuable resource for the investigation of physiological and pathological processes associated with disulfide redox activity.
Materials and Methods
Identifying redox protein-pairs in the PDB
The PDB16 was mined for pairs of structures with Cys in different redox states. First, all disulfide-containing structures were identified by querying their DSSP records,112 as SSBOND records in the PDB header were found to be unreliable. For each disulfide the atomic residue positions were mapped to the sequence position. The connectivity of the disulfides in the sequence was then used to assign a unique identifier, dMN score [Eq. (1)], to each protein.113 The dMN score allows direct comparison of the disulfide connectivity between structures as structures with the same disulfide bonding will have the same dMN score.
| (1) |
where i and j are the sequence positions of disulfide-bonded Cys.
All protein sequences in the PDB were clustered at 95% sequence identity using BlastClust.114 The 95% threshold was chose to capture the same protein with some natural and engineered mutations allowed. Some highly conserved orthologs from closely related species were also captured in the same cluster, for example, the NF-κB orthologs in human and mouse. Proteins within each group were compared to detect subgroups with different dMN scores-indicating alternative disulfide connectivity. For fully oxidized proteins, a set of “pseudo-reduced” dMNs were calculated by iteratively ignoring one or more disulfides. The set of pseudo-reduced dMNs for the oxidized protein were matched against dMNs for the reduced proteins in the dataset to select Redox Pairs. As an additional control, proteins were split into two groups based on whether some or all disulfide bonds in the protein were reduced. Proteins in which Cys residues were engineered into proteins were filtered from the list.
Analysis of gene ontology term enrichment
GO terms that are over and under-represented in the oxidized proteins in the Redox-Pairs dataset compared to the PDB as a whole were identified using Fisher's Exact Test, in the same way as applied in tools that analyze gene-expression data.115 GO annotations for the entire PDB were taken from the GOA resource, downloaded from the EBI.28
To ensure a fair comparison, the GO annotations of a nonredundant set of the oxidized structures (95% sequence identity) were compared to GO annotations of a nonredundant PDB data set (95% sequence identity). To improve overall annotation coverage, a representative PDB structure is assigned all the GO terms belonging to the PDBs within its cluster group.
Because GO is a hierarchical data structure, GO terms are set to a single level in the hierarchy to enable a fair comparison. Here we used GO level three. At this level there is a compromise between information quality and the number of annotations available.116 Where a GO term is below this level we move up the hierarchy until we reach the third level GO term(s). GO terms above level three cannot be used.
A P-value was calculated representing the probability that the observed number of each GO term (n) could have resulted from randomly selecting this GO term in the PDB as a whole (N). Fisher's Exact test was used in order to approximate this P-value. GO terms that are most specific to the Redox Pairs set will have P-values near zero, and those that are under-represented will be near one.
Analysis of protein conformational change
Protein conformational changes were examined using a variety of complementary approaches. We used ProFit (www.bioinf.org.uk/software/profit) to search for large conformational changes such as hinge movements. ProFit superimposes two structures using Cα atoms and was used to calculate RMSD and distances between residues. Large movements were indicated by the change in distance between the sulfur groups of the Cys residues in a disulfide.
Because RMSD is averaged over the entire molecule, large changes confined to a subdomain of protein may not be obvious using RMSD. Large conformational changes between structure-pairs corresponding to rearrangements of the polypeptide backbone were also identified using an implementation of a method by Flocco and Mowbray.117 For pairs of structures, the method compares the change in a pseudo-torsional angles defined by four consecutive Cα-atoms. Conformational changes were identified by running a smoothing window (five residues long) over the torsional-difference plot. Stretches of contiguous residues with seven or more pseudo-dihedral angles that exhibited individual movements in excess of 20° were initially filtered from the dataset. These conformational changes were further classified by comparing DSSP annotations, solvent accessibility and secondary structure, between the protein pairs. Proteins with conformational changes where the summed changes in the pseudo-torsional angles exceeded a threshold of 1000 degrees with accompanying secondary structure changes were classified as morphing. The presence or absence of multiple chains in the PDB structure could cause errors when calculating changes in solvent accessibility; therefore, DSSP was run on individual chains only. All solvent accessibility measures were normalized using values derived by Chothia.118 Each amino acid was assigned to one of two states; core or exposed. Core residues are those with a solvent accessibility of less than 10% and exposed residues are those with a solvent accessibility equal to 10% or more. Secondary structures were assigned to one of three states: helix, strand and coil.
Acknowledgments
The authors thank Dr. Kieran Scott for interesting discussions. Disulfide torsional energy calculations were performed at the NCI National Facility.
