

Abstract
Objective:
Posterior cortical atrophy (PCA) and logopenic progressive aphasia (LPA) are clinical syndromes associated with posterior brain atrophy. We compared PCA and LPA to each other and to an age-matched group of patients with early age at onset of Alzheimer disease (EO-AD). We hypothesized that these 3 syndromes are part of a single clinical and biologic continuum.
Methods:
Voxel-based morphometry (VBM) was used to assess atrophy in 14 PCA, 10 LPA, and 16 EO-AD patients compared to 65 healthy controls. Genetic analysis for APOE was conducted in 30 patients and 44 controls. Four patients came to autopsy. An additional 14 were studied with the beta-amyloid specific PET with tracer 11C-labeled Pittsburgh Compound-B (PIB).
Results:
VBM results demonstrated that, compared to controls, each patient group showed a large area of overlapping atrophy in bilateral parietal, occipital, precuneus, posterior cingulate, posterior temporal, and hippocampal regions. Surrounding this common area, group-specific atrophy was found in small, symptom-specific regions for each group: the right ventral-occipital and superior parietal regions in PCA, the left middle and superior temporal gyri in LPA, and the prefrontal cortex in EO-AD. APOE ε4 frequency was higher in all patient groups compared to controls. Four PCA, 5 LPA, and 8 EO-AD patients showed evidence of cortical amyloid at pathology (n = 3) or on PIB-PET (n = 14).
Conclusions:
Logopenic progressive aphasia and posterior cortical atrophy showed largely overlapping anatomic and biologic features with early age at onset of Alzheimer disease, suggesting that these clinical syndromes represent the spectrum of clinical manifestation of the nontypical form of Alzheimer disease that presents at an early age.
GLOSSARY
- AD
= Alzheimer disease;
- CBD
= corticobasal degeneration;
- EO-AD
= early age at onset of Alzheimer disease;
- LPA
= logopenic progressive aphasia;
- MAC
= Memory and Aging Center;
- PCA
= posterior cortical atrophy;
- PIB
= Pittsburgh Compound-B;
- PPA
= primary progressive aphasia;
- UCSF
= University of California San Francisco;
- VBM
= voxel-based morphometry.
It is well known that a progressive amnestic syndrome in an elderly subject is highly predictive of Alzheimer disease (AD) pathology. More surprising is that amyloid plaques and neurofibrillary tangles are also found in younger patients (under age 65) in association with atypical, focal, clinical syndromes in which a single cognitive domain, not related to memory, is predominantly affected. Examples of such syndromes are the logopenic variant of primary progressive aphasia (LPA)1,2 and posterior cortical atrophy (PCA).3-5 Both of these syndromes have been associated with a posterior pattern of atrophy, with relatively less involvement of the medial temporal lobe. This neuroimaging pattern differs from typical AD but has been described in sporadic early age at onset forms of AD (EO-AD).6,7
PCA is characterized by early impairment of visuospatial skills with less prominent memory loss3,8,9 and is associated with atrophy in parieto-occipital and posterior temporal cortices with right predominance.10 LPA is a clinical variant of primary progressive aphasia (PPA),11 characterized by slow speech with frequent word-finding pauses, and deficits in sentence repetition.12 LPA is associated with left-hemisphere predominant atrophy in posterior temporal cortex and inferior parietal areas.1,12,13 The term EO-AD applies to patients who meet criteria for AD,14 but show onset of symptoms before the age of 65. Clinically, EO-AD patients present with an early multidomain cognitive impairment including language, visuospatial, and executive functions difficulties. Memory deficits are variable, and often not predominant.15,16 Several recent imaging and pathologic studies of EO-AD demonstrated greater atrophy and pathologic burden in parietal and frontal cortices, with relative sparing of hippocampus, when compared to classic, late age at onset AD.7,17-19
In this study, we directly compared clinical, cognitive, neuroimaging, and genetic data and assessed for the presence of cortical β-amyloid in a group of patients with PCA, LPA, and EO-AD. We hypothesize that patients with PCA and LPA and an age-appropriate group of patients with clinically defined EO-AD would be more similar than previously thought, and may represent the spectrum of clinical manifestation of the same disease.
METHODS
Subjects.
