

Abstract
Objective:
To examine whether obesity during childhood, adolescence, or adulthood is associated with an increased risk of multiple sclerosis (MS).
Methods:
Women in the Nurses’ Health Study (n = 121,700) and Nurses’ Health Study II (n = 116,671) provided information on weight at age 18 and weight and height at baseline, from which body mass index was derived. Women also selected silhouettes representing their body size at ages 5, 10, and 20. Over the total 40 years of follow-up in both cohorts combined, we confirmed 593 cases of MS. Cox proportional hazards models, adjusting for age, latitude of residence, ethnicity, and cigarette smoking, were used to estimate the rate ratios and 95% confidence intervals (CI).
Results:
Obesity at age 18 (body mass index ≥30 kg/m2) was associated with a greater than twofold increased risk of MS (multivariate relative riskpooled = 2.25, 95% CI: 1.50-3.37, p trend <0.001). After adjusting for body size at age 20, having a large body size at ages 5 or 10 was not associated with risk of MS, whereas a large body size at age 20 was associated with a 96% increased risk of MS (95% CI: 1.33-2.89, p trend = 0.009). No significant association was found between adult body mass and MS risk.
Conclusions:
Obese adolescents have an increased risk of developing multiple sclerosis (MS). Although the mechanisms of this association remain uncertain, this result suggests that prevention of adolescent obesity may contribute to reduced MS risk.
GLOSSARY
- 25(OH)D
= 25-hydroxyvitamin D;
- BMI
= body mass index;
- CI
= confidence interval;
- MS
= multiple sclerosis;
- NHS
= Nurses’ Health Study;
- NHSII
= Nurses’ Health Study II;
- RR
= relative risk.
Current evidence suggests that the etiology of multiple sclerosis (MS) involves both environmental and genetic factors1–3 with childhood or adolescence thought to be a critical period.4 Elevated levels of serum 25-hydroxyvitamin D appear to decrease MS risk.5 Obese individuals have lower levels of vitamin D metabolites,6–8 including 25-hydroxyvitamin D, than normal weight individuals, and therefore obesity in childhood could be an important risk factor for MS. Further, obesity is associated with a low-grade chronic inflammatory state and release of cytokines that affect immune responses and possibly MS risk.9–11 Because the relation between obesity in early life and MS risk has not been previously investigated, using 2 large, longitudinal cohorts of US women, we examined whether being obese during different life periods was associated with MS risk, and whether among women with MS there was a detectable weight loss occurring after the onset of the disease.
METHODS
Participants.
The Nurses’ Health Study (NHS) began in 1976 when 121,700 female registered nurses, who were age 30 to 55, married, and living in one of 11 states, completed a lifestyle and medical history questionnaire. The Nurses’ Health Study II (NHSII) began in 1989 when 116,671 female registered nurses, who were 25 to 42 years old, married, and residing in one of 14 states, completed a similar questionnaire. Women in both cohorts update their health behavior and medical information via questionnaire every 2 years.
Standard protocol approvals, registrations, and patient consents.
This study was approved by the institutional review board of Brigham and Women’s Hospital.
Case ascertainment.
Women who self-reported an MS diagnosis were asked to provide permission for study investigators to contact their neurologists and obtain a copy of their medical records pertaining to the diagnosis. The neurologists were asked whether the diagnosis is definite, probable, or possible, and whether the results of laboratory tests (e.g., MRI, oligoclonal banding in the CSF) support the diagnosis. In a previous validation study, there was a 93% agreement between the treating neurologists’ diagnoses and those of a study neurologist who reviewed the medical records.12 Therefore, women were considered as having definite or probable MS if so reported by their neurologist or, in the absence of the treating neurologist’s diagnosis, if so determined after medical record review by our study neurologist. Between 1976 and June 2002 we have confirmed 241 cases (166 definite, 75 probable) of MS among women in the NHS and between 1989 and June 2003, 352 cases (278 definite, 74 probable) in the NHSII.
Body mass index and childhood body size.