References
- 1.Yang Y, Song Y, Loscalzo J. Regulation of the protein disulfide proteome by mitochondria in mammalian cells. Proc Natl Acad Sci USA. 2007;104:10813–10817. doi: 10.1073/pnas.0702027104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buchanan BB, Balmer Y. Redox regulation: a broadening horizon. Annu Rev Plant Biol. 2005;56:187–220. doi: 10.1146/annurev.arplant.56.032604.144246. [DOI] [PubMed] [Google Scholar]
- 3.Ahamed J, Versteeg HH, Kerver M, Chen VM, Mueller BM, Hogg PJ, Ruf W. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci USA. 2006;103:13932–13937. doi: 10.1073/pnas.0606411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matthias LJ, Yam PTW, Jiang XM, Vandegraaff N, Li P, Poumbourios P, Donoghue N, Hogg PJ. Disulfide exchange in domain 2 of CD4 is required for entry of HIV-I. Nat Immunol. 2002;3:727–732. doi: 10.1038/ni815. [DOI] [PubMed] [Google Scholar]
- 5.Park B, Lee S, Kim E, Cho K, Riddell SR, Cho S, Ahn K. Redox regulation facilitates optimal peptide selection by MHC class I during antigen processing. Cell. 2006;127:369–382. doi: 10.1016/j.cell.2006.08.041. [DOI] [PubMed] [Google Scholar]
- 6.Huber-Wunderlich M, Glockshuber R. A single dipeptide sequence modulates the redox properties of a whole enzyme family. Fold Des. 1998;3:161–171. doi: 10.1016/S1359-0278(98)00024-8. [DOI] [PubMed] [Google Scholar]
- 7.Krause G, Holmgren A. Substitution of the conserved tryptophan-31 in Escherichia coli thioredoxin by site-directed mutagenesis and structure-function analysis. J Biol Chem. 1991;266:4056–4066. [PubMed] [Google Scholar]
- 8.Lin TY, Kim PS. Urea dependence of thiol disulfide equilibria in thioredoxin—confirmation of the linkage relationship and a sensitive assay for structure. Biochemistry. 1989;28:5282–5287. doi: 10.1021/bi00438a054. [DOI] [PubMed] [Google Scholar]
- 9.Wunderlich M, Glockshuber R. Redox properties of protein disulfide isomerase (DsbA) from Escherichia coli. Protein Sci. 1993;2:717–726. doi: 10.1002/pro.5560020503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilbert HF. Molecular and cellular aspects of thiol disulfide exchange. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 11.Wiita AP, Ainavarapu SRK, Huang HH, Fernandez JM. Force-dependent chemical kinetics of disulfide bond reduction observed with single-molecule techniques. Proc Natl Acad Sci USA. 2006;103:7222–7227. doi: 10.1073/pnas.0511035103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wouters MA, George RA, Haworth NL. “Forbidden” disulfides: their role as redox switches. Curr Protein Pept Sci. 2007;8:484–495. doi: 10.2174/138920307782411464. [DOI] [PubMed] [Google Scholar]
- 13.Alphey MS, Gabrielsen M, Micossi E, Leonard GA, McSweeney SM, Ravelli RBG, Tetaud E, Fairlamb AH, Bond CS, Hunter WN. Tryparedoxins from Crithidia fasciculata and Trypanosoma brucei - photoreduction of the redox disulfide using synchrotron radiation and evidence for a conformational switch implicated in function. J Biol Chem. 2003;278:25919–25925. doi: 10.1074/jbc.M301526200. [DOI] [PubMed] [Google Scholar]
- 14.Roberts BR, Wood ZA, Jonsson TJ, Poole LB, Karplus PA. Oxidized and synchrotron cleaved structures of the disulfide redox center in the N-terminal domain of Salmonella typhimurium AhpF. Protein Sci. 2005;14:2414–2420. doi: 10.1110/ps.051459705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weik M, Ravelli RBG, Kryger G, McSweeney S, Raves ML, Harel M, Gros P, Silman I, Kroon J, Sussman JL. Specific chemical and structural damage to proteins produced by synchrotron radiation. Proc Natl Acad Sci USA. 2000;97:623–628. doi: 10.1073/pnas.97.2.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Messens J, Martins JC, Van Belle K, Brosens E, Desmyter A, De Gieter M, Wieruszeski JM, Willem R, Wyns L, Zegers I. All intermediates of the arsenate reductase mechanism, including an intramolecular dynamic disulfide cascade. Proc Natl Acad Sci USA. 2002;99:8506–8511. doi: 10.1073/pnas.132142799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Littler DR, Harrop SJ, Fairlie WD, Brown LJ, Pankhurst GJ, Pankhurst S, DeMaere MZ, Campbell TJ, Bauskin AR, Tonini R, Mazzanti M, Breit SN, Curmi PMG. The intracellular chloride ion channel protein CLIC1 undergoes a redox-controlled structural transition. J Biol Chem. 2004;279:9298–9305. doi: 10.1074/jbc.M308444200. [DOI] [PubMed] [Google Scholar]
