

Abstract
Frequent hallmarks of T-cell acute lymphoblastic leukemia (T-ALL) include aberrant NOTCH signaling and deletion of the CDKN2A locus, which contains 2 closely linked tumor suppressor genes (INK4A and ARF). When bone marrow cells or thymocytes transduced with a vector encoding the constitutively activated intracellular domain of Notch1 (ICN1) are expanded ex vivo under conditions that support T-cell development, cultured progenitors rapidly induce CD4+/CD8+ T-ALLs after infusion into healthy syngeneic mice. Under these conditions, enforced ICN1 expression also drives formation of T-ALLs in unconditioned CD-1 nude mice, bypassing any requirements for thymic maturation. Retention of Arf had relatively modest activity in suppressing the formation of T-ALLs arising from bone marrow–derived ICN1+ progenitors in which the locus is epigenetically silenced, and all resulting Arf +/+ tumors failed to express the p19Arf protein. In striking contrast, retention of Arf in thymocyte-derived ICN1+ donor cells significantly delayed disease onset and suppressed the penetrance of T-ALL. Use of cultured thymocyte-derived donor cells expressing a functionally null Arf-GFP knock-in allele confirmed that ICN1 signaling can induce Arf expression in vivo. Arf activation by ICN1 in T cells thereby provides stage-specific tumor suppression but also a strong selective pressure for deletion of the locus in T-ALL.
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) accounts for approximately 20% of all pediatric lymphoblastic leukemias and comprises a significant proportion of cancers affecting children and adolescents.1 Advances in treatment, focusing largely on dose intensification in multiagent therapeutic regimens, have produced remarkable cure rates, with overall survival of approximately 80%. Nonetheless, T-ALL is overly represented among relapsed ALL cases, and the dose-intensive therapy required to improve its cure imposes its own disease burden.
Activating mutations of NOTCH1 have been found in 50% of T-ALLs, making it one of the most commonly mutated genes in this disease.2 Notch is a cell-surface receptor expressed in many developing organ systems, in which Notch ligands function in conjunction with other morphogens, including Wnt, Hedgehog, and bone morphogenic proteins, to program cell fate decisions.3 The Notch receptor is formed by intracellular proteolytic cleavage of a single polypeptide chain, the 2 resulting subunits undergoing dimerization and transport to the plasma membrane.4 When bound by its ligands (members of the delta/jagged family), the transmembrane receptor is cleaved extracellularly by an ADAM protease and intracellularly by γ-secretase, thereby releasing the intracellular Notch (ICN) domain into the cytoplasm. After transport into the cell nucleus, the ICN complexes with DNA-binding partner proteins to activate target genes. Mutations in Notch may affect its heterodimerization domain, which reduces or eliminates ligand dependency of the receptor, or they may target the intracellular proline-, glutamic acid-, serine-, and threonine-rich domain, thereby stabilizing the active, intracellular signaling moiety.2,4,5 These 2 classes of mutations can occur concomitantly in up-regulating Notch target gene expression.
Notch transcriptional activity directs virtually every stage of T-cell development, from the earliest commitment of bone marrow–derived progenitors to the T-lymphoid lineage through stages of thymocyte maturation to double-positive (DP) CD4+/CD8+ cells.5,6 Although malignant thymocytes that typify Notch1-associated T-ALL are usually arrested at the DP stage, DP progenitors do not appear to be the tumor-initiating population. Instead, tumorigenic cells arise from more immature T-cell progenitors that ultimately generate monoclonal tumors expressing unique T-cell receptor (TCR)–β chains and diverse TCR-α chains.7 Although leukemogenesis is independent of the pre-TCR, per se, ICN1 overexpression cannot induce leukemia in cells that lack pre-TCR signaling, implying that malignant transformation occurs after pre-TCR signaling but before completion of α chain rearrangement.7–9 Although the extent to which NOTCH1 mutations represent founding oncogenic events in T-ALLs has been debated,10,11 robust mouse models of T-ALL driven by activated Notch1 implicate this pathway as an appealing target for therapeutic intervention.4,5,11–13
Occurring more frequently than mutation of NOTCH1, deletion of the CDKN2A (hereafter INK4A-ARF) locus, which encodes 2 functionally distinct but closely chromosomally linked tumor suppressors, p16INK4A and p14ARF (p19Arf in the mouse), affects more than 70% of T-ALL cases.14,15 Most cases of T-ALL that exhibit NOTCH1 mutations also sustain deletions of INK4A-ARF, arguing that functional interactions stemming from these different events may be an obligate part of the life history of the majority of T-ALLs. By inhibiting cyclin D–dependent kinases, p16Ink4a helps to maintain the retinoblastoma protein (RB) in its hypophosphorylated growth-suppressive form, thereby blocking entry into the DNA synthetic (S) phase of the cell-division cycle. Instead, p19Arf antagonizes the E3 ubiquitin ligase activity of the Mdm2 gene product to activate a p53-dependent transcriptional program that triggers either cell-cycle arrest or apoptosis, depending upon the cell type and collateral signal inputs.16 The products of the Ink4a-Arf locus are generally not expressed in normal mouse tissues but are activated by aberrantly elevated and sustained hyperproliferative stress signals. Subsequent expression of p16Ink4a and p19Arf triggers RB- and p53-dependent programs that eliminate incipient cancer cells. Conversely, deletion of the Ink4a-Arf locus simultaneously compromises the activities of both RB and p53 and is appreciated to be one of the most frequent events occurring in many forms of human cancer.17,18
The Ink4a-Arf locus is epigenetically silenced in bone marrow–derived adult hematopoietic stem cells,19–24 but it is remodeled during lymphoid development, becoming poised to respond to oncogenic stress signals during the maturation of the T- and B-cell lineages.25–27 Moreover, both Ink4a and Arf transcripts progressively accumulate in peripheral B-lymphoid cells as mice age, thus conferring resistance to lymphoid tumor development once the adaptive immune system has developed.26,27 Deletion of the Ink4a-Arf locus in the mouse germline counters these age-related effects, and by increasing the self-renewal potential of lymphoid cells, strongly predisposes to the early development of T-cell neoplasms.28,29 Using an adoptive transfer protocol that takes advantage of cultured Notch1-expressing T cells derived from progenitor pools enriched for different stages of the T-cell developmental program, we have now established a functional relationship between constitutive Notch1 signaling and Arf deletion in the generation of T-ALL.