Obesity during adolescence and adulthood was assessed using the body mass index (BMI). On the baseline questionnaire (1976, NHS; 1989, NHSII) women reported their current weight and height, and in 1980 (NHS) and 1989 (NHSII), their weight at age 18. Using current height as reported on the baseline questionnaires, we calculated their BMI at baseline and age 18 by dividing weight (in kilograms) by height (in meters) squared. We utilized the World Health Organization’s BMI definitions where <18.5 kg/m2 is considered underweight, 18.5-<25 kg/m2 normal weight, 25.0-<30 kg/m2 overweight, and ≥30 kg/m2 obese.13 We then further subcategorized normal and overweight BMI to determine if smaller variations were associated with MS risk. Therefore, women were categorized into 1 of 7 BMI categories (in kg/m2) for both time points: <18.5, 18.5-<21, 21.0-<23, 23.0-<25, 25.0-<27, 27.0-<30, and ≥30.
In 1988 (NHS) and 1989 (NHSII), women were asked to select 1 of 9 body silhouettes, ranging from very thin to extremely obese,14 that best represented their body size at ages 5, 10, and 20 (figure 1). Due to small sample sizes for the overweight/obese silhouettes, a “large body size” category was created by including all women who reported having a body size most similar to 1 of the 4 largest silhouettes.

Figure 1 Nine silhouettes women selected to best describe their body size at ages 5, 10, and 20
Covariates.
State of residence at ages 15 and 30 was reported in 1992 (NHS) and in 1993 (NHSII). Women were categorized into north, middle, and south tiers as previously described.12 Ancestry was reported in 1992 (NHS) and 1989 (NHSII) as Southern European/Mediterranean, Scandinavian, other Caucasian, African American, Hispanic, Asian, Native American, and/or other and women were categorized as previously described.12 Every 2 years women in both cohorts update their smoking status and current smokers report how many cigarettes they smoke per day. Pack-years of smoking were derived from this information. Women in both cohorts (1988 in NHS, 1989 in NHSII) also reported how often (never, 1-3 months/year, 4-6, 7-9, or 10-12) they participated in strenuous physical activity or sports at least twice per week between the ages of 18 and 22, and women in the NHSII additionally reported this information for their time in high school.
Statistical analysis.
Using previously collected data, women were followed from the date they returned the baseline questionnaire until the earliest of date of MS diagnosis, date of death, or June 2002 (NHS)/June 2003 (NHSII). In the NHSII, all main exposure variables were assessed at baseline; therefore, analyses in this cohort were completely prospective. In the NHS, however, BMI at age 18 was reported in 1980 and childhood body size in 1988. Therefore, in the NHS, we also conducted secondary analyses starting follow-up time using 1980 as baseline for BMI at age 18, which included 203 cases (84%) with diagnosis after 1980, and for childhood body size, starting follow-up in 1988, which included 127 cases (53%) diagnosed after 1988. Cox proportional hazards models, stratified by age in months and 2-year time periods, were used to estimate the rate ratios and 95% confidence intervals (CI) for the effect of BMI at age 18 and baseline, and childhood/adolescent body size on MS. In multivariate analyses, we further adjusted for latitude of residence at age 15, ancestry/ethnicity, and pack-years of smoking. Missing indicator variables were included in the models to account for missing data. Tests for linear trends across the categories of BMI were assessed by modeling the median BMI in each category as a continuous variable, and across categories of body size at age 5, 10, or 20 by modeling the silhouette variable as a continuous, ordinal variable. Analyses were conducted separately within each cohort and then pooled using the inverse of the variance of the risk estimates as the weight and a Q statistic to assess heterogeneity of the estimates.15 All analyses were also conducted restricted to the women whose first MS symptoms appeared after baseline (n = 141 [59%] in the NHS and 208 [59%] in the NHSII), and to definite cases. Analyses were conducted using SAS version 9. A p value <0.05 was considered significant.
Weight change analyses.
