

A hexagonal crystal of free methionine-(R)-sulfoxide reductase from S. aureus has been obtained using ammonium sulfate as a precipitant.
Keywords: methionine sulfoxide, free methionine, free methionine-(R)-sulfoxide reductase
Abstract
Free methionine-(R)-sulfoxide reductase (fRMsr) catalyzes the reduction of the free form of methionine-(R)-sulfoxide back to free methionine. The fRMsr protein from Staphylococcus aureus was overexpressed in Escherichia coli, purified and crystallized at 295 K using ammonium sulfate as a precipitant. Diffraction data were collected to 1.7 Å resolution from a native crystal using synchrotron radiation. The crystal belonged to the hexagonal space group P6122, with unit-cell parameters a = b = 89.84, c = 88.75 Å, α = β = 90, γ = 120°. Assuming the presence of one molecule in the asymmetric unit, the calculated Matthews coefficient value was 2.21 Å3 Da−1, with a solvent content of 57.1%.
1. Introduction
Various reactive oxygen and nitrogen species can damage cellular components, including lipids, DNA and proteins (Lin et al., 2007 ▶). Damage to proteins is likely to be a consequence of the oxidation of methionine and cysteine residues. Methionine sulfoxide (MetSO) occurs as two stereoisomers, methionine-S-sulfoxide (Met-S-SO) and methionine-R-sulfoxide (Met-R-SO) (Weissbach et al., 2005 ▶), and is associated with a variety of diseases and the aging process. The reduction of the S- and R-forms of MetSO in peptides and as free MetSO is catalyzed by methionine sulfoxide reductase A (MsrA) and methionine sulfoxide reductase B (MsrB) (Boschi-Muller et al., 2005 ▶; Kauffmann et al., 2005 ▶). Organisms possess MetSO-reduction systems that confer them with the ability to repair oxidation damage, with a consequent impact on their longevity in oxidative environments (Moskovitz, 2005 ▶; Weissbach et al., 2005 ▶). In addition, the MsrA and MsrB families (Msrs) have a role as virulence determinants in some bacterial pathogens (Moskovitz, 2005 ▶; Weissbach et al., 2005 ▶). The structure and catalytic mechanisms of Msrs from various organisms have been studied (Boschi-Muller et al., 2005 ▶; Kauffmann et al., 2005 ▶). The catalytic mechanism common to all Msrs involves the formation of the sulfenic acid intermediate followed by regeneration of the catalytic cysteine. In some cases, this regeneration requires the formation and then the reduction of an internal disulfide bond at the catalytic and resolving Cys (Boschi-Muller et al., 2008 ▶).
Recently, an enzyme specific to the reduction of the free form of Met-R-SO has been characterized and named fRMsr (Lin et al., 2007 ▶). The fRMsr sequence is conserved in bacteria and yeast but is not found in humans (Lin et al., 2007 ▶). The overall folds show that MsrA, MsrB and fRMsr are unrelated. To date, only one crystal structure of fRMsr, that from Escherichia coli, is available in the PDB (PDB code 1vhm) and its biochemical function and catalytic mechanism were reported to be unknown (Badger et al., 2005 ▶). The X-ray structure suggested that fRMsrs use Cys residues for catalysis and that the active site is enclosed in a small cavity (Lin et al., 2007 ▶). The enclosed cavity supports the apparent substrate specificity. Moreover, E. coli fRMsr is the first member of the GAF-domain family to show enzymatic activity. Although speculative, the reduction of free Met-R-SO by fRMsr may function as a key element in cell signalling in response to oxidative stress and nutrients (Zoraghi et al., 2004 ▶). Additional structural and enzymatic studies are essential to appraise the binding mode of the substrate and to determine the role of the active-site residues in the catalytic mechanism. The amino-acid sequence homology between Staphylococcus aureus and E. coli fRMsr is almost 60%. Although crystal structures of E. coli fRMsr and the GAF-domain family have been solved and (for the latter) extensively analyzed, a definitive cysteine catalytic mechanism has not been elucidated for fRMsr. To understand the cysteine catalytic mechanism and specific substrate binding in S. aureus fRMsr, we overexpressed and crystallized the S. aureus fRMsr protein and collected X-ray crystallographic data in order to determine its three-dimensional structure.