Methods
Expression vectors and retroviral production
A cDNA encoding the intracellular domain of human NOTCH1 (ICN1; amino acids 1761-2555 of the full-length protein) was expressed in a mouse stem cell virus–internal ribosome entry site–green fluorescent protein vector (MSCV-IRES-GFP)30 provided by J. Opferman (St Jude Children's Research Hospital). A re-engineered vector that produces cherry fluorescent protein (CFP) in lieu of GFP was used where indicated. Packaging of replication-incompetent, ecotropic retroviral particles was achieved by transient cotransfection of HEK-293T cells with vector and helper plasmids as previously described.31
Culture conditions
Marrow was obtained from the long bones of wild-type or syngeneic Arf-null C57BL/6 mice harboring targeted deletions of exon-1β29 either 4 days after treatment with 150 mg/kg 5-fluorouracil (5-FU; Abraxis) or from untreated donors. Cells from 5-FU–treated mice were incubated for 2 days at 37°C in Stemspan SFEM medium (StemCell Technologies) supplemented with 20 ng/mL recombinant mouse interleukin-3 (IL-3), 50 ng/mL recombinant human IL-6, 50 ng/mL recombinant mouse stem cell factor and 100 ng/mL recombinant mouse thrombopoietin (all from R&D Systems). Bone marrow cells or single-cell suspensions prepared from the thymi of mice from the same genotypes were then infected with retrovirus immobilized on retronectin (Takara Bio). Cells exposed to virus for a period of 24 to 48 hours were either directly inoculated into irradiated mice or placed into coculture with OP9 stromal cells.32,33 Cocultures on OP9 stroma were maintained in alpha-minimum essential medium (Invitrogen) supplemented with 10% fetal calf serum (Hyclone), 1mM sodium pyruvate (Sigma-Aldrich), penicillin/streptomycin, and recombinant mouse IL-7 and FMS-like tyrosine kinase 3 ligand (R&D Systems) both at 5 ng/mL. Cells were enumerated and passaged onto fresh stroma every 4 days. The immunophenotypes of GFP-positive cells in the cultures were determined by fluorescence-activated cell sorting (FACS) using fluorescently tagged antibodies directed to the mouse T-cell markers Thy1.2, TCRβ, CD4, and CD8.
Bone marrow transplantation and adoptive transfer of cultured cells
All mice were housed in an American Association of Laboratory Animal Care–accredited facility and were treated on protocols approved by St Jude Children's Hospital Animal Care and Use Committee in accordance with National Institutes of Health guidelines. Recipient male C57BL/6 or CD-1 nude mice (8-12 weeks old) were lethally irradiated with 11 Gy split into 2 equal doses. Each recipient mouse was injected via tail vein with a mixture of 5 × 105 unmanipulated bone marrow cells and half of the ICN1-transduced bone marrow from one 5-FU–treated donor mouse. Irradiated mice were maintained on water supplemented with enrofloxacin (Bayer) for 3 weeks after transplantation.
Bone marrow–derived or thymocyte-derived cells cultured for 12 days on OP9 stroma were harvested, and the vector-infected percentage was determined by FACS for the GFP or CFP marker. Recipient animals were injected via tail vein with a dose calculated to deliver 2 × 105 fluorescent cells. All recipient mice were observed daily for clinical signs of illness and killed when moribund.
Genomic PCR and immunoblotting
GFP-positive leukemic T cells were purified by FACS from single-cell suspensions prepared from the spleens or lymph nodes of moribund animals. Genomic DNA was subjected to polymerase chain reaction (PCR) for exon-1β and exon-2 of the Cdkn2a (Ink4a-Arf) locus. PCR reactions were carried out in 25-μL volumes using Taq polymerase, Q solution, and buffer (all from QIAGEN), 12.5 pmol forward and reverse primers, and 0.2mM deoxynucleotide triphosphates. The reaction mix was heated for 3 minutes at 94°C and then subjected to 30 cycles of 1-minute denaturation at 94°C, 1-minute annealing at 66°C, and 2-minute extension at 72°C. PCR products were visualized after electrophoresis on 2% agarose gels containing 0.5 ng/mL ethidium bromide. Primers for Arf exon-1β were (sense) 5′-AGTACAGCAGCGGGAGCATGG and (antisense) 5′-TTGAGGAGGACCGTGAAGCCG, and for Ink4a-Arf exon-2 were (sense) 5′-ATGATGATGGGCAACGTTC and (antisense) 5′-CCAATATCGCACGATGTC. Immunoblotting was performed as described.31 In brief, detergent lysates were prepared from purified GFP-positive leukemic cells and protein concentration was quantified by bicinchoninic acid assay (Pierce). Samples (50 μg protein per lane) were electrophoretically separated on 4% to 12% Bis-Tris NuPAGE gels (Invitrogen), transferred to polyvinylidene fluoride membranes (Millipore), and detected using a highly sensitive rat monoclonal antibody to p19Arf (5C3-1).34 Antibodies to nucleophosmin (NPM; Invitrogen) were used to control for protein loading.
Southern blotting
Genomic DNA (20 μg/lane) from FACS-sorted, GFP-marked lymph node cells from moribund mice was digested with HindIII, separated on a 0.7% agarose gel, and transferred to Nytran SuPerCharge nylon membrane (Whatman Inc). The membrane was prehybridized for 1 hour at 65°C with PerfectHyb Plus (Sigma-Aldrich) containing 100 μg/mL salmon sperm DNA. Radiolabeled DNA probes prepared by random priming with [α32P] deoxyadenosine triphosphate (Roche Diagnostics) were then added, and hybridization was continued for 16 hours. TCRβ rearrangements were visualized using a radiolabeled 2.3-kb EcoRI Jβ2 fragment.35 Membranes were briefly rinsed in 0-3M NaCl, 0.03M Na citrate containing0.1% sodium dodecyl sulfate, followed by a stringent wash in 15mM NaCl, 1.5mM Na citrate with 0.1% sodium dodecyl sulfate for 30 minutes at 55°C. Dried membranes were exposed to Kodak BioMax film (Kodak) at −80°C.