For each questionnaire cycle beginning at baseline (every 2 years from 1976 to 2004 for NHS, and 1989 to 2003 for NHSII), we calculated the time between the date of questionnaire return and date of diagnosis or first symptoms of MS, and what percent each cohort participant’s weight was of the average cohort weight (their percent weight). In the event a participant contributed more than one observation to a time period, we used the value closest to the midpoint of the time interval. Using generalized linear models, we regressed the percent weights on age (in 5-year categories), smoking (quintiles of pack-years), height, latitude of residence at age 30, and ethnicity separately for each cohort. Residuals from these models were saved and for each MS case reflect the difference between their percent weight of the cohort weight and their expected percent weight based on a certain age, smoking status, height, latitude of residence at age 30, ethnicity, and time period if participant did not have MS. The mean residuals for each time period were calculated in both NHS and NHSII cohorts combined. Generalized estimating equations, accounting for the repeated measures, were used to test the linearity of weight change overall, and before and after the diagnosis or onset of MS. We followed a similar strategy to determine the residual percent weight of women with MS at age 18.
RESULTS
Body size and MS risk.
The relation between BMI and other MS risk factors is shown in table 1. The most notable observation is that, in both cohorts, a higher percentage of women who were obese (BMI ≥30 kg/m2) at age 18 were ever smokers at baseline as compared to women with lower BMI.
Table 1 Selected characteristics of women in Nurses’ Health Study (NHS) and Nurses’ Health Study II (NHSII) by body mass index (BMI) at age 18
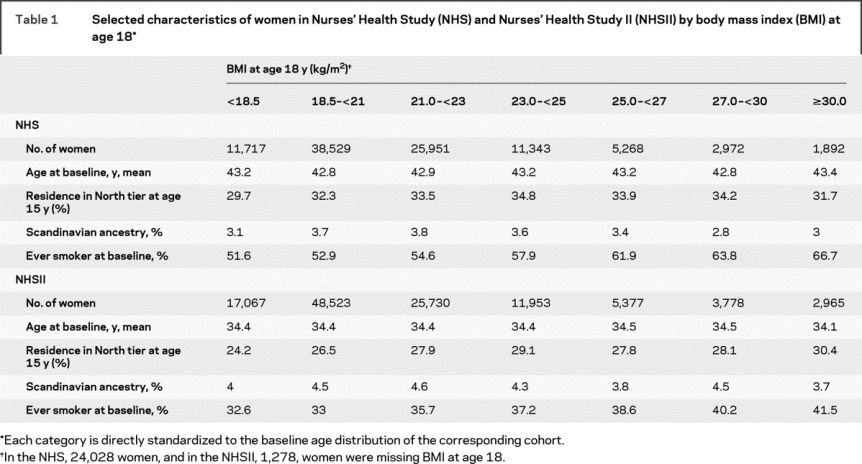
In age-adjusted analyses, women with a BMI ≥30 kg/m2 at age 18 had a greater than twofold risk of developing MS as compared to women with a BMI between 18.5 and 20.9 kg/m2 (table 2). This association persisted after further adjusting for smoking, latitude of residence at age 15, and ethnicity. MS risk among women who were overweight, but not obese, at age 18 (BMI 25-29.9 kg/m2) was only moderately increased (table 2). Results were similar when starting follow-up in 1980 in the NHS cohort or when restricted to definite cases (data not shown). Restricting to cases with onset of first symptoms after baseline, there was a slight attenuation of the relative risks (RRs): women with a BMI ≥30 kg/m2 at age 18 had a 71% increased risk of MS as compared to women with BMI between 18.5 and 20.9 kg/m2 (RRpooled = 1.71, 95% CI 0.96-3.05, p trend 0.36). As smoking was the main confounder of the association between obesity at age 18 and MS risk, we also restricted analyses to never smokers in the NHSII (there were too few never smokers in the NHS cohort to do a comparable analysis); the twofold increased risk of MS persisted among never smoking women with a BMI at age 18 ≥30 kg/m2 (RR = 2.34, 95% CI 1.21-4.53, p = 0.01). Adjusting for physical activity between ages 18 and 22 or during high school (NHSII only) or excluding women who were pregnant at age 18 also did not change the results (data not shown). Further, at age 18, weight among women with MS was on average 2.7% greater than weight among the non-cases (p = 0.0003) (figure 1).