Eligible subjects were identified by searching the database at the University of California San Francisco (UCSF) Memory and Aging Center (MAC) for patients meeting criteria for PCA, PPA, and EO-AD. Clinical diagnosis was prospectively obtained and based on multidisciplinary evaluation including history and neurologic examination by a neurologist, caregiver interview by a nurse, and neuropsychological test battery. The following diagnostic criteria were applied: Alladi et al.5 and McMonagle et al.9 for PCA; Gorno-Tempini et al.12 for LPA; McKhann et al.14 criteria and symptom onset prior to age 65 for EO-AD (table e-1 on the Neurology® Web site at www.neurology.org). First-symptom research questionnaires of each patient were reviewed by an expert neurologist (R.M.) to determine the presence or absence of specific clinical symptoms at disease onset (table 2). All patients with PCA endorsed severe visuospatial complaints that were the only problems impacting their everyday life. However, on detailed questioning they also endorsed mild episodic memory loss, and some word-finding and calculation deficits. At disease onset, patients with LPA only reported language complaints. Patients with EO-AD reported memory loss, difficulties in visuospatial and executive functions, as well as word-finding problems, and deficits in all domains were severe enough to impact their everyday functioning. In order to be included in the study, patients needed to have an MRI within 6 months of the first clinical evaluation. All cases were evaluated as part of an ongoing research project and identified based on clinical criteria. The cohort consisted of 14 patients with PCA, 10 patients with LPA, and 16 patients with EO-AD (table 1). The patients with EO-AD were consecutively selected within a larger sample in order to match PCA and LPA groups for gender, disease duration, and severity. No patient had a dominant family history of dementia or psychiatric diseases.
Table 2 Frequency of symptoms and signs at disease onset, at presentation, and at follow-up (mean interval of 21.3 months from disease onset) in patients with posterior cortical atrophy, logopenic/phonological progressive aphasia, and Alzheimer disease with early age at onset (formal cognitive testing scores were not considered)
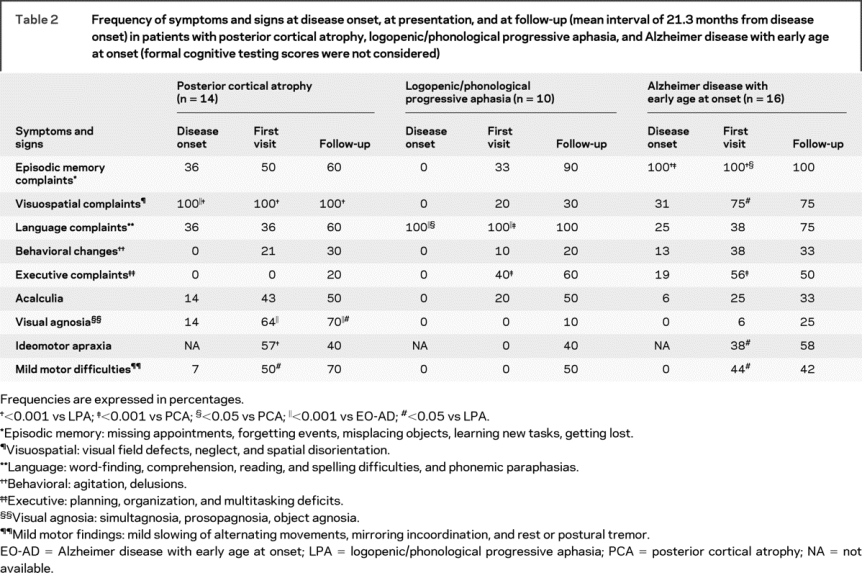
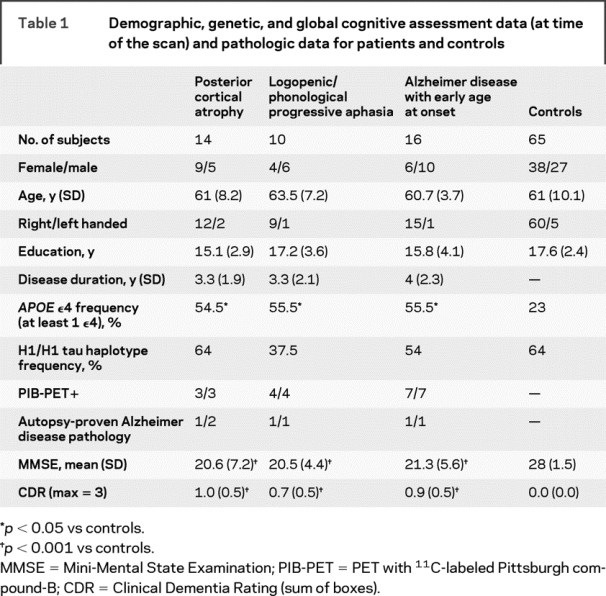
Table 1 Demographic, genetic, and global cognitive assessment data (at time of the scan) and pathologic data for patients and controls
Patient medical records were further reviewed to establish the presence or absence of specific clinical symptoms at presentation at the UCSF MAC, and at follow-up visit at 1 to 2 years (mean 21.3 months ± 5.2) (table 2).
Sixty-five age- and sex-matched healthy subjects, with no history of neurologic or psychiatric disorders, were used as controls for the MRI and the genetic study (38 women, 27 men; mean age = 61 ± 10 years). The study was approved by the UCSF committee on human research. All subjects provided written informed consent before participating.
Cognitive testing.