Results
Arf inactivation modestly accelerates ICN1-induced T-cell maturation ex vivo
Established mouse models of Notch1-driven leukemia rely on transplantation of lethally irradiated recipient mice with primitive bone marrow–derived progenitors engineered to constitutively express gain-of-function Notch1 mutations.8,11–13 Because the bone marrow transplantation (BMT) model of Notch1-driven leukemia is relatively, though not absolutely, dependent on the lethal irradiation of recipients, it diverges from the pathogenesis of the human disease, which arises in a hematologically and immune-competent host. Culturing of Bcr-Abl–transduced Arf-null bone marrow cells under conditions that favor the outgrowth of pre-B cells was previously revealed to efficiently enrich for leukemia-initiating cells (LICs) that rapidly induced fatal tumors in healthy, nonirradiated syngeneic recipient mice.36,37 Following an analogous strategy, we transduced bone marrow progenitors or thymocytes from Arf +/+ or Arf−/− mice with a vector encoding ICN1 and GFP and established short-term cocultures on OP9 stroma to support T-cell development through the DP stage.32 Although mutant Notch1 alleles can drive ectopic T-cell development, those most commonly found in human T-ALL fail to initiate disease on their own in the mouse BMT model.11 Therefore, to maximize the chances of inducing leukemia after infusion of cultured cells into naive recipient mice, we chose to use ICN1, a “strong” mutant allele that efficiently induces T-ALL in the BMT setting.
When transduced with the ICN1-GFP vector, cultured bone marrow and thymic progenitors passaged at 4-day intervals gave rise to T-cell populations that steadily expanded for at least a month (supplemental Figure 1, available on the Blood website; see the Supplemental Materials link at the top of the online article). Although Arf +/+ and Arf −/− bone marrow cells infected with a control vector encoding GFP alone were also able to proliferate, these populations expressed the B-cell markers B220 and CD19 and negligible levels of the T-cell marker Thy1.2. Arf +/+ donor thymocytes transduced with the control vector were completely incapable of initiating cultures on OP9 stroma, whereas their Arf −/− counterparts produced poorly growing T-cell colonies at very low efficiencies (data not shown). Thus, expression of ICN1 was required to support T-cell development ex vivo.
After 23 days of culture, almost all of the cultured cells from mice of both Arf genotypes expressed GFP, implying that ICN1 coexpression provided them with a strong selective advantage (Figure 1A). However, GFP-marked Arf −/− bone marrow–derived cells did not gain as strong a proliferative advantage initially (Figure 1A, day 12), reflecting the propensity for Arf inactivation alone to facilitate the development of competing unmarked bone marrow–derived B-lineage cells that dominated the cultures at day 12 but were outcompeted by GFP-marked T cells by day 23. By day 12, 80% to 90% of GFP-marked cells expressed Thy1.2 (Figure 1B). Expression of TCRβ, CD4, and CD8 increased as the cells were passaged, highlighting ongoing maturation (Figure 1B). In age-matched cultures, Arf−/− cells reproducibly yielded a greater fraction of cells expressing TCRβ, CD4, and CD8 than did Arf +/+ cells (Figure 1B), suggesting that Arf inactivation confers an increased propensity to generate DP thymocytes more rapidly.
Figure 1.

Emergence of GFP+ cells and their immunophenotypes on day 12 and day 23 of culture. (A) Cells derived from 5-FU–conditioned bone marrow (BM) or from thymocytes explanted from mice of the indicated Arf genotypes (labeled at bottom) were transduced with a vector encoding ICN1 and GFP in cis and cultured on OP9 stroma with cytokine support. Cultured cells expanded exponentially and underwent 25 to 30 population doublings over a period of 36 days (supplemental Figure 1). The percentage of GFP+ cells in the cultures was determined at day 12 and day 23. The results of 3 experiments are shown, and error bars indicate SDs. (B) After 12 and 23 days of culture (indicated at the top of the panels) the percentages of GFP+ cells that expressed CD4 and CD8 or Thy1.2 and TCRβ were determined using an automated cell sorter. The percentages of cells expressing each marker are indicated in at least 3 of the 4 quadrants of each panel. The origin of donor cells and their Arf genotypes are indicated to the left.
Cultured ICN1-expressing donor cells derived from bone marrow generate T-ALL in healthy nonirradiated animals
We next infused 2 × 105 GFP-positive cells derived from 12-day cultures of 5-FU–conditioned bone marrow into nonirradiated syngeneic mice. Recipients of these adoptively transferred ICN1-positive T cells developed a rapidly fatal T-ALL (Figure 2A), with bone marrow invasion, lymphocytosis, and bulky disseminated disease involving the spleen, liver, and lymph nodes, and variably invading the thymus, kidneys, and lungs (Figure 3A). Each of 15 recipients of ICN1+ Arf −/− donor cells and 18 of 20 recipients of ICN1+ Arf +/+ cells succumbed to disease (Figure 2A). Median survival times for healthy mice that had received GFP-marked Arf +/+ or Arf −/− bone marrow–derived cells were 34 and 29 days, respectively (Figure 2A). Notably, deletion of Ink4a in addition to Arf did not further accelerate disease onset (supplemental Figure 2A). Furthermore, these features paralleled the penetrance and latency periods observed in lethally irradiated recipient mice that received a transplant of acutely transduced donor cells together with an excess number of radioprotective, untransduced bone marrow progenitors (supplemental Figure 2B-C). The immunophenotype of the tumors confirmed their T-cell identity, with Thy1.2, CD4, CD8, and TCRβ all expressed (Figure 3B) and CD19, Mac-1, and Gr-1 absent (data not shown). Notably, the majority of leukemic cells were CD4+/CD8+ (Figure 3B) unlike the much less mature day-12 bone marrow–derived donor population that contained only approximately 1% DP cells (Figure 1B). The overall clinical signs of leukemic involvement in nonirradiated healthy recipients receiving cells by adoptive transfer were also similar to those observed in the BMT model (Figure 3C-D). The differences were the tendency for more frequent bleeding and greater cytopenia in the BMT setting, reflected by significantly lower hemoglobin concentrations and platelet counts (Figure 3C).
Figure 2.