Table 2 Pooled relative risks and 95% confidence intervals (CIs) for body mass index (BMI) at age 18 and baseline and risk of multiple sclerosis, Nurses’ Health Study (NHS) (1976-2002), and Nurses’ Health Study II (NHSII) (1989-2003)
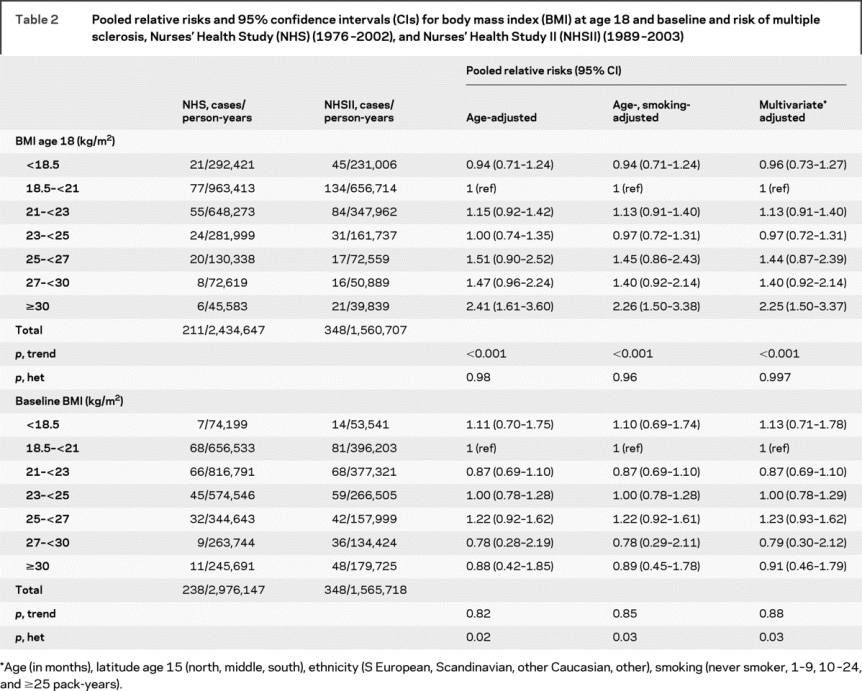
In contrast to the results for BMI at age 18, BMI at baseline (i.e., at age 30 to 55 in NHS and age 25 to 42 in NHSII) was not associated with MS risk in pooled analyses of both cohorts (table 2). However, the test for heterogeneity of the pooled results was significant (table 2). While there was no association between baseline BMI and MS risk in NHSII, the risk of MS among NHS participants decreased with increasing baseline BMI; relative to women with a BMI of 18.5-20.9 kg/m2, the RR of MS was 0.61 (95% CI 0.32-1.15, p = 0.13) among obese women (BMI ≥30 kg/m2), and 0.46 (95% CI 0.23-0.93, p = 0.03) among those overweight (BMI = 27-29.9 kg/m2). This unexpected result may be explained by the fact that many cases of MS in the NHS occurred before MRI was widely used, and the diagnosis was therefore often delayed with respect to the onset of symptoms. This decrease in MS risk in the NHS may thus be the spurious result of the decrease in weight that appears to occur after MS onset (see below). Results were unchanged after excluding women who were pregnant at baseline (data not shown).