2. Materials and methods
2.1. Expression and purification
The gene (UniProtKB/TrEMBL accession No. Q6GFY9) encoding fRMsr was cloned from S. aureus genomic DNA using polymerase chain reaction techniques. The oligonucleotide primers for the forward and reverse directions were 5′-ACAATAGCTAGCATGACAACAATTAACCCAACAAAC-3′ and 5′-ACAATACTCGAGTTATGCGAGTTGCTTTTCAAT-3′, containing NheI and XhoI restriction sites, respectively. The amplified DNA was inserted into the NheI/XhoI restriction endonuclease-digested expression vector pET-28a(+) (Novagen, USA) for the expression of N-terminally six-histidine-tagged proteins. The recombinant plasmid was transformed into E. coli strain BL21 (DE3) (Novagen, USA) for protein expression. The cells were grown in Luria–Bertani medium containing 50 µg ml−1 kanamycin at 310 K. When the density of the culture reached 0.45 at 600 nm, recombinant fRMsr protein was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). After 18 h at 291 K, the cells were harvested by centrifugation at 6000g for 30 min at 277 K. The cell pellet was resuspended in ice-cold lysis buffer (20 mM Tris–HCl pH 7.9, 500 mM NaCl, 5 mM imidazole, 1 mM phenylmethylsulfonyl fluoride) and the cells were disrupted by sonication. The crude lysate was centrifuged at 13 000g for 30 min at 277 K. The supernatant was loaded onto an Ni2+-chelated HiTrap chelating HP column (GE Healthcare, USA) which had previously been equilibrated with buffer A (20 mM Tris–HCl pH 7.9, 500 mM NaCl, 5 mM imidazole) and subsequently washed with buffer A containing 1 M imidazole. The protein was eluted with a gradient to 1 M imidazole in buffer A. Final purification of the protein was achieved by gel filtration on a HiLoad 16/60 Superdex 200 column (GE Healthcare, USA) that had been pre-equilibrated with buffer B (20 mM Tris–HCl pH 7.9, 5 mM DTT). The purified protein was concentrated using an Amicon Ultra-15 ultrafiltration device (Millipore, USA) to 20 mg ml−1 for crystallization. The homogeneity of the purified protein was determined by SDS–PAGE under denaturing conditions using 15%(v/v) polyacrylamide gels. The protein concentration was determined by the Bradford method using bovine serum albumin as a standard.
2.2. Crystallization and X-ray analysis
Initial crystallization screening was performed using Crystal Screens I and II (Hampton Research, USA) by the hanging-drop vapour-diffusion method in 24-well VDX plates at 295 K, mixing 1 µl protein solution and 1 µl reservoir solution. The first crystals were obtained after one week using reservoir solution consisting of 2 M ammonium sulfate, 10%(v/v) 2-propanol. The best crystallization conditions for fRMsr protein consisted of 2 M ammonium sulfate, 7%(v/v) 2-propanol. Crystal growth was scaled up using ammonium sulfate as precipitant. Single crystals were obtained within 5 d and were used for X-ray diffraction experiments. Several cryosolvents were tested for data collection under cryogenic conditions, including glycerol, ethylene glycol, PEG 400 and MPD, with ethylene glycol giving the best results. The crystal was transferred into well solution containing 25%(v/v) ethylene glycol before being flash-frozen in liquid nitrogen. X-ray diffraction data were collected from the cooled crystals using an ADSC Quantum CCD 210 detector on beamline 6C at Pohang Light Source (Pohang, South Korea). A total range of 360° was covered with 1.0° oscillation and 30 s exposure per frame. The wavelength of the synchrotron X-rays was 1.23986 Å. The crystal-to-detector distance was set to 130 mm. Diffraction data were collected to 1.7 Å resolution for the native crystal. The raw data for the native crystal were processed and scaled using the HKL-2000 software package.
