

The structure of the branched-chain amino-acid transaminase (IlvE) from M. tuberculosis has been resolved at 1.9 Å resolution. The structure, together with sequence conservation, identifies residues that are likely to be responsible for substrate specificity with mechanistic and potential therapeutic relevance.
Keywords: Mycobacterium tuberculosis, transaminases, pyridoxal 5′-phosphate, pyridoxamine 5′-phosphate, branched-chain amino acids
Abstract
Unlike mammals, bacteria encode enzymes that synthesize branched-chain amino acids. The pyridoxal 5′-phosphate-dependent transaminase performs the final biosynthetic step in these pathways, converting keto acid precursors into α-amino acids. The branched-chain amino-acid transaminase from Mycobacterium tuberculosis (MtIlvE) has been crystallized and its structure has been solved at 1.9 Å resolution. The MtIlvE monomer is composed of two domains that interact to form the active site. The biologically active form of IlvE is a homodimer in which each monomer contributes a substrate-specificity loop to the partner molecule. Additional substrate selectivity may be imparted by a conserved N-terminal Phe30 residue, which has previously been observed to shield the active site in the type IV fold homodimer. The active site of MtIlvE contains density corresponding to bound PMP, which is likely to be a consequence of the presence of tryptone in the crystallization medium. Additionally, two cysteine residues are positioned at the dimer interface for disulfide-bond formation under oxidative conditions. It is unknown whether they are involved in any regulatory activities analogous to those of the human mitochondrial branched-chain amino-acid transaminase.
1. Introduction
The branched-chain amino acids isoleucine, leucine and valine are essential amino acids in the mammalian diet but are biosynthesized in bacteria. The hydrophobic nature of the side chains of these residues assists in protein folding and maturation as well as in the formation of amphipathic helices, coiled coils and leucine zippers (Deng et al., 2008 ▶). Unlike mammals, bacteria can synthesize these amino acids, making these pathways attractive targets for the development of novel antibiotics. These pathways and their regulation can vary dramatically between bacterial species, but one common characteristic is a final transamination step in which the branched-chain amino acid is produced from an α-keto acid precursor by a branched-chain amino-acid transaminase (BCAT; EC 2.6.1.42; Venos et al., 2004 ▶; Huang et al., 1992 ▶; Magnus et al., 2006 ▶; Leyval et al., 2003 ▶; Mäder et al., 2004 ▶). The pyridoxal 5′-phosphate (PLP) dependent transaminases can be classified into five fold types, I–V, with BCATs belonging to type IV (Hirotsu et al., 2005 ▶; Grishin et al., 1995 ▶). To date, all observed type IV PLP transaminases consist of two domains with an interdomain loop (Goto et al., 2003 ▶) and can be l- or d-amino-acid donor specific (Okada et al., 2001 ▶). While humans lack the overall biosynthetic pathway, human mitochondrial and cytosolic isoforms (hBCATm and hBCATc) of BCAT exist which are orthologues of IlvE from Mycobacterium tuberculosis (MtIlvE; Conway et al., 2004 ▶; Yennawar et al., 2006 ▶). However, selective inhibition of MtIlvE appears to be possible since hBCATc is selectively inhibited compared with hBCATm by the adjunctive anti-epileptic molecule gabapentin (Goto et al., 2005 ▶). Thus, we have begun the detailed structural characterization of MtIlvE and have cloned, expressed and purified MtIlvE and solved its three-dimensional structure at 1.9 Å resolution.
2. Materials and methods
2.1. Cloning
The MtIlvE gene Rv2210c was cloned from M. tuberculosis genomic DNA using the forward primer 5′-ATCCCGCTGCTAGCGGCTCCCTTCAATTC-3′ and the reverse primer 3′-ATCCCGCTCTCGAGCTACCCCAGCCGCGCCAT-5′, which included NheI and XhoI digestion sites, respectively (bold). A band corresponding to the MtIlvE gene was observed on agarose-gel electrophoresis and was purified from the gel. This product and the pET28a vector were then both separately digested using the NheI and XhoI enzymes for 4 h at 310 K and then ligated together overnight at 289 K using T4DNA ligase. The ligated products were transformed into TOP10 cells. Purified plasmids were extracted using the Qiagen QIAprep Spin Miniprep Kit and screened using a combination of digestion tests and sequencing with the T7 forward and reverse primers for the production of a correct mutation-free plasmid.