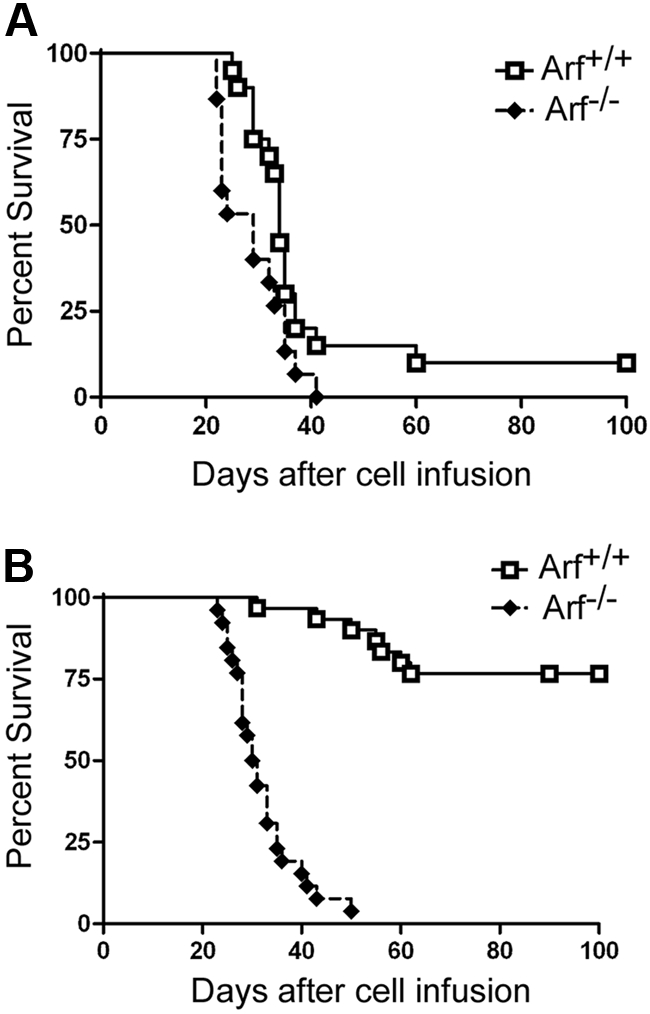
Survival curves of recipient mice developing T-ALL. Survival curves are shown for cohorts of healthy nonirradiated C57BL/6 mice injected with cultured (day-12) ICN1+ cells derived from the bone marrow of 5-FU–conditioned mice (A) or unfractionated thymocytes (B) of the indicated Arf genotypes. (A) Twenty recipients of Arf +/+ and 15 recipients of Arf −/− bone marrow–derived cells in 2 experiments had a median survival of 34 days and 29 days, respectively. The differences in latency are statistically significant (P = .02 by log-rank test). (B) In 3 experiments, all but 7 of 30 recipients of Arf +/+ thymus-derived cells did not develop T-ALL, whereas 25 of 26 recipients of Arf −/− cells died of disease with a median survival of 30.5 days. Differences in latency are highly significant (P < .001 by log-rank test).
Figure 3.

T-ALLs arising from cultured ICN1+ cells have similar disease phenotypes whether derived from bone marrow or thymocytes. (A) Moribund mice were killed, dissected, and photographed. Images were captured with a Nikon Coolpix 5200 camera (Nikon) in ambient room light and through a Tiffen 15 deep yellow filter (Tiffen) under fluorescent illumination provided by an Illumatool light source (Light Tools Research). The images were merged using Photoshop 7.0 software (Adobe Systems). Recipient animals developed T-ALL with hepatosplenomegaly and extensive invasion of lymph nodes with GFP-marked leukemic cells. Representative recipients of marrow-derived cells (left panel) and of thymocyte-derived cells (right panel) are depicted. (B) Like irradiated animals that received bone marrow transplants of ICN1-transduced, but uncultured, donor cells, the T-ALLs arising in nonirradiated mice largely coexpressed CD4 and CD8, though some T-ALLs had a CD8 single-positive subpopulation, as well. No consistent variations in representative immunophenotypes of leukemic cells taken from different tissues or derived from Arf +/+ or Arf −/− donors were seen. The remaining panels (C-D) compare clinical parameters of T-ALLs arising in irradiated animals undergoing conventional bone marrow transplantations (BMTs) with those of recipient mice that received cultured day-12 ICN1+ bone marrow–derived or thymus-derived cells. Clinical hematologic measurements including a comparison of total white blood cell counts, hemoglobin concentration, platelet count, and spleen weights are shown in panel C, whereas the percentages of GFP-marked cells in various tissues are shown in panel D. The broad horizontal bars indicate median values, the gray bars delineate the upper and lower quartiles, and the whiskers indicate the overall range of readings.
Groups of 5 nonirradiated mice receiving cells derived from 5-FU–conditioned bone marrow infected with the control vector expressing GFP alone and cultured either on OP9 or OP9-DL1 stroma producing the delta ligand did not develop disease during a 6-month observation period. Moreover, although ICN1-positive cells derived from unconditioned (non–5-FU–treated) bone marrow cells also grew readily in culture and exhibited characteristics largely indistinguishable from cultures derived from 5-FU–treated marrow, such cells were unable to generate leukemias after adoptive transfer into a cohort of 10 healthy mice. Thus, by enriching for immature progenitors, 5-FU pretreatment before culture establishment increased the likelihood of endowing explanted bone marrow cells with detectable leukemogenicity, a dependency also observed using the BMT model.12
In short, the nearly complete penetrance of tumor development, the tissues involved, and the immunophenotype of T-cell tumors derived from adoptive transfer of cultured ICN1-expressing bone marrow cells were virtually identical to those seen after directly transplanting ICN1-transduced bone marrow into irradiated recipients. The principle virtue of this T-ALL model is that ex vivo culture maintains the survival of ICN1+ leukemogenic cells that rapidly induce T-cell neoplasms in healthy unirradiated recipients. However, the significant but relatively modest acceleration of disease progression conferred by Arf inactivation in bone marrow progenitors, and the inability of the retained Arf gene to fully protect recipients from lethal disease, begs the question of why the CDKN2A locus is inactivated in human T-ALL.