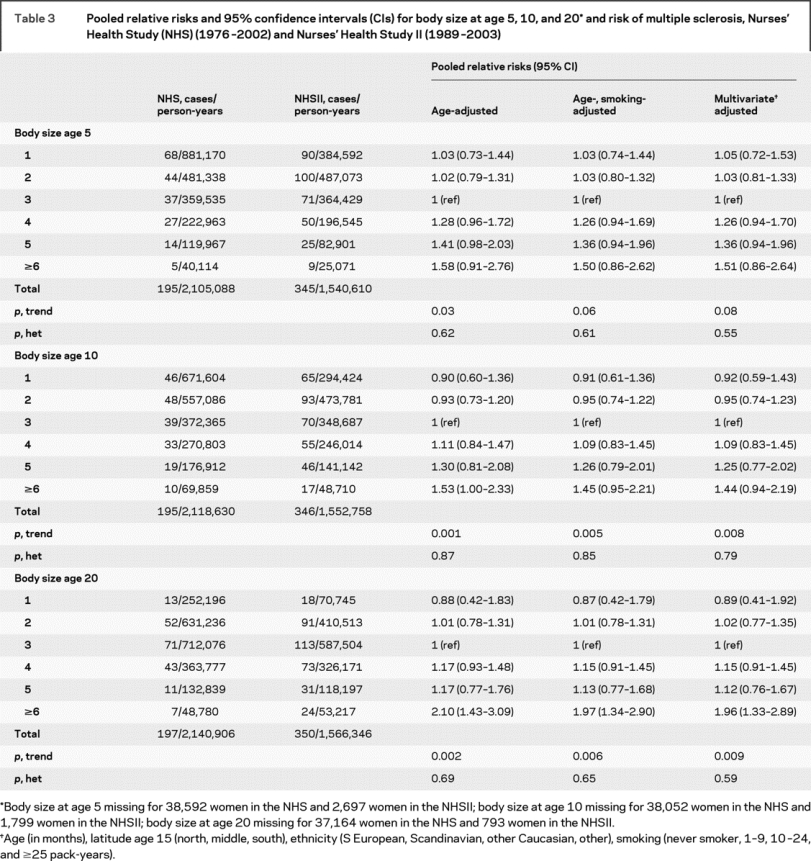
Women who reported having a larger body size, using the silhouettes, at age 20 also had a twofold increased risk of MS as compared to women who reported a thinner body size (table 3). There was also a suggestion of an increased risk of MS associated with having a larger body size during childhood at ages 5 or 10 (table 3). However, reported body size silhouette at ages 5, 10, and 20 years were highly correlated (NHS: age 5-10 r = 0.93, 5-20 r = 0.83, 10-20 r = 0.87; NHSII: 5-10 r = 0.80, 5-20 r = 0.49, 10-20 r = 0.59). In multivariate analyses simultaneously adjusting for body size at all 3 ages, having a larger body size at age 20 remained significantly associated with MS risk (RRpooled body size ≥6 vs 3 = 1.70, 95% CI 1.11-2.60, p = 0.02; p trend across all categories = 0.18), whereas there was no longer a suggestion of an increased risk with larger body sizes at ages 5 and 10 (age 5 RRpooled body size ≥6 vs 3 = 1.17 95% CI 0.57-2.38, p trend = 0.46; age 10 RRpooled = 1.15, 95% CI 0.65-2.04, p trend = 0.15).
Table 3 Pooled relative risks and 95% confidence intervals (CIs) for body size at age 5, 10, and 20* and risk of multiple sclerosis, Nurses’ Health Study (NHS) (1976-2002) and Nurses’ Health Study II (1989-2003)
Weight change among women with MS.
Consistent with the result that BMI at baseline was not associated with an increased risk of MS, the multivariate adjusted percent weight of women who developed MS was similar to the noncases prior to diagnosis (figure 2), and appeared to be stable before the onset of MS (change = 0.08% every 2 years; ptrend = 0.28). After onset, however, the percent relative weight among cases declined 0.26% every 2 years (ptrend < 0.0001).

Figure 2 Relative weight of women with multiple sclerosis at age 18 and by time of diagnosis, Nurses’ Health Study (1976-2004) and Nurses’ Health Study II (1989-2003)
Adjusted for age, latitude of residence, ethnicity, and height. Error bars are the 95% confidence intervals of the mean.
DISCUSSION
In this large, longitudinal study among US women, we found that obesity at age 18 was associated with a greater than twofold increased risk of MS. Further, after adjusting for body size at age 20, there was no increased risk of MS associated with having a large body size during childhood, though the twofold increased risk of MS associated with a large body size at age 20 persisted. Collectively, these results suggest that weight during adolescence, rather than childhood or adulthood, is critical in determining MS risk, consistent with the body of evidence identifying adolescence as an important period in MS etiology.
To our knowledge, the only previous study examining body size as a risk factor for MS was a Canadian case-control study.16 Among women, a 5-unit increase in BMI was associated with a 31% reduction in MS risk. However, cases were asked to report their weight and height at diagnosis (1 year before interview for controls); due to age and secular changes, it is questionable whether the weight of controls at 1 year prior to the interview is comparable to weight of cases at diagnosis. Further, a decrease in weight among the MS cases after onset may also explain the apparent protective effect of BMI. Consistent with previous studies,17,18 we found that women with MS weighed less on average than noncases, and that this decrease in relative weight occurred after the onset of the disease.