2.2. Expression
The product was transformed into the Escherichia coli Rosetta 2 expression strain. 4 l Luria Broth was inoculated with bacteria, grown at 310 K to an OD of ∼0.6 and induced using 0.1 mM IPTG. Induction proceeded at 291 K for ∼16 h. The cells were harvested by centrifugation at 4000g for 15 min. The cell pellets were collected and frozen at 253 K.
2.3. Purification
The cells were resuspended on ice in 20 mM HEPES pH 7.5 containing 200 units of DNase and Complete protease-inhibitor cocktail from Roche and lysed by a total of 5 × 30 s steps of sonication on ice. Cellular debris was spun down at 18 000 rev min−1 for 1 h using an SS-34 Beckman centrifuge rotor. The supernatant was gravity-loaded onto a Qiagen Ni+ column equilibrated in 20 mM HEPES pH 7.5 (buffer A). The column was washed with 70 ml buffer A containing 500 mM NaCl and 1 mM DTT. The protein was eluted using 15 ml buffer A containing 300 mM imidazole and 1 mM DTT. The eluted protein was observed at a molecular weight of ∼40 kDa on SDS–PAGE, which was in agreement with the theoretical value of 39.7 kDa. The eluted protein was dialyzed at 277 K overnight in 2 l buffer A containing 300 mM NaCl and 1 mM DTT. The dialyzed protein volume was reduced to 2 ml using an Amicon spin concentrator and the protein was subsequently loaded onto a S75 gel-filtration column (Pharmacia) in buffer A containing 300 mM NaCl. The protein eluted as a single peak. Selected fractions were combined after SDS–PAGE analysis. The purified MtIlvE protein was soluble and was stable for several days at 277 K and for extended periods stored in 50% glycerol at 253 K.
2.4. Analytical gel filtration
The Bio-Rad gel-filtration standard was run on a Superdex 200 column (Amersham Pharmacia Biotech) equilibrated in buffer A containing 300 mM NaCl. 20 µl 10 mg ml−1 MtIlvE was loaded onto the column with and without the protein standard. Measurements of the MtIlvE elution peak position resulted in a calculated molecular weight of ∼80 kDa, indicating that the protein exists as a dimer.
2.5. Crystallization
The purified protein was buffer-exchanged into buffer A and concentrated using Amicon Ultra Centrifugal filters to 43.4 mg ml−1 as measured using the Bradford protein assay. Sparse-matrix crystal screening was performed under oil for MtIlvE (5 mg ml−1 with 0.2 mM added PLP) with various commercially available crystallization screens in a 1 µl:1 µl ratio. Thin plates and rods were observed in 1%(w/v) tryptone, 50 mM HEPES pH 7.0 and 12%(w/v) polyethylene glycol 3350 after ∼3–4 weeks. Larger drops with a 5 µl:5 µl ratio yielded larger better diffracting crystals after one month of incubation.
2.6. Structure determination
Crystals were directly fished from the under-oil drops and snap-frozen in liquid nitrogen. Data were collected on beamline X12B at Brookhaven National Laboratory. The data had clear systematic absences in the h00 and 0k0 but not in the 00l reflections and best fitted in space group P21212, with unit-cell parameters a = 65.5, b = 85.4, c = 59.7 Å, α = β = γ = 90.0°. The data showed no evidence of twinning and indexed readily using MOSFLM (Leslie, 1992 ▶). Data-collection and refinement statistics are displayed in Table 1 ▶. The MtIlvE sequence was found to be most similar to that of human mitochondrial hBCATm (PDB code 2hhf), which was used to obtain initial phases via molecular replacement (MR) in MOLREP (Vagin & Teplyakov, 1997 ▶). An MR solution was only found in the above-mentioned space group using only chain A from 2hhf, which resulted in very poor initial phases, but when it was fed as input to the autobuilding SOLVE/RESOLVE scripts in the software suite PHENIX (Adams et al., 2002 ▶) together with sequence information it resulted in a dramatic improvement in the phases and map quality. The iterative threading algorithm resulted in a broken backbone as well as >50% of the side chains being fitted. Model building in Coot (Emsley & Cowtan, 2004 ▶) and structure refinement using REFMAC5 (Murshudov et al., 1997 ▶) in the software suite CCP4 (Potterton et al., 2003 ▶) resulted in a final model with an R work of 0.184 and an R free of 0.199 to 1.9 Å resolution. The enzyme crystallized with a monomer in the asymmetric unit; the homodimer was formed with a symmetry mate. The structure quality was determined using MolProbity (Davis et al., 2007 ▶). The monomer and dimer surface areas were calculated using AREAIMOL (Saff & Kuijlaars, 1997 ▶) in CCP4.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution bin.