Arf strongly suppresses leukemia initiated by thymocyte-derived, ICN1-expressing T cells
When similar experiments were performed with cultured T cells derived from ICN1-transduced thymocytes, we observed a striking effect of Arf in preventing tumorigenesis. Whereas ICN1-transduced Arf −/− donor cells produced T-cell leukemias in healthy syngeneic recipients with a median survival of 31 days in 25 of 26 mice surveyed in 3 separate experiments, their Arf+/+ counterparts initiated disease in only 7 of 30 mice observed for more than 4 months (Figure 2B). Considering only those recipients of thymocyte-derived ICN1+ Arf +/+ cells that succumbed to disease, their median survival was 56 days. Arf −/− thymocytes infected with a control GFP virus gave rise to Thy1.2-positive T cells when cultured on OP9-DL1 stroma expressing the delta ligand, but the GFP-marked cells were completely unable to initiate disease when infused into 10 recipients. Thus, Arf loss alone was incapable of producing T-ALL, even when the cells received developmental cues capable of directing T-cell differentiation through ligation of their endogenous Notch receptor.
Thymocyte-derived ICN1-expressing cells produced DP leukemic lymphoblasts with immunophenotypes and organ distributions indistinguishable from T-ALLs arising from cultured donor bone marrow cells (Figure 3A-D and other data not shown). The only consistent clinical difference between the T-cell neoplasms arising in mice receiving cultured ICN1-positive thymocyte-derived and bone marrow–derived cells was the tendency for Arf −/− thymocyte-derived cells to produce a more aggressive disease marked by unexpected deaths, hind limb paralysis, and ataxia. The latter signs indicated a greater propensity for central nervous system involvement in this group, which was documented at necropsy (supplemental Figure 3).
The intrinsic leukemogenic potential of thymocyte-derived T-ALL was confirmed by secondary transplantation of leukemic bone marrow from the affected animals. Bone marrow from 4 mice with thymocyte-induced Arf −/− T-cell neoplasms was infused into cohorts of 5 healthy, nonirradiated recipient mice at a dose of 105 GFP-positive cells per mouse. This produced an even more rapidly fatal leukemia in all recipients (supplemental Figure 4), which phenotypically recapitulated the primary disease (data not shown). Although the majority of marked retransplanted donor cells were a clonally derived CD4+/CD8+ population enriched for tumor-initiating cells, the LICs might still derive from a minor fraction of less mature donor T cells.
The rates of T-ALL induction in healthy recipient mice receiving cultured ICN1+ Arf−/− cells derived from bone marrow and from thymocytes (Figure 2) were not statistically different from one another (median survival of 29 and 31 days, respectively) and were similar to that observed in the BMT setting (33 days; supplemental Figure 2). This implies that the frequencies of leukemia-initiating cells in these different donor populations were comparable. Indeed, limiting dilution experiments performed with cultured ICN1+ cells showed efficient leukemia-initiating capacity at a dose of 2 × 104 marked cells derived from either Arf−/− thymocytes or from Arf +/+ bone marrow, but not at lower cell doses (supplemental Table 1). Hence, the number of leukemogenic progenitors generated from cultures of ICN1-expressing cells was lower than 1 in 2000. Southern blotting of genomic DNA from populations of T-ALLs derived from 2 × 105 ICN1+ Arf−/− thymocytes harbored 2 to 6 rearranged TCRβ gene fragments, and in some experiments, multiple tumors derived from a single donorculture shared the same bands (supplemental Figure 5). Together, these results argue that thymocyte-derived ICN1+ Arf−/− donor cells require additional genetic events to convert them to LICs.
The thymic microenvironment is dispensable for ICN1 induction of T-ALL
In the case of bone marrow–derived ICN1+ donor cells, only early progenitors (from 5-FU–conditioned donors) were capable of inducing disease, although the resulting tumors reproducibly displayed the DP immunophenotype and expressed surface TCRβ. Thus, in agreement with previous BMT studies,4,5,7,9,12 in vivo progression along the canonical T-cell developmental pathway to the DP stage appears to be a requisite feature of ICN1+ leukemogenesis. There is some evidence that Notch-dependent T-lineage commitment can proceed at extrathymic sites after BMT into irradiated recipients.38 Conceivably, constitutive ICN1 signaling coupled with cytokine and stromal support ex vivo might substitute for the thymic microenvironment in guaranteeing the development of malignant CD4+/CD8+ T cells. To address this issue directly, we repeated both BMT and adoptive transfer experiments using CD-1 nude recipient mice that lack a thymic epithelium and cannot support normal T-cell development. In these experiments, ICN1+ cells again produced a rapidly fatal leukemia with involvement of the bone marrow, spleen, lymph nodes, and peripheral blood (Figure 4 and data not shown). As in syngeneic C57BL/6 recipients, an intact Arf locus provided a survival benefit in irradiated (Figure 4A) or nonconditioned (Figure 4B) recipients when ICN1+ bone marrow donor cells were generated from 5-FU–treated mice. A relatively greater protective effect was seen using Arf +/+ versus Arf −/− thymocyte-derived donor cells (Figure 4C), although the disease was fully penetrant in immunodeficient recipients. The immunophenotype of all T-ALLs, regardless of Arf genotype or source tissue, showed strong marking with Thy1.2 and TCRβ, and a predominance of DP cells (Figure 4D–5F). Constitutive ICN1 expression is therefore sufficient to drive T-ALL–initiating cells to the DP stage in the absence of additional thymic function.
Figure 4.

Athymic nude mice develop CD4+/CD8+ T-ALLs when infused with ICN1-expressing cells. Survival curves are shown for cohorts of irradiated nude mice infused with noncultured ICN1+ bone marrow–derived cells (A; 5 mice/group; P = .03 by log-rank test), nonirradiated nude mice infused with ICN1+ bone marrow–derived cells after 12 days of culture (B; 9 Arf +/+, 8 Arf −/− recipient mice, P = .002), and nonirradiated nude mice infused with cultured ICN1+ thymus-derived cells (C; 3 mice/group, P = .02). Representative immunophenotypes of T-ALLs arising in each experiment are depicted in panels to the right of the corresponding survival curves (D-F). Although all leukemias had a dominant CD4+/CD8+ population, several also had CD8 single-positive subpopulations.
Figure 5.