The neuropsychological measures included in our bedside screening protocol have been described previously20 (table e-2). Patients meeting PPA criteria underwent a comprehensive language and speech evaluation as previously described.12
Neuroimaging study.
MRI scanning and analysis.
MRI scans were obtained on a 1.5 Tesla Magnetom VISION system (Siemens, Iselin, NJ). Structural MRI sequences included double spin echo sequence and a volumetric magnetization prepared rapid gradient echo sequence, as previously described.12 MRI analysis was conducted using standard methods of optimized VBM21,22 in the SPM2 software package (Wellcome Department of Imaging Neuroscience, London; http://www.fil.ion.ucl.ac.uk/spm). Ad hoc template and a priori images were created by averaging both normal control and patient scans, Jacobian modulation was performed, and GM images used for analysis were smoothed with a 12-mm FWHM isotropic Gaussian kernel. Age, gender, and total intracranial volume were entered into the design matrix as nuisance variables.
The following sets of linear contrasts were performed to identify regional GM atrophy: 1) each syndrome vs controls (PCA vs controls: [−1 0 0 1]; LPA vs controls: [0 −1 0 1]; EO-AD vs controls: [0 0 −1 1]); 2) all patient groups vs controls [−1 −1 −1 3]; 3) each syndrome vs the other 2 syndromes (PCA vs LPA and EO-AD [−1 1 0 0; −1 0 −1 0]; LPA vs PCA and EO-AD [1 −1 0 0; 0 −1 1 0], EO-AD vs PCA and LPA [1 0 −1 0; 0 1 −1 0]). First, direct comparisons were performed to identify GM atrophy in each syndrome vs controls (contrast set 1). To investigate regions of common atrophy across syndromes the results of contrast 2 were inclusively masked by all contrasts of set 1 (the inclusive masking procedure limits the main effect contrast to regions that are also present in each syndrome vs controls contrast). Then, regions of atrophy specific to each syndrome compared to the other syndromes were identified by masking the relevant contrast from set 1 (e.g., PCA vs controls) with the appropriate contrast from set 3 (e.g., PCA vs LPA and EO-AD). A significance threshold of p < 0.05 corrected for multiple comparisons (family-wise error) was accepted when comparing patients vs controls and of p < 0.001 when directly comparing between patient groups.
Genetic methods.
APOE ε4 is a known risk factor for AD.23 We also considered the microtubule-associated protein τ (MAPT) H1/H1 allele that was found in association with tauopathies, such as corticobasal degeneration (CBD) and progressive supranuclear palsy24 (for further details, see appendix e-1). Genetic analysis for APOE genotype was available for 29 patients (11 PCA, 9 LPA, and 9 EO-AD) and 44 controls, while MAPT haplotype analysis was available for 32 patients (11 PCA, 8 LPA, and 13 EO-AD) and 42 controls.
PIB-PET and pathologic data.
The PET with tracer 11C-labeled Pittsburgh Compound-B (PIB) study was conducted in a subgroup of patients: 3 PCA, 4 LPA, and 7 EO-AD, using the procedures previously described.25 The autopsy study was performed at UCSF for 4 additional patients: 2 PCA, 1 LPA, and 1 EO-AD, using a previously published protocol.26 The pathologic diagnosis was based on previously published criteria (AD27; CBD28).
Statistical analyses.
Group comparisons in continuous data were evaluated using analysis of variance and post hoc Tukey tests, while dichotomous variables were compared using the χ2 test. Statistical analyses were implemented using SPSS software (version 10.0.05 for windows; SPSS, Chicago, IL).
RESULTS
Demographic and clinical data.
There were no significant group differences in gender, age, handedness, disease duration, or Clinical Dementia Rating score (table 1). At time of first clinical evaluation (table 2), several subjects with PCA endorsed visual agnosia, ideomotor apraxia, and mild motor signs. Subjects with LPA also reported mild praxis and calculation difficulties, and some mild episodic memory and executive complaints, but these symptoms never became cause of significant functional impairment. Patients with EO-AD showed executive difficulties more frequently than patients with PCA, and demonstrated visuospatial deficits, ideomotor apraxia, and mild motor signs more frequently than patients with LPA (table 2). At time of follow-up evaluation, there were no differences in the prevalence of memory, language, executive, calculation, behavioral, praxic, and motor deficits in the 3 syndromes. However, LPA continued to show visuospatial deficits less frequently than PCA, and PCA still demonstrated visual agnosia more commonly than EO-AD and LPA. Furthermore, for PCA and LPA the main cause of functional impairment remained visuospatial and language, respectively (table 2).
Motor signs were always mild and never justified a diagnosis of CBD, progressive supranuclear palsy, or Lewy Body dementia.
Cognitive data.