| Data collection | |
| Resolution (Å) | 39.2–1.9 (2.0–1.9) |
| Completeness (%) | 99.6 (100) |
| Average redundancy | 4.9 (4.9) |
| I/σ(I) | 7.7 (1.9) |
| Rmerge† | 0.072 (0.386) |
| Space group | P21212 |
| Unit-cell parameters (Å, °) | a = 65.5, b = 85.4, c = 59.7, α = β = γ = 90.0 |
| Reflections | 26897 (3897) |
| Wilson B factor (Å2) | 22.9 |
| Refinement statistics | |
| Rwork | 0.184 (0.234) |
| Rfree | 0.199 (0.303) |
| Rwork reflections | 25546 |
| Rfree reflections | 1351 |
| No. of atoms | |
| Protein | 2567 [335 residues] |
| PLP | 16 |
| Solvent | 281 |
| Average B factors (Å2) | |
| Protein | 22.7 |
| PLP | 15.0 |
| Solvent | 31.1 |
| R.m.s.d. from ideal geometry‡ | |
| Bond lengths (Å) | 0.011 |
| Bond angles (°) | 1.4 |
| Ramachandran plot (%) | |
| Favored | 97.6 |
| Outliers | 0.0 |
| PDB code | 3ht5 |
R
merge = 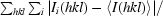
 .
.
Engh & Huber (1991 ▶).
3. Results
3.1. Overall structure
The overall structure of MtIlvE belongs to the fold-type IV class of PLP-dependent enzymes (Fig. 1 ▶ a). While the enzyme crystallized with one monomer in the asymmetric unit, MtIlvE forms a tight dimer with one of the symmetry mates. This homodimer superimposes with the human mitochondrial branched-chain amino-acid transaminase (hBCATm) dimer structure (Yennawar et al., 2006 ▶) with an r.m.s.d. of 1.27 Å. MtIlvE eluted as a dimer on analytical gel filtration, indicating that the biologically active form of MtIlvE is a dimer, as opposed to the trimer-of-dimers hexamer found for E. coli BCAT (eBCAT; Okada et al., 1997 ▶). In addition, the enzyme was shown to have in vitro transaminase activity with several branched-chain amino acids. All 368 residues of the native protein show clear density except for two regions: the 178–181 linker loop and the N-terminal 33 residues. The N-terminal residues were highly disordered with no observable density and were not included in the final MtIlvE model. A similar observation was made in the case of the mature form (lacking the mitochondrial targeting sequence) of hBCATm, in which the 33 N-terminal residues were highly flexible with no discernable secondary structure (Goto et al., 2005 ▶; Yennawar et al., 2002 ▶).
Figure 1.

(a) The MtIlvE homodimer displayed as rainbow coloring from the N-terminus (blue) to the C-terminus (red), with the PMP molecule displayed as black sticks. (b) The two domains of MtIlvE are shown in orange and green, with the PMP molecule colored black. The flexible linker loop is displayed in red and the connecting helix is displayed in blue. All secondary structure is labeled accordingly.
The MtIlvE monomer is composed of two domains which interact to form the active site (Fig. 1 ▶). Domain 1 (residues 34–177 and 364–368) is composed of both N- and C-terminal residues and contains α-helices 1–3 and β-strands 1–7 and 16. All eight β-strands of this domain participate in the formation of a core β-sheet, which is surrounded by α-helices 1–3. Domain 2 (residues 182–363) is composed of two β-sheets formed by β-strands 9–12 and by β-strands 8, 14 and 15, which are all surrounded by α-helices 4–7. The two domains are connected via α-helix 8 (residues 341–352) and a linker loop (residues 178–181). α-Helix 8 is a highly ordered helical rod which rigidly connects the two domains of the monomer. Furthermore, no evidence of interdomain movement upon ligand binding has been observed in the structures of orthologues (Peisach et al., 1998 ▶; Okada et al., 2001 ▶). The other interdomain motif is the linker loop, which is partially disordered owing to the presence of two glycine residues. These residues provide flexibility near the active-site entrance and allow both the substrate and product access into and out of the active site (Yennawar et al., 2002 ▶). A role of the flexible linker loop in substrate binding has previously been proposed for the eBCAT enzyme (Okada et al., 2001 ▶).