- 19.Bienert GP, Schjoerring JK, Jahn TP. Membrane transport of hydrogen peroxide. Biochim Biophys Acta. 2006;1758:994–1003. doi: 10.1016/j.bbamem.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 20.Conour JE, Graham WV, Gaskins HR. A combined in vitro/bioinformatic investigation of redox regulatory mechanisms governing cell cycle progression. Physiol Genomics. 2004;18:196–205. doi: 10.1152/physiolgenomics.00058.2004. [DOI] [PubMed] [Google Scholar]
- 21.Sun JP, Wang WQ, Yang H, Liu S, Liang F, Fedorov AA, Almo SC, Zhang ZY. Structure and biochemical properties of PRL-1, a phosphatase implicated in cell growth, differentiation, and tumor invasion. Biochemistry. 2005;44:12009–12021. doi: 10.1021/bi0509191. [DOI] [PubMed] [Google Scholar]
- 22.Buhrman G, Parker B, Sohn J, Rudolph J, Mattos C. Structural mechanism of oxidative regulation of the phosphatase Cdc25B via an intramolecular disulfide bond. Biochemistry. 2005;44:5307–5316. doi: 10.1021/bi047449f. [DOI] [PubMed] [Google Scholar]
- 23.Choi HJ, Kim SJ, Mukhopadhyay P, Cho S, Woo JR, Storz G, Ryu SE. Structural basis of the redox switch in the OxyR transcription factor. Cell. 2001;105:103–113. doi: 10.1016/s0092-8674(01)00300-2. [DOI] [PubMed] [Google Scholar]
- 24.Mao SS, Holler TP, Yu GX, Bollinger JM, Booker S, Johnston MI, Stubbe J. A model for the role of multiple cysteine residues involved in ribonucleotide reduction—amazing and still confusing. Biochemistry. 1992;31:9733–9743. doi: 10.1021/bi00155a029. [DOI] [PubMed] [Google Scholar]
- 25.Parsonage D, Karplus PA, Poole LB. Substrate specificity and redox potential of AhpC, a bacterial peroxiredoxin. Proc Natl Acad Sci USA. 2008;105:8209–8214. doi: 10.1073/pnas.0708308105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adam V, Royant A, Nivière V, Molina-Heredia FP, Bourgeois D. Structure of superoxide reductase bound to ferrocyanide and active site expansion upon X-ray-induced photo-reduction. Structure. 2004;12:1729–1740. doi: 10.1016/j.str.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 27.Wouters MA, Lau KK, Hogg PJ. Cross-strand disulphides in cell entry proteins: poised to act. BioEssays. 2004;26:73–79. doi: 10.1002/bies.10413. [DOI] [PubMed] [Google Scholar]
- 28.Camon E, Magrane M, Barrell D, Lee V, Dimmer E, Maslen J, Binns D, Harte N, Lopez R, Apweiler R. The Gene Ontology Annotation (GOA) Database: sharing knowledge in Uniprot with Gene Ontology. Nucleic Acids Res. 2004;32:D262–D266. doi: 10.1093/nar/gkh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conant CG, Stephens RS. Chlamydia attachment to mammalian cells requires protein disulfide isomerase. Cell Microbiol. 2007;9:222–232. doi: 10.1111/j.1462-5822.2006.00783.x. [DOI] [PubMed] [Google Scholar]
- 30.Jain S, McGinnes LW, Morrison TG. Thiol/disulfide exchange is required for membrane fusion directed by the Newcastle disease virus fusion protein. J Virol. 2006;81:2328–2339. doi: 10.1128/JVI.01940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gallina A, Hanley TM, Mandel R, Trahey M, Broder CC, Viglianti GA, Ryser HJP. Inhibitors of protein-disulfide isomerase prevent cleavage of disulfide bonds in receptor-bound glycoprotein 120 and prevent HIV-1 entry. J Biol Chem. 2002;277:50579–50588. doi: 10.1074/jbc.M204547200. [DOI] [PubMed] [Google Scholar]
- 32.Barbouche R, Miquelis R, Jones IM, Fenouillet E. Protein-disulfide isomerase-mediated reduction of two disulfide bonds of HIV envelope glycoprotein 120 occurs post-CXCR4 binding and is required for fusion. J Biol Chem. 2003;278:3131–3136. doi: 10.1074/jbc.M205467200. [DOI] [PubMed] [Google Scholar]
- 33.Katz BA, Kossiakoff A. The crystallographically determined structures of atypical strained disulfides engineered into subtilisin. J Biol Chem. 1986;261:5480–5485. [PubMed] [Google Scholar]