MMSE scores at time of diagnosis were not significantly different in the 3 groups (table 1). Patients with PCA scored lower on visuospatial tasks than both patients with LPA and patients with EO-AD, and lower than patients with LPA on visual memory and executive tasks that involve visuospatial material, such as design fluency. Patients with LPA performed better than other groups on non-language-based tests. They did not show worse performance on the naming and word generation tasks (such as verbal fluency) included in the general neuropsychological screening battery. Patients with EO-AD performed worse than patients with LPA on a visual memory task (table e-2).
Neuroimaging data.
Areas involved in each syndrome vs controls (figure 1 and table e-3): When compared to controls, PCA showed GM atrophy in temporo-parietal-occipital regions and hippocampus, bilaterally, with right-sided predominance. Small clusters were found in the precentral and middle frontal gyri, bilaterally. LPA showed bilateral GM atrophy at the temporo-parietal junctions, with left-hemisphere predominance, as well as small clusters in precentral and inferior frontal gyri. EO-AD demonstrated GM atrophy in left hippocampus and bilateral medial (precuneus, posterior cingulate) and lateral parietal and temporal cortex (middle and superior temporal gyri, superior and inferior parietal lobule) and in bilateral middle and inferior frontal gyri. Figure e-1 shows examples of native MRIs of 3 representative patients.
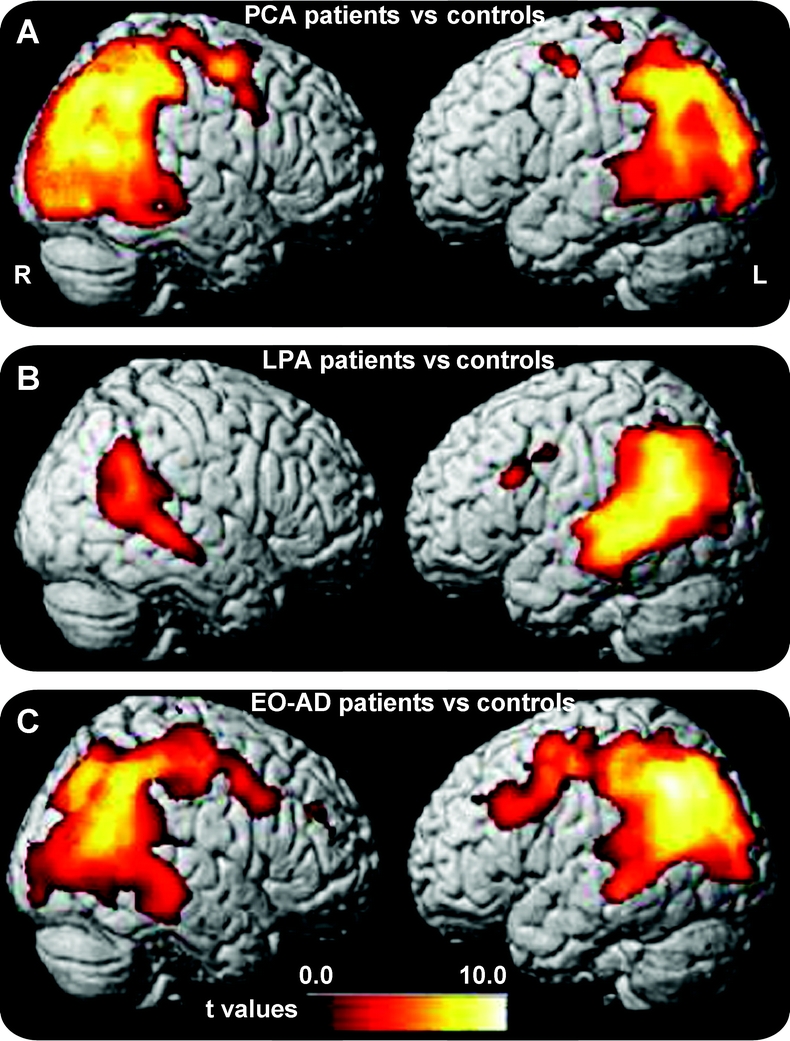
Figure 1 Gray matter areas significantly atrophied in each patient group vs controls
(A) Posterior cortical atrophy (PCA), (B) logopenic/phonological progressive aphasia (LPA), and (C) early age at onset AD (EO-AD). Regions of gray matter (GM) atrophy are shown on the 3-dimensional rendering of the Montreal Neurological Institute standard brain. Results are shown at a threshold of p < 0.05 corrected.
Areas common to all syndromes (figure 2, A and B, and table e-3): GM atrophy was found in bilateral middle occipital gyrus, bilateral posterior cingulate, left precuneus, bilateral inferior parietal lobule, posterior portions of superior and middle temporal gyri, and left hippocampus. Smaller clusters were also found in bilateral precentral gyrus, and the middle and inferior frontal gyri bilaterally. In the left panel of figure e-2, volumes of the most significant GM voxels are plotted for common areas.