3.2. The active site
The active site of MtIlvE contains density consistent with a pyridoxamine 5′-phosphate (PMP) molecule bound in the active site. This is displayed in Fig. 2 ▶(a) using F o − F c OMIT density (green cage) contoured at 4σ (0.26 e Å−3). In crystal structures of PLP-dependent enzymes the PLP molecule is often observed as an internal aldimine formed via Schiff-base bond formation with the conserved active-site lysine residue. In MtIlvE, the electron density unambiguously shows the absence of Schiff-base bond formation with Lys204. In addition, when refined under the restraints of PMP versus PLP, the difference density favored the bond angles associated with sp 3 hybridization of the C4′ PMP C atom over the sp 2 hybridization of the C4′ PLP carbonyl C atom.
Figure 2.

(a) The PMP molecule bound in the active site is displayed using green F o − F c OMIT density contoured at 4σ (0.26 e Å−3). The surrounding active site is shown using blue 2F 0 − F c electron density contoured at 1σ (0.28 e Å−3) and a few important active-site residues are labeled. (b) The hydrogen-bonding network of PMP within the active site. PMP atoms are numbered according to convention.
The PMP molecule, while not covalently bound in the active site, forms an extensive network of hydrogen bonds to MtIlvE (Fig. 2 ▶ b). The three phosphate O atoms of PMP make multiple contacts with residues in the highly conserved phosphate-binding pocket of MtIlvE (Fig. 2 ▶ b). These contacts include interactions with residues Arg101, Ile271, Thr272 and Thr314 as well as waters W388, W423 and W448. Additional short hydrogen bonds are made between the phenolic moiety of the cofactor and Tyr209 (2.4 Å) and the pyridine ring N atom and Glu240 (2.7 Å). The side chain of the conserved catalytic Lys204 residue is poised at a distance of 2.7 Å from the PMP amine N atom, a distance suitable for formation of the PLP Schiff base.
4. Discussion
The branched-chain amino-acid pathways are highly regulated in bacterial species (Huang et al., 1992 ▶; Magnus et al., 2006 ▶; Leyval et al., 2003 ▶; Mäder et al., 2004 ▶). For example, in M. tuberculosis the first committed step in the synthesis of l-leucine catalyzed by α-isopropylmalate synthase is strongly inhibited by l-leucine (de Carvalho et al., 2005 ▶). The existence of the IlvE/BCAT transaminase gene was inferred from complete sequencing of the M. tuberculosis genome (Cole et al., 1998 ▶). A previous study found this enzyme to be active with isoleucine, leucine, valine, phenylalanine and glutamate (Venos et al., 2004 ▶). This suggests that MtIlvE may catalyze the final amination of the keto acids corresponding to each of the branched-chain amino acids at the expense of l-glutamate.
The three-dimensional structures of a number of IlvE orthologs have been solved (Hirotsu et al., 2005 ▶; Okada et al., 1997 ▶; Yennawar et al., 2002 ▶; Goto et al., 2005 ▶), but no structural data exist for the MtIlvE enzyme. The PLP-dependent transaminases are classified into five fold types: I–V (Grishin et al., 1995 ▶). The structures of branched-chain amino-acid transaminases determined all exhibit a type IV fold (Hirotsu et al., 2005 ▶). All type IV PLP-dependent transaminases consist of two domains with an interdomain loop and perform chemistry characterized by proton transfer on the re face of the PLP cofactor (Goto et al., 2003 ▶). Enzymes exhibiting the type IV fold can be l- or d-amino-acid donor specific (Okada et al., 2001 ▶). E. coli BCAT (eBCAT) is a homohexamer, while the human cytosolic and mitochondrial isoforms (hBCATc and hBCATm) are homodimers with ∼40 kDa subunits roughly the same size as MtIlvE. MtIlvE shares 24.0% sequence identity with eBCAT and 30.4% sequence identity with hBCATm.