- 34.Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, Profeta S, Weiner P. A new force-field for molecular mechanical simulation of nucleic-acids and proteins. J Am Chem Soc. 1984;106:765–784. [Google Scholar]
- 35.Wells JA, Powers DB. In vivo formation and stability of engineered disulfide bonds in subtilisin. J Biol Chem. 1986;261:6564–6570. [PubMed] [Google Scholar]
- 36.Wetzel R. Harnessing disulfide bonds using protein engineering. Trends Biochem Sci. 1987;12:478–482. [Google Scholar]
- 37.Richardson JS. The anatomy and taxonomy of protein structure. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 38.Thornton JM. Disulfide bridges in globular proteins. J Mol Biol. 1981;151:261–287. doi: 10.1016/0022-2836(81)90515-5. [DOI] [PubMed] [Google Scholar]
- 39.Numata T, Ikeuchi Y, Fukai S, Suzuki T, Nureki O. Snapshots of tRNA sulphuration via an adenylated intermediate. Nature. 2006;442:419–424. doi: 10.1038/nature04896. [DOI] [PubMed] [Google Scholar]
- 40.Maret W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid Redox Signal. 2006;8:1419–1441. doi: 10.1089/ars.2006.8.1419. [DOI] [PubMed] [Google Scholar]
- 41.Mesecke N, Terziyska N, Kozany C, Baumann F, Neupert W, Hell K, Herrmann JM. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell. 2005;121:1059–1069. doi: 10.1016/j.cell.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 42.Evans JC, Huddler DP, Jiracek J, Castro C, Millian NS, Garrow TA, Ludwig ML. Betaine-homocysteine methyltransferase: zinc in a distorted barrel. Structure. 2002;10:1159–1171. doi: 10.1016/s0969-2126(02)00796-7. [DOI] [PubMed] [Google Scholar]
- 43.Rouhier N, Villarejo A, Srivastava M, Gelhaye E, Keech O, Droux M, Finkemeier I, Samuelsson G, Dietz KJ, Jacquot JP. Identification of plant glutaredoxin targets. Antioxid Redox Signal. 2005;7:919–929. doi: 10.1089/ars.2005.7.919. and others. [DOI] [PubMed] [Google Scholar]
- 44.Leichert LI, Jakob U. Protein thiol modifications visualized in vivo. PLoS Biol. 2004;2:1723–1737. doi: 10.1371/journal.pbio.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Auerbach G, Herrmann A, Bracher A, Bader G, Gutlich M, Fischer M, Neukamm M, Garrido-Franco M, Richardson J, Nar H, Huber R, Bacher A. Zinc plays a key role in human and bacterial GTP cyclohydrolase I. Proc Natl Acad Sci USA. 2000;97:13567–13572. doi: 10.1073/pnas.240463497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hansen JM, Go Y-M, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu Rev Pharmacol Toxicol. 2006;46:215–234. doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- 47.Rosenzweig AC, Huffman DL, Hou MY, Wernimont AK, Pufahl RA, O'Halloran TV. Crystal structure of the Atx1 metallochaperone protein at 1.02 Å resolution. Structure. 1999;7:605–617. doi: 10.1016/s0969-2126(99)80082-3. [DOI] [PubMed] [Google Scholar]
- 48.Dimasi N, Pasquo A, Martin F, Di Marco S, Steinkuhler C, Cortese R, Sollazzo M. Engineering, characterization and phage display of hepatitis C virus NS3 protease and NS4A cofactor peptide as a single-chain protein. Protein Eng Des Sel. 1998;11:1257–1265. doi: 10.1093/protein/11.12.1257. [DOI] [PubMed] [Google Scholar]
- 49.Miyanaga A, Fushinobu S, Ito K, Shoun H, Wakagi T. Mutational and structural analysis of cobalt-containing nitrile hydratase on substrate and metal binding. Eur J Biochem. 2004;271:429–438. doi: 10.1046/j.1432-1033.2003.03943.x. [DOI] [PubMed] [Google Scholar]
- 50.Li H, Raman CS, Glaser CB, Blasko E, Young TA, Parkinson JF, Whitlow M, Poulos TL. Crystal structures of Zinc-free and -bound heme domain of human inducible nitric-oxide synthase: implications for dimer stability and comparison with endothelial nitric-oxide synthase. J Biol Chem. 1999;274:21276–21284. doi: 10.1074/jbc.274.30.21276. [DOI] [PubMed] [Google Scholar]
- 51.Black S. Reversible interconversion of two forms of a valyl-tRNA synthetase-containing protein complex. Science. 1986;234:1111–1114. doi: 10.1126/science.3535073. [DOI] [PubMed] [Google Scholar]