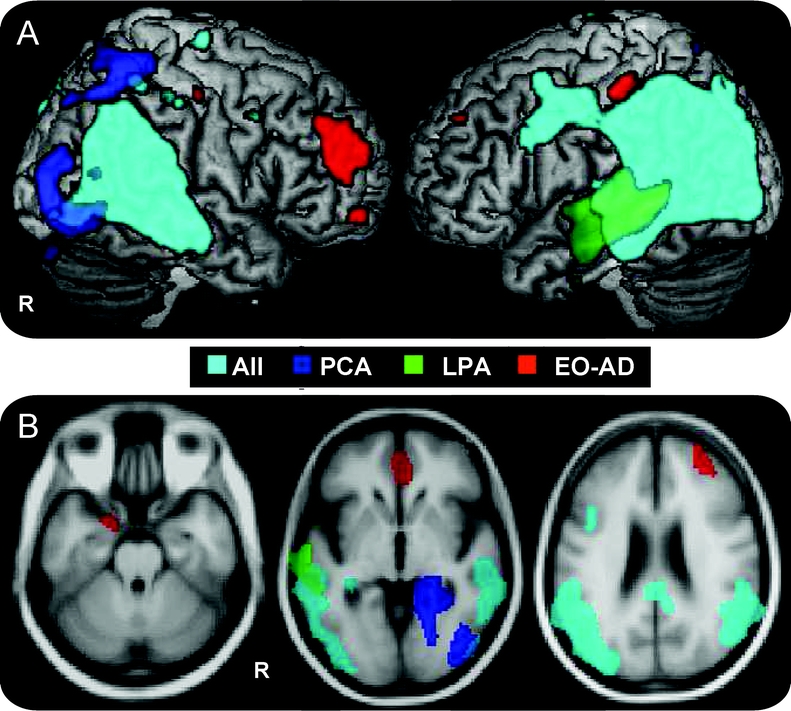
Figure 2 Common and specific areas of gray matter atrophy
Gray matter (GM) atrophy common to all patients vs controls is shown in cyan, while GM regions specifically atrophied in each group compared with controls are indicated in blue for posterior cortical atrophy (PCA), green for logopenic/phonological progressive aphasia (LPA), and red for early age at onset AD (EO-AD). Results are superimposed on the 3-dimensional rendering of the Montreal Neurological Institute standard brain and on axial sections of the mean image of the scans used to obtain the template image. For display purposes, results are shown at a threshold of p < 0.05 corrected for common and of p < 0.001 uncorrected for specific areas.
Areas specifically affected in each syndrome (figure 2, A and B, and table e-3): GM loss specific to PCA included right middle occipital gyrus, bilateral lingual and fusiform gyri, right superior parietal gyrus, and right hippocampus. LPA-specific GM atrophy occurred in the middle third of the left middle temporal gyrus and left superior temporal sulcus. EO-AD-specific GM atrophy was medially distributed in the right anterior cingulate, right middle frontal gyrus, and left hippocampus. In the right panel of figure e-2, volumes of the most significant GM voxels are plotted for syndrome-specific areas.
Genetic data.
The frequency of the APOE ε4 genotype was higher in patients than controls (p < 0.05), but was not different between patient groups. Frequency of the H1/H1 τ haplotype was not different across groups and compared to controls.
PIB and pathologic data.
All patients studied with PIB-PET showed elevated cortical tracer retention on visual inspection, showing presence of amyloid deposition. Three patients had AD pathology at autopsy: 1 PCA, 1 LPA, and 1 EO-AD. One PCA patient received the pathologic diagnosis of CBD.
Post hoc imaging analysis of autopsy-proven and PIB-positive subgroup.
Four patients with PCA, 5 patients with LPA, and 8 patients with EO-AD with evidence of cortical amyloid were compared to a control group. Contrasts were identical to the general analysis but the statistical threshold was p < 0.001 uncorrected because of the small number of subjects. Common and specific areas were comparable to the larger group analysis (figure 3).

Figure 3 Pattern of gray matter atrophy common to all autopsy-proven and PIB-positive patient groups vs controls
Results are superimposed on the 3-dimensional rendering of the Montreal Neurological Institute standard brain and on coronal section of the mean image of the scans used to obtain the template image. Results are shown at a threshold of p < 0.001 uncorrected.
DISCUSSION
We investigated the clinical, neuroimaging, and biologic features associated with PCA, LPA, and EO-AD. Our findings show that 1) PCA and LPA, initially very distinct, subsequently share some symptoms and signs with EO-AD, although maintaining greater impairment in the visuospatial or language domain; 2) all 3 groups are associated with a largely overlapping pattern of atrophy centered in the temporoparietal region, surrounded by smaller syndrome-specific areas; and 3) all groups share common genetic, pathologic, and PIB-PET features suggestive of AD etiology. We discuss these results, proposing a framework in which PCA and LPA are 2 possible clinical presentations of the variant of AD that presents below 65 years of age (EO-AD).