A structural alignment between MtIlvE and hBCATm (PDB entry 1kt8) highlights several key features (Fig. 3 ▶ a) and reveals significant structural conservation. The MtIlvE and hBCATm dimers share a similar interface between the monomer subunits and have comparable buried surface areas. In both instances, each monomer introduces a loop (MtIlvE154–160) into the active site of the other monomer. Within this loop, Val158 of MtIlvE is positioned nearly identically to an equivalent valine in hBCATm (Fig. 3 ▶ b). Not surprisingly, this is a strictly conserved residue in BCAT enzymes (Fig. 4 ▶). The structure of hBCATm (PDB entry 1kt8) is the complex of the ketimine form with the substrate isoleucine and positions this conserved hydrophobic residue 3.6 Å from the branched hydrophobic side-chain C atom of the ketimine complex. Thus, these loops positioned at the dimer interface form an adjacent portion of the partner’s active site and house the strictly conserved valine residue for interaction with the hydrophobic branched chain of the substrate. Thus, the loop functions as a substrate-specificity determinant with selectivity towards the branched-chain side chain of the substrate amino acid.
Figure 3.

Structural alignment of the MtIlvE (blue) and hBCATm (PDB code 1kt8; orange) dimers. The green loop is donated from the partner molecule and contributes a conserved valine which stabilizes the external ketimine in the active site of PDB entry 1kt8. The N-terminus of the 1kt8 structure (black loop) shows the conserved phenylalanine residue shielding the active site from bulk water molecules.
Figure 4.

The sequence alignment between MtIlvE, human orthologs and orthologs from several other bacterial pathogens. Conserved phosphate-binding residues are marked with a square below the alignment and the conserved substrate-selectivity residues are marked with a diamond. The Cys169 residue position is marked by a black circle above the residue. Secondary structure is indicated above the sequence alignment.
Apart from the overall structural similarity between these two enzymes, there are a few notable differences. The N-terminal portion of the MtIlvE enzyme is disordered with no electron density, whereas the hBCATm–ketimine complex has clear N-terminal density. In particular, the hBCATm N-terminal residue Phe30 is positioned 5.1 Å from the branched chain of the isoleucine–PLP complex (Fig. 3 ▶ b). This shields bulk solvent from the active site and further stabilizes the hydrophobic substrate within the active site for transamination. It is likely that a similar mechanism of ligand binding is employed by MtIlvE, but is not observed in our structure. The Phe30 position is strictly conserved in all orthologues except for that from E. coli, indicating this may be an additional ligand-binding mechanism, especially amongst the homodimeric BCAT enzymes.
One difference between MtIlvE and other orthologues is the presence of a cysteine (Cys196) residue positioned adjacent to the identical symmetry-mate cysteine at the homodimer interface. While the electron density between these two residues does not support a disulfide bond, the position and proximity of these two residues certainly allows putative disulfide-bond formation (Fig. 5 ▶). Two tryptophan residues reside in the equivalent positions at the homodimer interface of hBCATm. In Fig. 4 ▶, the primary sequence alignment indicates that the cysteine is not conserved amongst orthologues. This unique feature may provide the active MtIlvE dimer with added stability, especially under the harsh oxidative conditions found in macrophages. Regulation of activity via disulfide-bond formation has previously been reported for hBCATm (Yennawar et al., 2006 ▶). Although the MtIlvE cysteine residues are positioned very differently (residing at the dimer interface) from those in hBCATm, these residues could be involved in the regulation of enzymatic activity via dimer stabilization under oxidative conditions.
Figure 5.

Two cysteine residues shown using 2F o − F c electron density contoured at 1σ (0.28 e Å−3) reside at the homodimer interface and are reduced with no disulfide formation. However, the distance (shown in Å) between these indicates the possibility of disulfide formation under oxidative conditions, which may confer additional protein stability.
5. Conclusions
The MtIlvE enzyme is a PLP-dependent transaminase with activity specific for branched-chain amino acids such as isoleucine, leucine, valine and glutamate, which is the likely common amine donor. The MtIlvE monomer is composed of two domains which interact to form the active site. The biologically active form of MtIlvE is a homodimer in which each monomer contributes a substrate-specificity loop to the partner molecule. This loop contains a strictly conserved valine residue which interacts with the hydrophobic side chain of the substrate molecule. Additional substrate selectivity may be imparted by a conserved N-terminal Phe30 residue, which was previously observed to shield the active site in the type IV fold homodimer. The active site of MtIlvE contains density that suggests the presence of a PMP molecule. This form of the cofactor was observed owing to the presence of tryptone in the crystallization conditions. Two cysteine residues are positioned for possible disulfide-bond formation to stabilize the dimer under oxidizing conditions.