- 52.Lindahl M, Florencio FJ. Thioredoxin-linked processes in cyanobacteria are as numerous as in chloroplasts, but targets are different. Proc Natl Acad Sci USA. 2003;100:16107–16112. doi: 10.1073/pnas.2534397100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kullik I, Toledano MB, Tartaglia LA, Storz G. Mutational analysis of the redox-sensitive transcriptional regulator OxyR: regions important for oxidation and transcriptional activation. J Bacteriol. 1995;177:1275–1284. doi: 10.1128/jb.177.5.1275-1284.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baena-Gonzalez E, Baginsky S, Mulo P, Summer H, Aro E-M, Link G. Chloroplast transcription at different light intensities. Glutathione-mediated phosphorylation of the major RNA polymerase involved in redox-regulated organellar gene expression. Plant Physiol. 2001;127:1044–1052. doi: 10.1104/pp.010168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang X, Mukhopadhyay P, Wood MJ, Outten FW, Opdyke JA, Storz G. Mutational analysis to define an activating region on the redox-sensitive transcriptional Regulator OxyR. J Bacteriol. 2006;188:8335–8342. doi: 10.1128/JB.01318-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vallee BL, Auld DS. Zinc coordination, function, and structure of zinc enzymes and other proteins. Biochemistry. 1990;29:5647–5659. doi: 10.1021/bi00476a001. [DOI] [PubMed] [Google Scholar]
- 57.Wouters MA, Husain A. Changes in zinc ligation promote remodeling of the active site in the zinc hydrolase superfamily. J Mol Biol. 2001;314:1191–1207. doi: 10.1006/jmbi.2000.5161. [DOI] [PubMed] [Google Scholar]
- 58.Maynard AT, Covell DG. Reactivity of zinc finger cores: analysis of protein packing and electrostatic screening. J Am Chem Soc. 2001;123:1047–1058. doi: 10.1021/ja0011616. [DOI] [PubMed] [Google Scholar]
- 59.Hofmann A, Grella M, Botos I, Filipowicz W, Wlodawer A. Crystal structures of the semireduced and inhibitor-bound forms of cyclic nucleotide phosphodiesterase from Arabidopsis thaliana. J Biol Chem. 2002;277:1419–1425. doi: 10.1074/jbc.M107889200. [DOI] [PubMed] [Google Scholar]
- 60.Kim SO, Merchant K, Nudelman R, Beyer WF, Keng T, DeAngelo J, Hausladen A, Stamler JS. OxyR: a molecular code for redox-related signaling. Cell. 2002;109:383–396. doi: 10.1016/s0092-8674(02)00723-7. [DOI] [PubMed] [Google Scholar]
- 61.Haworth NL, Gready JE, George RA, Wouters MA. Evaluating the stability of disulfide bridges in proteins: a torsional potential energy surface for diethyl disulfide. Mol Simul. 2007;33:475–485. doi: 10.1080/08927020701361876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chiadmi M, Navaza A, Miginiac-Maslow M, Jacquot JP, Cherfils J. Redox signalling in the chloroplast: structure of oxidized pea fructose-1,6-bisphosphate phosphatase. EMBO J. 1999;18:6809–6815. doi: 10.1093/emboj/18.23.6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wenderoth I, Scheibe R, vonSchaewen A. Identification of the cysteine residues involved in redox modification of plant plastidic glucose-6-phosphate dehydrogenase. J Biol Chem. 1997;272:26985–26990. doi: 10.1074/jbc.272.43.26985. [DOI] [PubMed] [Google Scholar]
- 64.Xu S-Z, Sukumar P, Zeng F, Li J, Jairaman A, English A, Naylor J, Ciurtin C, Majeed Y, Milligan CJ, Bahnasi YM, Al-Shawaf E, Porter KE, Jiang L-H, Emery P, Sivaprasadarao A, Beech DJ. TRPC channel activation by extracellular thioredoxin. Nature. 2008;451:69–72. doi: 10.1038/nature06414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crow A, Acheson RM, Le Brun NE, Oubrie A. Structural basis of redox-coupled protein substrate selection by the cytochrome c biosynthesis protein ResA. J Biol Chem. 2004;279:23654–23660. doi: 10.1074/jbc.M402823200. [DOI] [PubMed] [Google Scholar]
- 66.Toral-Barza L, Zhang WG, Huang X, McDonald LA, Salaski EJ, Barbieri LR, Ding WD, Krishnamurthy G, Yong BH, Lucas J, Bernan VS, Cai P, Levin JI, Mansour TS, Gibbons JJ, Abraham RT, Yu K. Discovery of lactoquinomycin and related pyranonaphthoquinones as potent and allosteric inhibitors of AKT/PKB: mechanistic involvement of AKT catalytic activation loop cysteines. Mol Cancer Ther. 2007;6:3028–3038. doi: 10.1158/1535-7163.MCT-07-0211. [DOI] [PubMed] [Google Scholar]