While the strengths of this study include the large sample size, and longitudinal design, there are limitations to consider. First, women in the NHS cohort reported information on weight at age 18 4 years after baseline, and body size in childhood and age 20 12 years after baseline. Excluding women who were diagnosed prior to reporting these exposures did not meaningfully alter the results, suggesting there was little or no differential recall among these cases, and they were thus retained in the analysis.
Another limitation of this study is the reliance on self-report of weight, height, and body size, and medical record confirmation of the MS diagnosis. A validation study among women in the NHSII found high correlations between adult recall of weight at age 18 and actual weight as recorded in nursing school entrance physical examination records and between self-reported body size silhouette at age 20 and BMI at age 18 (either self-reported or from records).19 Women tended to slightly underreport their weight at age 18, but we would expect any underreporting of weight in our study to be nondifferential and attenuate the association between obesity at age 18 and MS risk. Among women in the Third Harvard Growth Study, recall of body size at ages 5 and 10 in adulthood, using the silhouettes, correlated well with measured BMI at these ages.20 While it is possible some cases of MS in our study were misdiagnosed, participants in our cohorts are all registered nurses, and have access to health care, as suggested by the 89% of cases diagnosed after 1990 with MRI findings consistent with MS. Further, the expected increased risk of MS associated with the HLA-DRB1*1501 allele that we found in these cohorts21 lends support that the diagnosis is correct for the majority of cases. Finally, we cannot completely exclude unknown confounding as an explanation of these results. Physical activity during high school or ages 18-22 was not associated with MS risk and therefore did not confound the association we observed with BMI at age 18. We controlled for all other major risk factors for MS except for Epstein-Barr virus antibody titers, vitamin D status, and HLA-DRB1 genotype. We could not control for these factors as this information is not available for all the women in this study, but it appears from the data available that neither anti-Epstein-Barr virus antibodies nor the HLA-DRB1 genotype are associated with obesity. Further, it is not clear that adjusting for vitamin D would be appropriate, because vitamin D may be on the causal pathway between obesity and MS, as discussed below. An additional limitation is that this study includes only women, and whether the associations reported here apply to men as well requires further study. Further, over 95% of the women in these cohorts are white, and our results may not be generalizable to other racial groups.
The biologic mechanism relating obesity to MS is not known. However, it is well-established that obese individuals, including obese children, have lower circulating 25-hydroxyvitamin D [25(OH)D] levels, a marker of vitamin D status, than nonobese individuals,6–8,22–25 and that total body fat is inversely related to 25(OH)D levels.26 Vitamin D is a known immunomodulator,27 and has been found to reduce the incidence and progression of an animal model of MS,28–30 likely through enhancement of regulatory T-cell function,27,31 which has been shown to be reduced in patients with MS.32 Further, high levels of circulating blood 25(OH)D have been associated with a reduced risk of MS in early adulthood.5 The higher risk of MS among women who were obese during adolescence is thus consistent with a protective effect of vitamin D. Although the average difference in blood levels of 25(OH)D between obese and lean individuals tends to be moderate (<15 ng/mL),6–8,22,23 it could be important if it occurs at an age that is critical for MS development. An alternative, or perhaps complementary, hypothesis is that the effects of obesity are mediated by a low-grade chronic inflammatory state. Adipose tissue secretes numerous adipokines and cytokines that influence immune system function, including leptin9 and interleukin-6,10 both of which have been shown to reduce regulatory T-cell activity,33,34 and elevated leptin levels were inversely correlated with frequency of regulatory T cells in patients with MS.11
Confirmation of these results in other populations is necessary.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Dr. Kassandra L. Munger and Dr. Alberto Ascherio.
ACKNOWLEDGMENT
The authors thank Dr. Eilis O’Reilly and Leslie Unger for technical support.