Clinical diagnosis of PCA and LPA were established prospectively at UCSF and patients presented at disease onset with distinct clinical syndromes with a clear predominance of visuospatial or language complaint. Retrospective analysis of specific symptoms and signs showed that few patients with PCA also reported other mild cognitive complaints at disease onset, while most had language and memory deficits at presentation at a tertiary memory clinic (a mean of 3.5 years from onset). On the other hand, no patients with LPA had non-language symptoms at onset and only a third reported mild episodic memory and apraxic and calculation problems at diagnosis. It is interesting that limb apraxia and acalculia have been considered the only non-language symptoms compatible with a diagnosis of PPA, even at onset.11 In general, the greater focality of the clinical presentation in LPA might depend on the fact that mild language deficits are noticed earlier and are more functionally significant than mild visuospatial difficulties.
Partially overlapping clinical features suggest the possibility of shared posterior anatomic involvement in PCA, LPA, and EO-AD. In this study, we directly compared the specific distribution of atrophy in these 3 syndromes at presentation and showed largely overlapping damage to the temporoparietal region. Around this large region of common atrophy, small distinctive regions were associated with each of the 3 syndromes. Consistently with their visual disturbance, patients with PCA showed greater atrophy in the right temporo-occipital and dorsal parietal regions previously associated with the ventral and dorsal visual pathways.9,29 LPA showed greater left posterior middle and superior temporal atrophy compatible with their greater language comprehension and production deficits.30 Finally, EO-AD showed greater frontal volume loss, in keeping with their greater executive difficulties.31 Our study is therefore consistent with previous MRI results that considered each of these syndromes separately,7,10,12 and previous fluorodeoxyglucose PET findings on atypical language and visuospatial AD presentation.32,33
No previous study had directly compared the specific clinical syndromes considered here, because they were originally considered as separate entities from each other and from EO-AD. In particular, both PCA and LPA (as a variant of PPA) were classically considered as being more often caused by non-AD pathologic changes. Instead, more recent evidence on PCA and LPA biology suggests that AD might be the most common underlying pathology.2,4,5 Therefore, we performed such direct comparison by entering images from all 3 groups in the same analysis, to investigate commonalities and differences. Results showed that there was a large region of overlapping atrophy in all patient groups in the temporoparietal network. This network is known to be preferentially affected pathologically, structurally, and functionally in AD, especially in younger patients.7,17-19,34,35 Although location of atrophy is not an absolute marker of pathology, it does increase the probability of a certain underlying pathologic process, since different brain regions seem to be more vulnerable to specific diseases.36 For this reason, site of atrophy has been included in the most recent criteria for AD.37 In our patient populations, the presence of common atrophy in temporoparietal regions suggests a high probability that the underlying biologic process is AD in all 3 clinical syndromes. Our biologic findings also support this view and suggest that PCA and LPA are most often clinical variants of EO-AD.
The genetic study showed that the 3 groups demonstrated higher than normal frequency of the APOE ε4 allele, and thus had increased genetic risk for AD pathology. On the other hand, none of our clinical groups showed increased risk for a tauopathy, since H1/H1 MAPT haplotype was comparable to controls. This result is particularly relevant since PCA was once thought to be frequently associated with CBD. MAPT haplotype data were not available for the PCA case with CBD at pathology. Consistent with the anatomic and genetic findings, molecular imaging and pathologic results in a subgroup of 18 patients showed evidence of AD-related changes in all cases of LPA (5/5) and EO-AD (8/8), and in all but 1 case of PCA (4/5). For 14 patients, PIB-PET data but not final pathologic specimens were available. While recent studies confirmed that the presence of cortical PIB-PET in vivo signal is highly correlated with the beta-amyloidosis of AD on autopsy,38,39 the presence of copathology cannot be excluded.25,40 Our subject sample with pathologic and PIB data is relatively small, although comparable to other pathologic series.
Taken together, our results and previous findings sustain that AD is the most common pathology underlying 2 different clinical syndromes associated with asymmetric posterior atrophy. PCA and LPA can thus be considered variants of EO-AD, just as progressive nonfluent aphasia and behavioral variant frontotemporal dementia are associated with asymmetric frontal atrophy and are considered clinical variants of frontotemporal lobar degeneration spectrum pathology. It remains unclear why posterior brain regions are particularly vulnerable to AD, especially in younger subjects, and why different individuals present with such asymmetric patterns of cognitive deficits and atrophy. Future studies are needed to identify how genetic, developmental, and environmental factors interact to determine the different vulnerability of the language or visuospatial networks in different subjects. Current research criteria emphasize memory loss in AD, and may miss early age at onset cases that present with isolated clinical syndromes associated with posterior brain involvement. Thus, extending the clinical phenotype of EO-AD to include visuospatial and language presentations is critical, considering that these patients could be good candidates for emerging anti-β-amyloid therapies.