- 67.Huang X, Begley M, Morgenstern KA, Gu Y, Rose P, Zhao H, Zhu X. Crystal structure of an inactive Akt2 kinase domain. Structure. 2003;11:21–30. doi: 10.1016/s0969-2126(02)00937-1. [DOI] [PubMed] [Google Scholar]
- 68.Chen W-J, Lee IS, Chen C-Y, Liao T-H. Biological functions of the disulfides in bovine pancreatic deoxyribonuclease. Protein Sci. 2004;13:875–883. doi: 10.1110/ps.03438204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Korolainen MA, Nyman TA, Nyyssonen P, Hartikainen ES, Pirttila T. Multiplexed proteomic analysis of oxidation and concentrations of cerebrospinal fluid proteins in Alzheimer disease. Clin Chem. 2007;53:657–665. doi: 10.1373/clinchem.2006.078014. [DOI] [PubMed] [Google Scholar]
- 70.Leslie NR. The redox regulation of PI 3-kinase-dependent signaling. Antioxid Redox Signal. 2006;8:1765–1774. doi: 10.1089/ars.2006.8.1765. [DOI] [PubMed] [Google Scholar]
- 71.Lincecum TL, Tukalo M, Yaremchuk A, Mursinna RS, Williams AM, Sproat BS, Van Den Eynde W, Link A, Van Calenbergh S, Grøtli M, Martinis SA, Cusack S. Structural and mechanistic basis of pre- and posttransfer editing by leucyl-tRNA synthetase. Mol Cell. 2003;11:951–963. doi: 10.1016/s1097-2765(03)00098-4. [DOI] [PubMed] [Google Scholar]
- 72.Tukalo M, Yaremchuk A, Fukunaga R, Yokoyama S, Cusack S. The crystal structure of leucyl-tRNA synthetase complexed with tRNALeu in the post-transfer-editing conformation. Nat Struct Mol Biol. 2005;12:923–930. doi: 10.1038/nsmb986. [DOI] [PubMed] [Google Scholar]
- 73.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 74.Oubrie A, Rozeboom HJ, Kalk KH, Olsthoorn AJJ, Duine JA, Dijkstra BW. Structure and mechanism of soluble quinoprotein glucose dehydrogenase. EMBO J. 1999;18:5187–5194. doi: 10.1093/emboj/18.19.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Phannachet K, Huang RH. Conformational change of pseudouridine 55 synthase upon its association with RNA substrate. Nucleic Acids Res. 2004;32:1422–1429. doi: 10.1093/nar/gkh287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Romero P, Obradovi Z, Kissinger C, Villafranca JE, Dunker AK. Identifying disordered regions in proteins from amino acid sequence. IEEE Int Conf Neural Networks. 1997;1:90–95. [Google Scholar]
- 77.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 78.Head JF, Swamy N, Ray R. Crystal structure of the complex between actin and human vitamin D-binding protein at 2.5 Å resolution. Biochemistry. 2002;41:9015–9020. doi: 10.1021/bi026054y. [DOI] [PubMed] [Google Scholar]
- 79.Verboven C, Rabijns A, De Maeyer M, Van Baelen H, Bouillon R, De Ranter C. A structural basis for the unique binding features of the human vitamin D-binding protein. Nat Struct Mol Biol. 2002;9:131–136. doi: 10.1038/nsb754. [DOI] [PubMed] [Google Scholar]
- 80.Oubrie A, Rozeboom HJ, Kalk KH, Duine JA, Dijkstra BW. The 1.7 A crystal structure of the apo form of the soluble quinoprotein glucose dehydrogenase from Acinetobacter calcoaceticus reveals a novel internal conserved sequence repeat. J Mol Biol. 1999;289:319–333. doi: 10.1006/jmbi.1999.2766. [DOI] [PubMed] [Google Scholar]
- 81.Xu L, Benson SD, Butcher SJ, Bamford DH, Burnett RM. The receptor binding protein P2 of PRD1, a virus targeting antibiotic-resistant bacteria, has a novel fold suggesting multiple functions. Structure. 2003;11:309–322. doi: 10.1016/s0969-2126(03)00023-6. [DOI] [PubMed] [Google Scholar]
- 82.Nathan RZ, May AP, Robinson RC, Burtnick LD, Crocker PR, Brossmer R, Kelm S, Jones EY. Crystallographic and in silico analysis of the sialoside-binding characteristics of the Siglec sialoadhesin. J Mol Biol. 2007;365:1469–1479. doi: 10.1016/j.jmb.2006.10.084. [DOI] [PubMed] [Google Scholar]