DISCLOSURE
Dr. Munger has received funding for travel and speaker honoraria from the Consortium of Multiple Sclerosis Centers and received speaker honoraria from The National Multiple Sclerosis Society. Dr. Chitnis has received speaker honoraria from the Consortium of Multiple Sclerosis Centers, the Child Neurology Society, and the Cambridge Health Alliance; has served as a consultant to Biogen Idec, Merck Serono, Teva Pharmaceutical Industries Ltd., and Bayer Schering Pharma (Berlex); serves on the speakers’ bureau of Merck Serono; and receives research support from the NIH/NINDS [K08 NS 047669-01 (PI)] and the National Multiple Sclerosis Society. Dr. Ascherio serves on the editorial board of Neurology®; serves on a scientific advisory board for the Michael J. Fox Foundation; received speaker honoraria from Merck Serono; and receives research support from the US Department of Defense (Army) [W81XWH-05-1-0117 (PI)], the NIH [R01 NS045893 (PI), R01 NS047467 (PI), R01 NS48517 (PI), NINDS R01 NS042194 (PI), and R01 NS046635 (PI)], and the Michael J. Fox Foundation (Co-I).
Address correspondence and reprint requests to Dr. Kassandra L. Munger, Harvard School of Public Health, Department of Nutrition, 655 Huntington Ave, Building 2, 3rd floor, Rm 312, Boston, MA 02115 kgorham@hsph.harvard.edu
Supported by grant NS047467 from the National Institute of Neurological Diseases and Stroke.
Disclosure: Author disclosures are provided at the end of the article.
Received May 12, 2009. Accepted in final form July 27, 2009.
REFERENCES
- 1.Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis: part I: the role of infection. Ann Neurol 2007;61:288–299. [DOI] [PubMed] [Google Scholar]
- 2.Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis: part II: noninfectious factors. Ann Neurol 2007;61:504–513. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt H, Williamson D, Ashley-Koch A. HLA-DR15 haplotype and multiple sclerosis: a HuGE review. Am J Epidemiol 2007;165:1097–1109. [DOI] [PubMed] [Google Scholar]
- 4.Gale CR, Martyn CN. Migrant studies in multiple sclerosis. Prog Neurobiol 1995;47:425–448. [PubMed] [Google Scholar]
- 5.Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA 2006;296:2832–2838. [DOI] [PubMed] [Google Scholar]
- 6.Bell NH, Epstein S, Greene A, Shary J, Oexmann MJ, Shaw S. Evidence for alteration of the vitamin D-endocrine system in obese subjects. J Clin Invest 1985;76:370–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liel Y, Ulmer E, Shary J, Hollis BW, Bell NH. Low circulating vitamin D in obesity. Calcif Tissue Int 1988;43:199–201. [DOI] [PubMed] [Google Scholar]
- 8.Wortsman J, Matsuoka LY, Chen TC, Lu Z, Holick MF. Decreased bioavailability of vitamin D in obesity. Am J Clin Nutr 2000;72:690–693. [DOI] [PubMed] [Google Scholar]
- 9.Maffei M, Halaas J, Ravussin E, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med 1995;1:1155–1161. [DOI] [PubMed] [Google Scholar]
- 10.Mohamed-Ali V, Goodrick S, Rawesh A, et al. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J Clin Endocrinol Metab 1997;82:4196–4200. [DOI] [PubMed] [Google Scholar]
- 11.Matarese G, Carrieri PB, La Cava A, et al. Leptin increase in multiple sclerosis associates with reduced number of CD4(+)CD25+ regulatory T cells. Proc Natl Acad Sci USA 2005;102:5150–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hernan MA, Olek MJ, Ascherio A. Geographic variation of MS incidence in two prospective studies of US women. Neurology 1999;53:1711–1718. [DOI] [PubMed] [Google Scholar]
- 13.World Health Organization. Obesity: Preventing and Managing the Global Epidemic: Report of a WHO Consultation. WHO Technical Report Series 894. Geneva: World Health Organization; 2000. [PubMed] [Google Scholar]
- 14.Stunkard AJ, Sorensen T, Schulsinger F. Use of the Danish Adoption Register for the study of obesity and thinness. Res Publ Assoc Res Nerv Ment Dis 1983;60:115–120. [PubMed] [Google Scholar]
