

The preliminary X-ray analysis of β-glucosidase (BglI) from K. marxianus, which belongs to glycoside hydrolase family 3, is described.
Keywords: β-glucosidases, Kluyveromyces marxianus NBRC1777, glycoside hydrolase family 3
Abstract
The intracellular β-glucosidase from Kluyveromyces marxianus NBRC1777 (KmBglI) belongs to glycoside hydrolase family 3 and has a unique domain architecture. Selenomethionine-labelled KmBglI was purified and crystallized by the hanging-drop vapour-diffusion method using the purified enzyme at 30 mg ml−1, 0.04 M potassium dihydrogen phosphate pH 5.1, 16%(w/v) PEG 8000 and 20%(v/v) glycerol. The crystal belonged to space group C2, with unit-cell parameters a = 245.8, b = 148.7, c = 119.9 Å, β = 112.9°. Multiple-wavelength anomalous dispersion data were collected at 2.4 and 2.5 Å resolution. A tetramer was assumed to be present in the asymmetric unit, which gave a Matthews coefficient of 2.6 Å3 Da−1.
1. Introduction
Cellulose is the most abundant renewable bioresource on earth and its efficient conversion into biofuels could be the key to replacing fossil fuels and hence maintaining a sustainable ecosystem. Simultaneous saccharification and fermentation (SSF) of cellulose using microorganisms is a promising approach to achieving this goal and has been studied by various groups, primarily using the ethanologenic yeast Saccharomyces cerevisiae (van Maris et al., 2006 ▶). In addition to S. cerevisiae, Kluyveromyces marxianus has recently attracted significant attention as this yeast has many beneficial properties for the development of an efficient SSF process: thermotolerance, a high growth rate, the utilization of a wide range of sugar types and easy genetic manipulation (Fonseca et al., 2008 ▶). The advantages of operating SSF at high temperatures include a lower energy cost for cooling, higher enzymatic activity, continuous ethanol distillation and a reduced risk of contamination.
We have recently succeeded in constructing a recombinant K. marxianus strain that heterologously expresses thermostable cellulases and have shown that this strain is able to use carboxymethyl cellulose as a sole carbon source (Hong et al., 2007 ▶). During the course of the study, we found that K. marxianus is able to grow, albeit slowly, on a medium containing cellobiose. The same observation was recently reported by Nonklang et al. (2008 ▶). Obtaining a better understanding of the cellobiose metabolism in K. marxianus cells would aid our efforts towards improving the efficiency of the SSF process.
A database search (http://www.cazy.org/; Cantarel et al., 2009 ▶) revealed the presence of a glycoside hydrolase family 3 (GH3) β-glucosidase in K. marxianus. Although its enzymatic characteristics have not been examined, this finding suggests the involvement of this β-glucosidase in the digestion of cellobiose (Raynal & Guerineau, 1984 ▶). The β-glucosidase from K. marxianus consists of 845 amino-acid residues containing three Pfam domains (Finn et al., 2006 ▶): an N-terminal GH3 domain (amino-acid residues 30–245), a C-terminal GH3 domain (residues 321–408 and 560–700) and a PA14 domain (residues 409–559). The striking feature of this enzyme is that the PA14 domain is inserted into the catalytic core of the GH3 domain. The PA14 domain, which is assumed to be involved in carbohydrate binding, is found in various protein structures, but the insertion of this domain into the core domain is only found in C-terminal GH3 domains (Rigden et al., 2004 ▶). Many glycosidases are known to have a modular structure that is comprised of a GH catalytic domain, a carbohydrate-binding domain and an Ig-like domain; however, these domains are generally connected in tandem. Consequently, it is rare for a particular domain to be inserted into the catalytically important conserved domain. Of the ∼1700 members of GH family 3, about 130 have PA14 insertions. This suggests the possibility that the PA14 domain plays a role in the protein structure or in catalysis. As such, elucidation of the enzymatic and structural characteristics of this enzyme is important.
Here, we describe the expression, purification, crystallization and preliminary X-ray analysis of β-glucosidase (BglI) from K. marxianus NBRC1777. The results will provide valuable information on the role of the PA14 domain and in understanding the catalytic mechanism in more detail, as only three members of GH family 3 that do not contain PA14 domains have been structurally analyzed: barley exo-β1,3–1,4-glucanase (ExoI; PDB code 1ex1; Varghese et al., 1999 ▶), Vibrio cholerae β-N-acetylhexosaminidase (NagZ; PDB codes 3gs6 and 3gsm; Balcewich et al., 2009 ▶) and Bacillus subtilis YbbD (PDB code 3bmx; A. Litzinger, S. Fischer, W. Welte, K. Diederichs & C. Mayer, unpublished work).