This study has the limitation that the number of patients who underwent autopsy or PIB-PET is relatively small. For this reason, observations based on neuropathologic data should be read considering this constraint.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Dr. Migliaccio, Dr. Agosta, and Dr. Rascovsky.
ACKNOWLEDGMENT
The authors thank William Jagust (Helen Wills Neuroscience Institute, University of California, Berkeley), Nina Dronkers (VA Northern California Health Care System, Martinez), Adam Boxer (UCSF–MAC, San Francisco, CA), Stephen Wilson (UCSF–MAC, San Francisco, CA), and Patrizia Montella (University of Naples, Italy) for their comments. They also thank all the study participants and their families for their support of our research.
DISCLOSURE
Dr. Migliaccio, Dr. Agosta, Dr. Rascovsky, and A. Karydas report no disclosures. Dr. Bonasera has received honoraria for lectures or educational activities not funded by industry and receives research support from the NIH [R21 AG26043 (PI) and K08 MH065983 (PI)], the UCSF REAC Investigator Program, and the Stephen D. Bechtel Foundation. Dr. Rabinovici receives research support from the NIH [NIA K23-AG031861 (PI)], the Alzheimer’s Association, and the John Douglas French Alzheimer’s Foundation. Dr. Miller has served on speakers’ bureaus for Novartis and Pfizer Inc. and served on an editorial advisory board for Neurocase. Dr. Gorno-Tempini receives research support from the NIH [R01 NS050915 (P1)].
Supplementary Material
Address correspondence and reprint requests to Dr. Maria Luisa Gorno-Tempini, Memory and Aging Center, Department of Neurology, University of California, San Francisco, 350 Parnassus Avenue, Suite 905, San Francisco, CA 94143-1207 marilu@memory.ucsf.edu
Supplemental data at www.neurology.org
Supported by the NIH (NINDS R01 NS050915, NIA P50 AG03006, NIA P01 AG019724); State of California (DHS 04-35516); Alzheimer’s Disease Research Center of California (03-75271 DHS/ADP/ARCC); John Douglas French Alzheimer’s Foundation; Alzheimer’s Association (NIRG-07-59422); Larry L. Hillblom Foundation; Koret Family Foundation; and McBean Family Foundation. PIB-PET scans were supported by NIA AG027859.
Disclosure: Author disclosures are provided at the end of the article.
Received March 3, 2009. Accepted in final form August 7, 2009.
REFERENCES
- 1.Josephs KA, Whitwell JL, Duffy JR, et al. Progressive aphasia secondary to Alzheimer disease vs FTLD pathology. Neurology 2008;70:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mesulam M, Wicklund A, Johnson N, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol 2008;63:709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang-Wai DF, Graff-Radford NR, Boeve BF, et al. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology 2004;63:1168–1174. [DOI] [PubMed] [Google Scholar]
- 4.von Gunten A, Bouras C, Kovari E, Giannakopoulos P, Hof PR. Neural substrates of cognitive and behavioral deficits in atypical Alzheimer’s disease. Brain Res Rev 2006;51:176–211. [DOI] [PubMed] [Google Scholar]
- 5.Alladi S, Xuereb J, Bak T, et al. Focal cortical presentations of Alzheimer’s disease. Brain 2007;130:2636–2645. [DOI] [PubMed] [Google Scholar]
- 6.Frisoni GB, Testa C, Sabattoli F, Beltramello A, Soininen H, Laakso MP. Structural correlates of early and late onset Alzheimer’s disease: voxel based morphometric study. J Neurol Neurosurg Psychiatry 2005;76:112–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frisoni GB, Pievani M, Testa C, et al. The topography of grey matter involvement in early and late onset Alzheimer’s disease. Brain 2007;130:720–730. [DOI] [PubMed] [Google Scholar]
- 8.Benson DF, Davis RJ, Snyder BD. Posterior cortical atrophy. Arch Neurol 1988;45:789–793. [DOI] [PubMed] [Google Scholar]
- 9.McMonagle P, Deering F, Berliner Y, Kertesz A. The cognitive profile of posterior cortical atrophy. Neurology 2006;66:331–338. [DOI] [PubMed] [Google Scholar]
- 10.Whitwell JL, Jack CR, Jr., Kantarci K, et al. Imaging correlates of posterior cortical atrophy. Neurobiol Aging 2007;28:1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mesulam MM. Primary progressive aphasia. Ann Neurol 2001;49:425–432. [PubMed] [Google Scholar]
- 12.Gorno-Tempini ML, Dronkers NF, Rankin KP, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol 2004;55:335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knibb JA, Xuereb JH, Patterson K, Hodges JR. Clinical and pathological characterization of progressive aphasia. Ann Neurol 2006;59:156–165. [DOI] [PubMed] [Google Scholar]