T-ALLs arising from cultured ICN1+Arf+/+ donor cells retain the gene but do not express p19Arf protein. (A) Leukemic GFP+ splenocytes from 8 mice that had received cultured (day-12) thymocyte-derived donor cells of the indicated Arf genotypes (top) were purified by FACS. Genomic DNA prepared from these cells was used as a template for PCR performed with primers specific for exon-1β and exon-2 of the Ink4a-Arf locus. Exon-1β encodes the unique N-terminus of p19Arf, whereas exon-2 encodes C-terminal portions of p19Arf and p16Ink4a from alternative reading frames. DNAs extracted from unmanipulated Arf +/+ bone marrow cells and from mouse NIH-3T3 cells that had deleted the Ink4a-Arf locus during the process of immortalization were used as the positive and negative controls, respectively. The fact that no exon-1β signal was revealed in samples 7 and 8 generated from Arf −/− donor cells indicates that the purified leukemic cells were uncontaminated by normal Arf +/+ cells. (B) Although all genotyped leukemias retained the Arf locus, immunoblotting of electrophoretically separated proteins extracted from robustly GFP-positive lymph nodes taken from the same recipients of Arf +/+ donor cells failed to reveal p19Arf expression. Arf expression is also silenced in normal bone marrow (BM) cells. Cells engineered to express Arf conditionally under the control of a metallothionein promoter (MT-Arf cells) were induced with zinc. The immunoblot was developed with a highly sensitive monoclonal antibody generated to mouse p19Arf (Bertwistle et al34), and the film was purposely overexposed in an attempt to reveal p19Arf expression. Nucleophosmin (NPM), an abundant nucleolar protein, was used as the loading control. (C) PCR analysis as in panel A. Samples 1 to 4 were taken from irradiated recipients that had received a transplant of uncultured ICN1-expressing bone marrow from 5-FU–conditioned donors. Samples 5 to 10 were taken from nonirradiated recipients that had received 5-FU–conditioned bone marrow transduced with ICN1 and cultured on OP9 stroma before their adoptive transfer. Control PCR reactions were performed using DNAs extracted from unmanipulated Arf +/+ bone marrow cells, NIH-3T3 cells lacking the Ink4a-Arf locus, bone marrow cells from Arf −/− mice in which exon-1β is disrupted, bone marrow cells from p16Ink4a-null mice in which the unique Ink4a exon-1α was disrupted, and from mice in which Ink4a-Arf exon-2 was disrupted. (D) Immunoblotting for p19Arf was performed on bone marrow taken from the mice analyzed in panel C.
Activation of Arf gene expression during T-cell leukemogenesis
The p19Arf protein must be expressed at least transiently in vitro or in vivo to limit the leukemogenicity of ICN1+ Arf +/+ thymocytes. Yet, we were unable to detect p19Arf expression in thymocytes cultured on OP9 stroma. Moreover, all T-ALLs that were induced by cultured ICN1+ Arf +/+ thymocyte-derived or bone marrow–derived cells and purified by FACS from moribund animals retained the Arf gene but failed to express the p19Arf protein (Figure 5). Thus, we assumed that any T cells that expressed Arf were rapidly eliminated, thereby forestalling tumor development, whereas those that did not were able to generate T-ALL.
In an attempt to establish that Arf can be induced during T-ALL development, we used donor T cells derived from a syngeneic mouse strain in which a cDNA encoding GFP was knocked into the cellular Arf locus at the expense of p19Arf coding sequences.39 The genomic Arf-Gfp allele is functionally null, so that homozygous Arf Gfp/Gfp thymocytes should be as susceptible as Arf −/− cells to ICN1-induced leukemogenesis. Importantly, the cellular Arf promoter and 5′ untranslated sequences remain intact and drive GFP expression in oncogene-induced tumors, but not in surrounding normal cells. We transduced thymocytes from Arf Gfp/Gfp mice with a retroviral vector expressing ICN1 and CFP, cultured the cells on OP9 stroma for 12 days, and infused 2 × 105 CFP-marked cells into nonirradiated syngeneic mice. Recipient animals developed CFP-positive T-ALLs that were indistinguishable from those previously generated using the ICN1-GFP vector. Notably, although less than 1% of ICN1-transduced T cells expressed Arf-Gfp after 12 days of culture, GFP was detected in a substantial percentage of CFP-marked T-ALL cells that arose in moribund animals (Figure 6A). Indeed, approximately one-third of CFP-marked T-ALL cells harvested from different hematopoietic tissues stably expressed Arf-Gfp (Figure 6B). These experiments provide direct evidence that the Arf promoter can be activated by ICN1 in a stage-specific manner during T-ALL development.
Figure 6.

Activation of the Arf promoter during T-ALL development. Healthy syngeneic C57BL/6 mice received donor T cells derived from either control Arf −/− or “knock-in” Arf Gfp/Gfp (also functionally null) thymocytes infected with an ICN1-CFP vector. (A) Cells from day-12 cultured thymocytes (left panels) or from spleens of moribund mice (right panels) were studied by FACS for coexpression of vector-coded CFP (ordinate) and cellular Arf-encoded GFP (abscissa). (B) Hematopoietic tissues from 17 such mice were similarly analyzed and contained a significant proportion of CFP-marked cells that coexpressed GFP (ordinate). Error bars indicate the SD from the mean.
Discussion
We have used short-term culture conditions to foster the outgrowth of ICN1-transduced progenitor cells that rapidly induce T-ALL in healthy syngeneic recipients. The cardinal characteristics of these T-ALLs were phenotypically indistinguishable from those induced with a standard BMT protocol,12 highlighting the fact that ablating the hematopoietic and immune systems of recipient animals is not mandatory for T-ALL development. Moreover, transduction of ICN1 into T-cell progenitors not only can command the T-cell developmental program32,33 but also is able to efficiently initiate peripheral CD4+/CD8+ T-ALLs in CD-1 nude mice that lack thymic function. These latter findings reinforce previous observations that Notch-dependent T-lineage commitment can proceed at extrathymic sites after BMT into irradiated recipients.38
Although activating Notch mutations and CDKN2A deletions commonly go hand in hand in human T-ALL, inactivation of Arf had no effect on disease frequency, phenotype, or clinical signs, and only a modest effect on disease latency when cultured ICN1-positive bone marrow–derived cells were used to trigger leukemogenesis. Inactivation of Ink4a in addition to Arf was without effect. Notably, although the Ink4a-Arf locus remained intact, p19Arf was not detectably expressed in leukemic blasts generated from these precursors. Abundant evidence indicates that the Ink4a-Arf locus is epigenetically silenced by polycomb group complexes in hematopoietic stem cells and in early progenitors that constitute the putative target cells in the donor bone marrow of 5-FU–treated mice.19–24 The failure of the locus to undergo deletion in more mature DP T cells that made up the bulk of the induced tumors implies that, under the experimental conditions used, silencing of the locus was maintained during ICN1-driven tumor progression. Hence, in not expressing p19Arf, these T-ALLs effectively phenocopied ICN1+ Arf −/− leukemias.