3. Results and discussion
The gene encoding fRMsr protein from S. aureus genomic DNA was cloned into pET-28a(+) vector and transformed into E. coli strain BL21 (DE3). fRMsr consists of 154 amino acids and has a molecular weight of 18 kDa. The fRMsr protein from S. aureus was overexpressed in E. coli, purified and crystallized for structural studies. The best crystallization conditions for fRMsr protein consisted of 2 M ammonium sulfate, 7% 2-propanol. Crystals suitable for X-ray diffraction analysis grew to maximum dimensions of approximately 0.5 × 0.5 × 0.2 mm within 5 d (Fig. 1 ▶). The crystals diffracted to 1.7 Å resolution at the Pohang Light Source synchrotron facility. The native crystal belonged to the hexagonal space group P6122, with unit-cell parameters a = b = 89.84, c = 88.75 Å, α = β = 90, γ = 120°. Assuming the presence of one molecule in the asymmetric unit, the calculated Matthews coefficient (V M) value was 2.21 Å3 Da−1 and the solvent content was 57.1% by volume (Matthews, 1968 ▶). Data-collection statistics are given in Table 1 ▶. The crystal structure of S. aureus fRMsr was solved by the molecular-replacement method using the program CNS (Brünger et al., 1998 ▶) with the coordinates of E. coli fRMsr (PDB code 1vhm; Badger et al., 2005 ▶) as a search model. Further refinement of the model structure is in progress.
Figure 1.

Native crystals of free methionine-(R)-sulfoxide reductase from S. aureus for X-ray data collection. The crystal dimensions are approximately 0.5 × 0.5 × 0.2 mm.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Wavelength (Å) | 1.23986 |
| Space group | P6122 |
| Resolution range (Å) | 50.0–1.7 (1.76–1.7) |
| Observed reflections | 479682 |
| Unique reflections | 23645 |
| Redundancy | 20.3 (7.6) |
| Completeness (%) | 99.1 (94.8) |
| Rmerge† (%) | 6.6 (28.7) |
| I/σ(I) | 16.0 (5.5) |
R
merge = 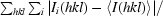
 , where Ii(hkl) is the observed intensity, 〈I(hkl)〉 is the average intensity and i counts through all symmetry-related reflections.
, where Ii(hkl) is the observed intensity, 〈I(hkl)〉 is the average intensity and i counts through all symmetry-related reflections.
Acknowledgments
We thank the staff for assistance during data collection at beamline 6C of Pohang Light Source, South Korea.
References
- Badger, J. et al. (2005). Proteins, 60, 787–796. [DOI] [PubMed]
- Boschi-Muller, S., Gand, A. & Branlant, G. (2008). Arch. Biochem. Biophys.474, 266–273. [DOI] [PubMed]
- Boschi-Muller, S., Olry, A., Antoine, M. & Branlant, G. (2005). Biochem. Biophys. Acta, 1703, 231–238. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Kauffmann, B., Aubry, A. & Favier, F. (2005). Biochim. Biophys. Acta, 1703, 249–260. [DOI] [PubMed]
- Lin, Z., Johnson, L. C., Weissbach, H., Brot, N., Lively, M. O. & Lowther, W. T. (2007). Proc. Natl Acad. Sci. USA, 104, 9597–9602. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Moskovitz, J. (2005). Biochim. Biophys. Acta, 1703, 213–219. [DOI] [PubMed]
- Weissbach, H., Resnick, L. & Brot, N. (2005). Biochim. Biophys. Acta, 1703, 203–212. [DOI] [PubMed]
- Zoraghi, R., Corbin, J. D. & Francis, S. H. (2004). Mol. Pharmacol.65, 267–278. [DOI] [PubMed]