- 15.DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials 1986;7:177–188. [DOI] [PubMed] [Google Scholar]
- 16.Ghadirian P, Jain M, Ducic S, Shatenstein B, Morisset R. Nutritional factors in the aetiology of multiple sclerosis: a case-control study in Montreal, Canada. Int J Epidemiol 1998;27:845–852. [DOI] [PubMed] [Google Scholar]
- 17.Nortvedt MW, Riise T, Maeland JG. Multiple sclerosis and lifestyle factors: the Hordaland Health Study. Neurol Sci 2005;26:334–339. [DOI] [PubMed] [Google Scholar]
- 18.Formica CA, Cosman F, Nieves J, Herbert J, Lindsay R. Reduced bone mass and fat-free mass in women with multiple sclerosis: effects of ambulatory status and glucocorticoid use. Calcif Tissue Int 1997;61:129–133. [DOI] [PubMed] [Google Scholar]
- 19.Troy LM, Hunter DJ, Manson JE, Colditz GA, Stampfer MJ, Willett WC. The validity of recalled weight among younger women. Int J Obes Relat Metab Disord 1995;19:570–572. [PubMed] [Google Scholar]
- 20.Must A, Willett WC, Dietz WH. Remote recall of childhood height, weight, and body build by elderly subjects. Am J Epidemiol 1993;138:56–64. [DOI] [PubMed] [Google Scholar]
- 21.De Jager PL, Simon KC, Munger KL, Rioux JD, Hafler DA, Ascherio A. Integrating risk factors: HLA-DRB1*1501 and Epstein-Barr virus in multiple sclerosis. Neurology 2008;70:1113–1118. [DOI] [PubMed] [Google Scholar]
- 22.Compston JE, Vedi S, Ledger JE, Webb A, Gazet JC, Pilkington TR. Vitamin D status and bone histomorphometry in gross obesity. Am J Clin Nutr 1981;34:2359–2363. [DOI] [PubMed] [Google Scholar]
- 23.Hey H, Stokholm KH, Lund B, Sorensen OH. Vitamin D deficiency in obese patients and changes in circulating vitamin D metabolites following jejunoileal bypass. Int J Obes 1982;6:473–479. [PubMed] [Google Scholar]
- 24.Reinehr T, de Sousa G, Alexy U, Kersting M, Andler W. Vitamin D status and parathyroid hormone in obese children before and after weight loss. Eur J Endocrinol 2007;157:225–232. [DOI] [PubMed] [Google Scholar]
- 25.Alemzadeh R, Kichler J, Babar G, Calhoun M. Hypovitaminosis D in obese children and adolescents: relationship with adiposity, insulin sensitivity, ethnicity, and season. Metabolism 2008;57:183–191. [DOI] [PubMed] [Google Scholar]
- 26.Arunabh S, Pollack S, Yeh J, Aloia JF. Body fat content and 25-hydroxyvitamin D levels in healthy women. J Clin Endocrinol Metab 2003;88:157–161. [DOI] [PubMed] [Google Scholar]
- 27.Hayes CE, Nashold FE, Spach KM, Pedersen LB. The immunological functions of the vitamin D endocrine system. Cell Mol Biol 2003;49:277–300. [PubMed] [Google Scholar]
- 28.Lemire JM, Archer DC. 1,25-dihydroxyvitamin D3 prevents the in vivo induction of murine experimental autoimmune encephalomyelitis. J Clin Invest 1991;87:1103–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cantorna MT, Hayes CE, DeLuca HF. 1,25-Dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc Natl Acad Sci USA 1996;93:7861–7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spach KM, Hayes CE. Vitamin D3 confers protection from autoimmune encephalomyelitis only in female mice. J Immunol 2005;175:4119–4126. [DOI] [PubMed] [Google Scholar]
- 31.Nashold FE, Hoag KA, Goverman J, Hayes CE. Rag-1-dependent cells are necessary for 1,25-dihydroxyvitamin D(3) prevention of experimental autoimmune encephalomyelitis. J Neuroimmunol 2001;119:16–29. [DOI] [PubMed] [Google Scholar]
- 32.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med 2004;199:971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Rosa V, Procaccini C, Cali G, et al. A key role of leptin in the control of regulatory T cell proliferation. Immunity 2007;26:241–255. [DOI] [PubMed] [Google Scholar]
- 34.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 2003;299:1033–1036. [DOI] [PubMed] [Google Scholar]