2. Materials and methods
2.1. Plasmid construction and preparation of the recombinant protein
The cDNA of the β-glucosidase gene (bglI) was obtained by the 5′ RACE and 3′ RACE techniques. In brief, mRNAs were isolated using the RiboPure-Yeast kit (Ambion) from K. marxianus NBRC1777 grown in YPD medium and a cDNA was synthesized using the FirstChoice RLM-RACE kit (Ambion). For 5′ RACE, oligonucleotides 1558-r (first PCR) and 1497-r (second PCR) (see Table 1 ▶) were used as the reverse primers. The primers were designed based on the DNA sequence of β-glucosidase from K. marxianus ATCC12424 (GenBank accession No. X05918; Raynal & Guerineau, 1984 ▶). The forward primers were supplied by the manufacturer. In the nested PCR for 3′ RACE, oligonucleotides 1338-f (first PCR) and 1398-f (second PCR) were used as the forward primers and the reverse primers were supplied by the manufacturer. The amplified fragments were separately sequenced and combined in silico. Finally, the bglI cDNA was obtained by high-fidelity PCR involving KOD-Plus polymerase (Toyobo, Japan). The primers used are listed in Table 1 ▶ (1-f and 2734-r). The DNA sequence was determined from both strands and was deposited in GenBank under accession No. FJ811961. To construct a KmBglI expression vector, the coding region of the bglI gene was amplified using the primer pair BglI-f (forward) and BglI-r (reverse). The forward and reverse primers contained NdeI and BamHI sites, respectively. The amplified fragment was digested with the restriction enzymes and inserted into the corresponding sites of the pET-3a (Novagen) vector. After sequence confirmation, the resulting plasmid was introduced into Escherichia coli B834 (DE3) (Novagen) for the expression of selenomethionine (SeMet) labelled protein. The transformants were grown in LeMaster medium (LeMaster & Richards, 1985 ▶) containing 25 mg l−1 l-SeMet (Wako Pure Chemical Industries, Japan), a vitamin mixture (0.5 mg l−1 thiamine, 1 mg l−1 pyridoxine hydrochloride, 1 mg l−1 calcium pantothenate and 0.5 mg l−1 biotin) and 50 mg l−1 ampicillin at 291 K. Isopropyl β-d-1-thiogalactopyranoside was added to a final concentration of 0.1 mM to induce protein expression when the optical density at 600 nm reached 0.5. Following incubation for a further 20 h, the cells were harvested by centrifugation, suspended in 20 mM Tris–HCl buffer pH 8.0 and disrupted by sonication. Ammonium sulfate was added to the soluble fraction to 60% saturation and the supernatant was recovered and dialyzed against 100 mM potassium phosphate buffer pH 7.0 containing 1 M ammonium sulfate. The dialysate was applied onto a Butyl Sepharose 4 Fast Flow column (GE Healthcare) and the protein was eluted with a linear gradient of 1–0 M ammonium sulfate in 100 mM potassium phosphate buffer pH 7.0. The active fractions were collected, dialyzed against 20 mM Tris–HCl buffer pH 8.0 and subjected to Mono Q 5/50 GL column chromatography (GE Healthcare), in which the protein was eluted by a linear gradient of 0–0.5 M NaCl in 20 mM Tris–HCl buffer pH 8.0. The enzyme was further purified by size-exclusion chromatography on a Superdex 200 10/300 GL column (GE Healthcare). The purified recombinant protein was precipitated by 90% saturated ammonium sulfate, dissolved in 5 mM HEPES buffer pH 7.0 and extensively dialyzed against the same buffer. The protein concentration was calculated from the absorbance at 280 nm using an absorption coefficient of 105 825 M −1 cm−1, which was estimated from the amino-acid composition of KmBglI (Pace et al., 1995 ▶).