- 14.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 15.Suribhatla S, Baillon S, Dennis M, et al. Neuropsychological performance in early and late onset Alzheimer’s disease: comparisons in a memory clinic population. Int J Geriatr Psychiatry 2004;19:1140–1147. [DOI] [PubMed] [Google Scholar]
- 16.Licht EA, McMurtray AM, Saul RE, Mendez MF. Cognitive differences between early- and late-onset Alzheimer’s disease. Am J Alzheimers Dis Other Demen 2007;22:218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shiino A, Watanabe T, Maeda K, Kotani E, Akiguchi I, Matsuda M. Four subgroups of Alzheimer’s disease based on patterns of atrophy using VBM and a unique pattern for early onset disease. Neuroimage 2006;33:17–26. [DOI] [PubMed] [Google Scholar]
- 18.Marshall GA, Fairbanks LA, Tekin S, Vinters HV, Cummings JL. Early-onset Alzheimer’s disease is associated with greater pathologic burden. J Geriatr Psychiatry Neurol 2007;20:29–33. [DOI] [PubMed] [Google Scholar]
- 19.Karas G, Scheltens P, Rombouts S, et al. Precuneus atrophy in early-onset Alzheimer’s disease: a morphometric structural MRI study. Neuroradiology 2007;49:967–976. [DOI] [PubMed] [Google Scholar]
- 20.Kramer JH, Jurik J, Sha SJ, et al. Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol 2003;16:211–218. [DOI] [PubMed] [Google Scholar]
- 21.Ashburner J, Friston KJ. Voxel-based morphometry: the methods. Neuroimage 2000;11:805–821. [DOI] [PubMed] [Google Scholar]
- 22.Good CD, Scahill RI, Fox NC, et al. Automatic differentiation of anatomical patterns in the human brain: validation with studies of degenerative dementias. Neuroimage 2002;17:29–46. [DOI] [PubMed] [Google Scholar]
- 23.Bird TD. Genetic aspects of Alzheimer disease. Genet Med 2008;10:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldman JS, Adamson J, Karydas A, Miller BL, Hutton M. New genes, new dilemmas: FTLD genetics and its implications for families. Am J Alzheimers Dis Other Demen 2007;22:507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rabinovici G, Furst A, Miller BL, Jagust WJ, Gorno-Tempini ML. [11C] PIB PET in three variants of primary progressive aphasia. Neurology 2007;68:A60. [Google Scholar]
- 26.Forman MS, Farmer J, Johnson JK, et al. Frontotemporal dementia: clinicopathological correlations. Ann Neurol 2006;59:952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging 1997;18:S1–S2. [PubMed] [Google Scholar]
- 28.Josephs KA, Petersen RC, Knopman DS, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 2006;66:41–48. [DOI] [PubMed] [Google Scholar]
- 29.Ungerleider LG MM. Two cortical visual system. In: Ingle DJ, Mansfiled RJW, Goodale MD, eds. The Analysis of Visual Behavior. Cambridge, MA: MIT Press; 1982:549–586. [Google Scholar]
- 30.Price CJ. The anatomy of language: contributions from functional neuroimaging. J Anat 2000;197:335–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Damasio AR. The frontal lobes. In: Heilman KM, Valenstein E, eds. Clinical Neuropsychology, 2nd ed. New York: Oxford University Press; 1985. [Google Scholar]
- 32.Haxby JV, Grady CL, Koss E, et al. Longitudinal study of cerebral metabolic asymmetries and associated neuropsychological patterns in early dementia of the Alzheimer type. Arch Neurol 1990;47:753–760. [DOI] [PubMed] [Google Scholar]
- 33.Martin A, Brouwers P, Lalonde F, et al. Towards a behavioral typology of Alzheimer’s patients. J Clin Exp Neuropsychol 1986;8:594–610. [DOI] [PubMed] [Google Scholar]
- 34.Bigio EH, Hynan LS, Sontag E, Satumtira S, White CL. Synapse loss is greater in presenile than senile onset Alzheimer disease: implications for the cognitive reserve hypothesis. Neuropathol Appl Neurobiol 2002;28:218–227. [DOI] [PubMed] [Google Scholar]
- 35.Ishii K, Kawachi T, Sasaki H, et al. Voxel-based morphometric comparison between early- and late-onset mild Alzheimer’s disease and assessment of diagnostic performance of z score images. AJNR Am J Neuroradiol 2005;26:333–340. [PMC free article] [PubMed] [Google Scholar]
- 36.Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron 2009;62:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol 2007;6:734–746. [DOI] [PubMed] [Google Scholar]
- 38.Bacskai BJ, Frosch MP, Freeman SH, et al. Molecular imaging with Pittsburgh Compound B confirmed at autopsy: a case report. Arch Neurol 2007;64:431–434. [DOI] [PubMed] [Google Scholar]
- 39.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain 2008;131:1630–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol 2004;55:306–319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.