In striking contrast, when cultures were initiated using ICN1-transduced thymocytes instead of 5-FU–conditioned bone marrow, T cells derived from Arf +/+ mice were much less efficient than Arf −/− donor cells in inducing T-ALL. Because Arf restrains tumors initiated by cultured ICN1+ Arf +/+ thymocytes, p19Arf expression must efficiently cull out incipient tumor cells. Thus, once bone marrow progenitor cells home to the thymus and undergo further T-cell maturation, the Arf locus must be epigenetically remodeled to allow its induction, unmasking its tumor-suppressive activity. The polycomb components Bmi1 and Mel-18 initially silence the Ink4a-Arf locus and maintain Notch-induced Hes1 expression to allow the rapid expansion of viable double-negative (DN) CD4−/CD8− thymic T cells through DN1 (CD44+/CD25−) to DN3 (CD44−/CD25+) stages of development.19,40,41 Conversely, Bmi1 deficiency in the thymus results in precocious p19Arf and p16Ink4a up-regulation. This promotes the p53-dependent death of activated pre-T cells and their complete failure to undergo a transition to the DP state,25 the stage at which malignant transformation is thought to occur.7–9 Notably, Arf, but not Ink4a, inactivation counters these effects of Bmi1 loss of function in T cells and allows further development to proceed.25 Arf inactivation would therefore be expected to compromise the p53 checkpoint, preventing the elimination of rare pre-T cells that harbor abortive receptor rearrangements25 and predisposing to the generation of clonal DP T-ALLs triggered by abnormal Notch expression.
The few tumors that arose from ICN1-transduced Arf +/+ thymocytes after a longer latency period might have been expected to sustain Arf deletions. Instead, like the T-ALLs induced by ICN1-expressing bone marrow progenitors, the locus was retained, and p19Arf was not detectably expressed. This subset of tumors may therefore have arisen from rare early DN cells present in the unfractionated thymocyte preparations used to initiate cocultures, in which epigenetic silencing of Arf is maintained. On the other hand, when functionally null Arf Gfp/Gfp donor thymocytes transduced with an ICN1-CFP vector were used to initiate T-ALL, a significant fraction of the CFP-marked T-ALL cells coexpressed GFP, providing the first direct evidence that the cellular Arf gene can be activated by ICN signaling at this stage of disease progression. However, this does not imply that an ICN1-containing complex directly binds and transactivates the Arf promoter, and indeed, genome-wide chromatin immunoprecipitation and microarray analyses failed to provide evidence that NOTCH1 activates CDKN2A.42 Moreover, although NOTCH1 induces c-MYC,42,43 a well-established inducer of ARF,31,39 Myc has not been demonstrated to bind directly to the Arf promoter either.16 Thus, we infer that both ICN1 and Myc regulate Arf through indirect mechanisms.
Deletion of Arf, Ink4a, or both genes also accelerates the onset of leukemia and increases disease penetrance in lck-tal1 transgenic mice, in which expression of the oncogenic Tal-1 transcription factor is limited to thymic and later stages of T-cell development.44 However, in another transgenic mouse model, in which an activated Notch1 allele is conditionally expressed broadly in hematopoietic cells,45 Arf is not induced.46 These seemingly incompatible results are consistent with our findings that engagement of the Arf-p53 pathway upon oncogenic stress is developmentally stage specific. Arf is induced most readily in later thymic stages of T-cell development, and thus functions as an effective tumor suppressor in the lck-tal1 system, whereas expression of ICN1 in more primitive hematopoietic cells generates leukemias that escape elimination by Arf, even though they can ultimately acquire characteristics of more differentiated T cells.
The very high frequency of INK4A-ARF deletion in human T-ALL implies that these tumors arise from similarly differentiated T-cell precursors, in which the INK4A-ARF gene is poised to respond to oncogenic insults. If silencing were maintained during the evolution of these tumors, as it is in murine bone marrow–derived T-ALL, no selective pressure would drive deletion of the locus. In further support of this hypothesis, human T-ALLs that delete INK4A-ARF are predominantly CD4+/CD8+, whereas an immature DN T-ALL subgroup characterized by expression of LYL1, CD45, and Bcl2 retains the locus.14
Most activated NOTCH1 alleles detected in human T-ALL are only weakly oncogenic in murine bone marrow transplantation assays, suggesting that they may arise as secondary mutations in the majority of tumors that harbor them.11 Nonetheless, γ-secretase treatment of murine T-ALLs driven primarily by mutant K-ras11 or by ectopic Tal-147 abrogates tumor growth, indicating that Notch signaling is indispensable for tumor maintenance. In turn, constitutive Notch signaling per se may be insufficient to activate INK4A-ARF in the majority of human T-ALLs, in which other activated oncogenes might be required to contribute to its induction and eventual elimination. In contrast, the supraphysiologic Notch signal provided by ICN1 not only can drive T-cell development and maturation but also can elicit a fully penetrant neoplastic disease. Nonetheless, even in the case of ICN1 expression and Arf deletion, the emerging T-ALLs are monoclonal or oligoclonal, based on studies of TCRβ gene rearrangements, implying that additional genetic events are required to guarantee disease.
The adoptive transfer model of Notch1-driven T-ALL provides significant advantages as a tool both for developing therapeutics and understanding disease mechanisms. By producing disease in healthy, immune-competent mice, the model eliminates the potential for confounding findings resulting from the effects of radiation. Moreover, the thymocyte-derived disease closely phenocopies pediatric human T-ALL in its distribution, stage-specific cooperation between Notch1 mutation and Arf deletion, and central nervous system involvement, a frequent cause of relapse in children.