Table 1. Oligonucleotides used in this study.
| Name | Sequence |
|---|---|
| 1558-r (for 5′ RACE) | 5′-AGTAGAACAAACCAGAACCATAAAC-3′ |
| 1497-r (for 5′ RACE) | 5′-CACGTACTGTCCGGTTAAGGTTA-3′ |
| 1338-f (for 3′ RACE) | 5′-CAAGTTTTACTCCAATCCAGTAGAA-3′ |
| 1398-f (for 3′ RACE) | 5′-CAAAGTCAATAGATCCAATGTTCAC-3′ |
| 1-f (for full-length PCR) | 5′-AGTAGAGTAGTGGTAGCAAAAGCCG-3′ |
| 2734-r (for full-length PCR) | 5′-ATAACGTGCAATGACCCATCATAAC-3′ |
| BglI-f (for expression-vector construction) | 5′-CCCCATATGTCTAAATTTGATGTTGAACAGT-3′ |
| BglI-r (for expression-vector construction) | 5′-CCCGGATCCTTACAAGCCTTTCCAATACAATTCT-3′ |
2.2. Crystallization
Crystallization was performed by the hanging-drop vapour-diffusion method using 48-well VDX plates (Hampton Research) at 293 K. The initial conditions were screened with the Crystal Screen Cryo formulation (Hampton Research). Each drop was prepared by mixing 1 µl purified protein solution (30 mg ml−1) with the same volume of reservoir solution and was equilibrated against 200 µl reservoir solution. Crystals were obtained in drops containing solution No. 42 of the kit [0.04 M potassium dihydrogen phosphate pH 5.1, 16%(w/v) PEG 8000, 20%(v/v) glycerol]. Unlabelled native protein was initially used for crystallization, but suitable crystals were obtained of SeMet-labelled protein and grew after 20 d incubation (Fig. 1 ▶).
Figure 1.

Crystal of KmBglI. The crystal was grown in a buffer consisting of 0.04 M potassium dihydrogen phosphate pH 5.1, 16%(w/v) PEG 8000 and 20%(v/v) glycerol.
2.3. Data collection
Multiwavelength anomalous dispersion (MAD) data were collected from the SeMet-labelled protein crystal using a charge-coupled device camera on beamline BL-17A of the Photon Factory (PF), Tsukuba, Japan. The crystal was flash-cooled in a nitrogen stream at 100 K. The reservoir solution contained 20%(v/v) glycerol as a cryoprotectant. A total of 720 frames were collected for each data set with 0.5° oscillation and 1 s exposures. Diffraction images were indexed, integrated and scaled with the HKL-2000 program suite (Otwinowski & Minor, 1997 ▶) and the statistics are shown in Table 2 ▶.
Table 2. Crystallographic data-collection statistics for the selenomethionine-labelled crystal.
Values in parentheses are for the highest resolution shell.
| Peak | Edge | Remote | |
|---|---|---|---|
| Wavelength (Å) | 0.97831 | 0.97928 | 0.96405 |
| Beamline | PF BL-17A | ||
| Space group | C2 | ||
| Unit-cell parameters | |||
| a (Å) | 245.8 | ||
| b (Å) | 148.7 | ||
| c (Å) | 119.9 | ||
| β (°) | 112.9 | ||
| Resolution (Å) | 50.00–2.40 (2.49–2.40) | 50.00–2.40 (2.49–2.40) | 50.00–2.50 (2.59–2.50) |
| Measured reflections | 1150410 | 1142105 | 1021559 |
| Unique reflections | 154893 | 154939 | 137340 |
| Completeness (%) | 99.7 (97.4) | 99.7 (97.1) | 99.8 (97.9) |
| Redundancy | 7.5 (5.8) | 7.4 (5.6) | 7.5 (5.9) |
| Mean I/σ(I) | 20.2 (3.2) | 19.9 (3.0) | 18.8 (3.1) |
| Rmerge† (%) | 10.3 (27.6) | 11.7 (29.1) | 11.2 (28.5) |
R
merge = 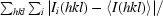
 , where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean value of Ii(hkl) for all i measurements.
, where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean value of Ii(hkl) for all i measurements.
3. Results and discussion
The cDNA of the β-glucosidase gene (bglI) was cloned from K. marxianus NBRC1777. The gene encodes a protein of 845 amino-acid residues with a calculated molecular mass of 94 kDa. The amino-acid sequence was 98% identical to that of β-glucosidase from K. marxianus ATCC12424, the gene for which was isolated from a genomic library (Raynal & Guerineau, 1984 ▶). KmBglI does not have a typical signal peptide and as suggested by Fiol (1970 ▶) this protein should be an intracellular enzyme. The purified protein migrated as a single band with an apparent molecular mass of 95 kDa on SDS–PAGE. Size-exclusion chromatography indicated that the molecular mass of the protein was 390 kDa, strongly suggesting the presence of the tetrameric form of the enzyme in solution (data not shown). The crystal of KmBglI belonged to the monoclinic space group C2 and diffracted to 2.4 Å resolution. Assuming the presence of one tetramer in the asymmetric unit, the V M value (Matthews, 1968 ▶) and solvent content were determined to be 2.6 Å3 Da−1 and 52.5%, respectively.