- 83.Galang CK, Hauser CA. Cooperative DNA binding of the human HoxB5 (Hox-2.1) protein is under redox regulation in vitro. Mol Cell Biol. 1993;13:4609–4617. doi: 10.1128/mcb.13.8.4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun XZ, Vinci C, Makmura L, Han SB, Tran D, Nguyen J, Hamann M, Grazziani S, Sheppard S, Gutova M, Zhou FM, Thomas J, Momand J. Formation of disulfide bond in p53 correlates with inhibition of DNA binding and tetramerization. Antioxid Redox Signal. 2003;5:655–665. doi: 10.1089/152308603770310338. [DOI] [PubMed] [Google Scholar]
- 85.Buchanan BB. Role of light in the regulation of chloroplast enzymes. Annu Rev Plant Physiol. 1980;31:341–374. [Google Scholar]
- 86.Fermani S, Sparla F, Falini G, Martelli PL, Casadio R, Pupillo P, Ripamonti A, Trost P. Molecular mechanism of thioredoxin regulation in photosynthetic A2B2-glyceraldehyde-3-phosphate dehydrogenase. Proc Natl Acad Sci USA. 2007;104:11109–11114. doi: 10.1073/pnas.0611636104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Oesterhelt C, Klocke S, Holtgrefe S, Linke V, Weber APM, Scheibe R. Redox regulation of chloroplast enzymes in Galdieria sulphuraria in view of eukaryotic evolution. Plant Cell Physiol. 2007;48:1359–1373. doi: 10.1093/pcp/pcm108. [DOI] [PubMed] [Google Scholar]
- 88.Shivapurkar N, Stastny V, Okumura N, Girard L, Xie Y, Prinsen C, Thunnissen FB, Wistuba II, Czerniak B, Frenkel E, Roth JA, Liloglou T, Xinarianos G, Field JK, Minna JD, Gazdar AF. Cytoglobin, the newest member of the globin family, functions as a tumor suppressor gene. Cancer Res. 2008;68:7448–7456. doi: 10.1158/0008-5472.CAN-08-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sugimoto H, Makino M, Sawai H, Kawada N, Yoshizato K, Shiro Y. Structural basis of human cytoglobin for ligand binding. J Mol Biol. 2004;339:873–885. doi: 10.1016/j.jmb.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 90.Mehta RA, Fawcett TW, Porath D, Mattoo AK. Oxidative stress causes rapid membrane translocation and in vivo degradation of ribulose-1,5-bisphosphate carboxylase/oxygenase. J Biol Chem. 1992;267:2810–2816. [PubMed] [Google Scholar]
- 91.Cámara-Artigas A, Hirasawa M, Knaff DB, Wang M, Allen JP. Crystallization and structural analysis of GADPH from Spinacia oleracea in a new form. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:1087–1092. doi: 10.1107/S174430910604200X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hui JO, Woo G, Chow DT, Katta V, Osslund T, Haniu M. The intermolecular disulfide bridge of human glial cell line-derived neurotrophic factor: its selective reduction and biological activity of the modified protein. J Protein Chem. 1999;18:585–593. doi: 10.1023/a:1020607501910. [DOI] [PubMed] [Google Scholar]
- 93.Garcia-Diaz M, Bebenek K, Krahn JM, Kunkel TA, Pedersen LC. A closed conformation for the Polλ catalytic cycle. Nat Struct Mol Biol. 2005;12:97–98. doi: 10.1038/nsmb876. [DOI] [PubMed] [Google Scholar]
- 94.Hink-Schauer C, Estebanez-Perpina E, Kurschus FC, Bode W, Jenne DE. Crystal structure of the apoptosis-inducing human granzyme A dimer. Nat Struct Mol Biol. 2003;10:535–540. doi: 10.1038/nsb945. [DOI] [PubMed] [Google Scholar]
- 95.Wang Y, Jiang Y, Meyering-Voss M, Sprinzl M, Sigler PB. Crystal structure of the EF-Tu.EF-Ts complex from Thermus thermophilus. Nat Struct Mol Biol. 1997;4:650–656. doi: 10.1038/nsb0897-650. [DOI] [PubMed] [Google Scholar]
- 96.Muto T, Tsuchiya D, Morikawa K, Jingami H. Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proc Natl Acad Sci USA. 2007;104:3759–3764. doi: 10.1073/pnas.0611577104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tiwari A, Haward TJ. Mutant SOD1 instability: implications for toxicity in amyotrophic lateral sclerosis. Neurodegener Dis. 2005;2:115–127. doi: 10.1159/000089616. [DOI] [PubMed] [Google Scholar]