Supplementary Material
PDB reference: MtIlvE, 3ht5, r3ht5sf
Acknowledgments
We would like to thank Dr Matt Vetting and all the beamline staff at X12B at BNL for their assistance during data collection. We also express our gratitude to Dr Meng-Chiao Ho, whose critical input resulted in the phasing of our data. We would also like to thank Dr Steven Roderick for his assistance throughout the model building. This work was supported by NIH grant AI33696 (to JSB).
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Carvalho, L. P. de, Argyrou, A. & Blanchard, J. S. (2005). J. Am. Chem. Soc.127, 10004–10005. [DOI] [PubMed]
- Cole, S. T. et al. (1998). Nature (London), 393, 537–544.
- Conway, M. E., Poole, L. B. & Hutson, S. M. (2004). Biochemistry, 43, 7356–7364. [DOI] [PubMed]
- Davis, I. W., Leaver-Fay, A., Chen, V. B. & Richardson, D. C. (2007). Nucleic Acids Res.35, W375–W383. [DOI] [PMC free article] [PubMed]
- Deng, Y., Liu, J., Zheng, Q., Li, Q., Kallenbach, N. R. & Lu, M. (2008). Chem. Biol.15, 908–919. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Engh, R. A. & Huber, R. (1991). Acta Cryst. A47, 392–400.
- Goto, M., Miyahara, I., Hayashi, H., Kagamiyama, H. & Hirotsu, K. (2003). Biochemistry, 42, 3725–3733. [DOI] [PubMed]
- Goto, M., Miyahara, I., Hirotsu, K., Conway, M., Yennawar, N., Islam, M. M. & Hutson, S. M. (2005). J. Biol. Chem.280, 37246–37256. [DOI] [PubMed]
- Grishin, N. V., Phillips, M. A. & Goldsmith, E. J. (1995). Protein Sci.4, 1291–1304. [DOI] [PMC free article] [PubMed]
- Hirotsu, K., Goto, M., Okamoto, A. & Miyahara, I. (2005). Chem. Rec.5, 160–172. [DOI] [PubMed]
- Huang, F., George, C. & Calhoun, D. (1992). J. Bacteriol.174, 4871–4877. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Leyval, D., Uy, D., Delaunay, S., Goergen, J. L. & Engasser, J. M. (2003). J. Biotechnol.104, 241–252. [DOI] [PubMed]
- Mäder, U., Hennig, S., Hecker, M. & Homuth, G. (2004). J. Bacteriol.186, 2240–2252. [DOI] [PMC free article] [PubMed]
- Magnus, J. B., Hollwedel, D., Oldiges, M. & Takors, R. (2006). Biotechnol. Prog.22, 1071–1083. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Okada, K., Hirotsu, K., Hayashi, H. & Kagamiyama, H. (2001). Biochemistry, 40, 7453–7463. [DOI] [PubMed]
- Okada, K., Hirotsu, K., Sato, M., Hayashi, H. & Kagamiyama, H. (1997). J. Biochem.121, 637–641. [DOI] [PubMed]
- Peisach, D., Chipman, D. M., Van Ophem, P. W., Manning, J. M. & Ringe, D. (1998). Biochemistry, 37, 4958–4967. [DOI] [PubMed]
- Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. (2003). Acta Cryst. D59, 1131–1137. [DOI] [PubMed]
- Saff, E. B. & Kuijlaars, A. B. J. (1997). Math. Intell.19, 5–11.
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
- Venos, E., Knodel, M. H., Radford, C. L. & Berger, B. J. (2004). BMC Microbiol.4, 39. [DOI] [PMC free article] [PubMed]
- Yennawar, N. H., Conway, M. E., Yennawar, H. P., Farber, G. K. & Hutson, S. M. (2002). Biochemistry, 41, 11592–11601. [DOI] [PubMed]
- Yennawar, N. H., Islam, M. M., Conway, M. E., Wallin, R. & Hutson, S. M. (2006). J. Biol. Chem.281, 39660–39671. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: MtIlvE, 3ht5, r3ht5sf
PDB reference: MtIlvE, 3ht5, r3ht5sf