The characteristic feature of KmBglI is the insertion of the PA14 domain into the catalytic core of the C-terminal GH3 domain. Of the members of GH family 3, about 130 have similar domain architectures, and no structures with an inserted PA14 domain are available. Three crystal structures of GH family 3 enzymes are currently available: those of barley ExoI (630 residues; PDB code 1ex1; Varghese et al., 1999 ▶), NagZ from V. cholerae (330 residues; PDB codes 3gs6 and 3gsm; Balcewich et al., 2009 ▶) and B. subtilis YbbD (PDB code 3bmx). Recently, preliminary X-ray diffraction analysis was reported for Thermotoga neapolitana β-glucosidase B (Turner et al., 2007 ▶). The overall sequence identity of KmBglI to ExoI, NagZ and YbbD was 17, 13 and 15%, respectively. Consequently, owing to the low sequence identity and potential structural discrepancies between the four proteins, the KmBglI crystal could not be phased by the molecular-replacement method using the above structures as search templates. MAD phasing is currently in progress. Crystallization and X-ray diffraction analysis of the unlabelled native protein will be described in a separate paper together with its enzymatic characteristics.
Acknowledgments
We wish to thank the staff of the Photon Factory, KEK for data collection. This work was supported in part by a Grant-in-Aid from the New Energy and Industrial Technology Development Organization (NEDO), Japan.
References
- Balcewich, M. D., Stubbs, K. A., He, Y., James, T. W., Davies, G. J., Vocadlo, D. J. & Mark, B. L. (2009). Protein Sci.18, 1541–1551. [DOI] [PMC free article] [PubMed]
- Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V. & Henrissat, B. (2009). Nucleic Acids Res.37, D233–D238. [DOI] [PMC free article] [PubMed]
- Finn, R. D., Mistry, J., Schuster-Bockler, B., Griffiths-Jones, S., Hollich, V., Lassmann, T., Moxon, S., Marshall, M., Khanna, A., Durbin, R., Eddy, S. R., Sonnhammer, E. L. & Bateman, A. (2006). Nucleic Acids Res.34, D247–D251. [DOI] [PMC free article] [PubMed]
- Fiol, J. B. (1970). Ann. Inst. Pasteur, 118, 697–708. [PubMed]
- Fonseca, G. G., Heinzle, E., Wittmann, C. & Gombert, A. K. (2008). Appl. Microbiol. Biotechnol.79, 339–354. [DOI] [PubMed]
- Hong, J., Wang, Y., Kumagai, H. & Tamaki, H. (2007). J. Biotechnol.130, 114–123. [DOI] [PubMed]
- LeMaster, D. M. & Richards, F. M. (1985). Biochemistry, 24, 7263–7268. [DOI] [PubMed]
- Maris, A. J. van, Abbott, D. A., Bellissimi, E., van den Brink, J., Kuyper, M., Luttik, M. A., Wisselink, H. W., Scheffers, W. A., van Dijken, J. P. & Pronk, J. T. (2006). Antonie Van Leeuwenhoek, 90, 391–418. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Nonklang, S., Abdel-Banat, B. M., Cha-aim, K., Moonjai, N., Hoshida, H., Limtong, S., Yamada, M. & Akada, R. (2008). Appl. Environ. Microbiol.74, 7514–7521. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pace, C. N., Vajdos, F., Fee, L., Grimsley, G. & Gray, T. (1995). Protein Sci.4, 2411–2423. [DOI] [PMC free article] [PubMed]
- Raynal, A. & Guerineau, M. (1984). Mol. Gen. Genet.195, 108–115. [DOI] [PubMed]
- Rigden, D. J., Mello, L. V. & Galperin, M. Y. (2004). Trends Biochem. Sci.29, 335–339. [DOI] [PubMed]
- Turner, P., Pramhed, A., Kanders, E., Hedström, M., Karlsson, E. N. & Logan, D. T. (2007). Acta Cryst. F63, 802–806. [DOI] [PMC free article] [PubMed]
- Varghese, J. N., Hrmova, M. & Fincher, G. B. (1999). Structure, 7, 179–190. [DOI] [PubMed]