- 98.Furukawa Y, Fu R, Deng HX, Siddique T, O'Halloran T. Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc Natl Acad Sci USA. 2006;103:7148–7153. doi: 10.1073/pnas.0602048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ilbert M, Horst J, Ahrens S, Winter J, Graf PCF, Lilie H, Jakob U. The redox-switch domain of Hsp33 functions as dual stress sensor. Nat Struct Mol Biol. 2007;14:556–563. doi: 10.1038/nsmb1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Castro C, Millian NS, Garrow TA. Liver betaine-homocysteine S-methyltransferase activity undergoes a redox switch at the active site zinc. Arch Biochem Biophys. 2008;472:26–33. doi: 10.1016/j.abb.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Millian NS, Garrow TA. Human betaine-homocysteine methyltransferase is a zinc metalloenzyme. Arch Biochem Biophys. 1998;356:93–98. doi: 10.1006/abbi.1998.0757. [DOI] [PubMed] [Google Scholar]
- 102.Mayo KH, Barker S, Kuranda MJ, Hunt AJ, Myers JA, Maione TE. Molten globule monomer to condensed dimer: role of disulfide bonds in platelet factor-4 folding and subunit association. Biochemistry. 1992;31:12255–12265. doi: 10.1021/bi00163a040. [DOI] [PubMed] [Google Scholar]
- 103.Vucetic S, Brown CJ, Dunker AK, Obradovic Z. Flavors of protein disorder. Proteins. 2003;52:573–584. doi: 10.1002/prot.10437. [DOI] [PubMed] [Google Scholar]
- 104.Bombarda E, Morellet N, Cherradi H, Spiess B, Bouaziz S, Grell E, Roques BP, Mely Y. Determination of the pKa of the four Zn2+-coordinating residues of the distal finger motif of the HIV-1 nucleocapsid proteconsequences on the binding of Zn2+ J Mol Biol. 2001;310:659–672. doi: 10.1006/jmbi.2001.4770. [DOI] [PubMed] [Google Scholar]
- 105.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza hemagglutinin at the pH of membrane-fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 106.Tuinstra RL, Peterson FC, Kutlesa S, Elgin ES, Kron MA, Volkman BF. Interconversion between two unrelated protein folds in the lymphotactin native state. Proc Natl Acad Sci USA. 2008;105:5057–5062. doi: 10.1073/pnas.0709518105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mapelli M, Massimiliano L, Santaguida S, Musacchio A. The Mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell. 2007;131:730–743. doi: 10.1016/j.cell.2007.08.049. [DOI] [PubMed] [Google Scholar]
- 108.Murzin AG. Metamorphic proteins. Science. 2008;320:1725–1726. doi: 10.1126/science.1158868. [DOI] [PubMed] [Google Scholar]
- 109.Yeates TO. Protein structure: evolutionary bridges to new folds. Curr Biol. 2007;17:R48–R50. doi: 10.1016/j.cub.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 110.Milbradt AG, Boulegue C, Moroder L, Renner C. The two cysteine-rich head domains of minicollagen from Hydra nematocysts differ in their cystine framework and overall fold despite an identical cysteine sequence pattern. J Mol Biol. 2005;354:591–600. doi: 10.1016/j.jmb.2005.09.080. [DOI] [PubMed] [Google Scholar]
- 111.Xu M, Arulandu A, Struck DK, Swanson S, Sacchettini JC, Young R. Disulfide isomerization after membrane release of its SAR domain activates P1 lysozyme. Science. 2005;307:113–117. doi: 10.1126/science.1105143. [DOI] [PubMed] [Google Scholar]
- 112.Kabsch W, Sander C. Dictionary of protein secondary structure—pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 113.van Vlijmen HWT, Gupta A, Narasimhan LS, Singh J. A novel database of disulfide patterns and its application to the discovery of distantly related homologs. J Mol Biol. 2004;335:1083–1092. doi: 10.1016/j.jmb.2003.10.077. [DOI] [PubMed] [Google Scholar]
- 114.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 115.Khatri P, Draghici S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 2005;21:3587–3595. doi: 10.1093/bioinformatics/bti565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mateos A, Herrero J, Tamames J, Dopazo J. Supervised neural networks for clustering conditions in DNA array data after reducing noise by clustering gene expression profiles. In: Lin S, Johnson K, editors. Methods of microarray data analysis. Boston: Kluwer Academic Publishers; 2002. pp. 91–103. [Google Scholar]
- 117.Flocco MM, Mowbray SL. Ca-based torsion angles: a simple tool to analyze protein conformational changes. Protein Sci. 1995;4:2118–2122. doi: 10.1002/pro.5560041017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP—a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]