Acknowledgments
We thank Richard A. Ashmun and Ann-Marie Easton-Hamilton for performing flow cytometry, Deborah Yons for help with animal husbandry, Jeffrey Morrison and Heather Briley for technical assistance, and members of the Sherr/Roussel laboratory for constructive suggestions and criticisms throughout the course of these studies.
This work was supported in part by Cancer Center Core grant CA-21765 and by American Lebanese Syrian Associated Charities of St Jude Children's Research Hospital. E.J.V. was supported by training grant CA070089. C.J.S. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: E.J.V. designed and performed research, generated new reagents, analyzed data, and wrote the paper; R.T.W. designed research, analyzed data, and wrote the paper; and C.J.S. oversaw the project, designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Charles J. Sherr, Department of Genetics & Tumor Cell Biology, Howard Hughes Medical Institute, St Jude Children's Research Hospital, 262 Danny Thomas Pl, Memphis, TN 38105; e-mail: sherr@stjude.org.
References
- 1.Pui C-H, Robison LL, Look AT. Acute lymphoblastic leukemia. Lancet. 2008;371(9617):1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- 2.Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 3.Irvine KD, Rauskolb C. Boundaries in development: formation and function. Annu Rev Cell Dev Biol. 2001;17:189–214. doi: 10.1146/annurev.cellbio.17.1.189. [DOI] [PubMed] [Google Scholar]
- 4.Aster JC, Pear WS, Blacklow SC. Notch signaling in leukemia. Annu Rev Pathol. 2008;3:587–613. doi: 10.1146/annurev.pathmechdis.3.121806.154300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukemia. Nat Rev Cancer. 2006;6(5):347–359. doi: 10.1038/nrc1880. [DOI] [PubMed] [Google Scholar]
- 6.Tanigaki K, Honjo T. Regulation of lymphocyte development by Notch signaling. Nat Immunol. 2007;8(5):451–456. doi: 10.1038/ni1453. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Gounari F, Protopopov A, Khazaie K, von Boehmer H. Oncogenesis of T-ALL and nonmalignant consequences of overexpressing intracellular NOTCH1. J Exp Med. 2008;205(12):2851–2861. doi: 10.1084/jem.20081561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allman D, Karnell FG, Punt JA, et al. Separation of Notch1 promoted lineage commitment and expansion/transformation in developing T cells. J Exp Med. 2001;194(1):99–106. doi: 10.1084/jem.194.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maillard I, Tu L, Sambandam A, et al. The requirement for Notch signaling at the β-selection checkpoint in vivo is absolute and independent of the pre-T cell receptor. J Exp Med. 2006;203(10):2239–2245. doi: 10.1084/jem.20061020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mansour MR, Duke V, Foroni L, et al. Notch-1 mutations are secondary events in some patients with T-cell acute lymphoblastic leukemia. Clin Cancer Res. 2007;13(23):6964–6969. doi: 10.1158/1078-0432.CCR-07-1474. [DOI] [PubMed] [Google Scholar]
- 11.Chiang MY, Xu L, Shestova O, et al. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J Clin Invest. 2008;118(9):3181–3194. doi: 10.1172/JCI35090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pear WS, Aster JC, Scott ML, et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med. 1996;183(5):2283–2291. doi: 10.1084/jem.183.5.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Neil J, Calvo J, McKenna K, et al. Activating Notch1 mutations in mouse models of T-ALL. Blood. 2006;107(2):781–785. doi: 10.1182/blood-2005-06-2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrando AA, Neuberg DS, Staunton J, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1(2):75–87. doi: 10.1016/s1535-6108(02)00018-1. [DOI] [PubMed] [Google Scholar]
- 15.Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukemia is characterized by the deletion of Ikaros. Nature. 2008;453(7191):110–114. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 16.Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13(1):77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 17.Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. BBA Rev Cancer. 1998;1378(2):F115–F177. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- 18.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2(2):103–112. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 19.Jacobs JJL, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397(6715):164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 20.Park I-K, Qian D, Kiel M, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423(6937):302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 21.Iwama A, Oguro H, Negishi M, et al. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. 2004;21(6):843–851. doi: 10.1016/j.immuni.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Pardal R, Molofsky AV, He S, Morrison SJ. Stem cell self-renewal and cancer cell proliferation are regulated by common networks that balance the activation of proto-oncogenes and tumor suppressors. Cold Spring Harb Symp Quant Biol. 2005;70:177–185. doi: 10.1101/sqb.2005.70.057. [DOI] [PubMed] [Google Scholar]
- 23.Oguro H, Iwama A, Morita Y, Kamijo T, van Lohuizen M, Nakauchi H. Differential impact of Ink4a and Arf on hematopoietic stem cells and their bone marrow microenvironment in Bmi1-deficient mice. J Exp Med. 2006;203(10):2247–2253. doi: 10.1084/jem.20052477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bracken AP, Kleine-Kohlbrecher D, Dietrich N, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21(5):525–530. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyazaki M, Miyazaki K, Itoi M, et al. Thymocyte proliferation induced by pre-T cell receptor signaling is maintained through polycomb gene product Bmi-1 mediated Cdkn2a repression. Immunity. 2008;28(2):231–245. doi: 10.1016/j.immuni.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 26.Signer RA, Montecino-Rodriguez E, Witte ON, Dorshkind K. Aging and cancer resistance in lymphoid progenitors are linked processes conferred by p16Ink4a and Arf. Genes Dev. 2008;22(22):3115–3120. doi: 10.1101/gad.1715808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Signer RA, Montecino-Rodriguez E, Witte ON, McLaughlin J, Dorschkind K. Age-related defects in B lymphopoiesis underlie the myeloid dominance of adult leukemia. Blood. 2007;110(6):1831–1839. doi: 10.1182/blood-2007-01-069401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serrano M, Lee H-W, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85(1):27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 29.Kamijo T, Zindy F, Roussel MF, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91(5):649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 30.Hawley RG, Lieu FH, Fong AZ, Hawley TS. Versatile retroviral vectors for potential use in gene therapy. Gene Ther. 1994;1(2):136–138. [PubMed] [Google Scholar]
- 31.Zindy F, Eischen CM, Randle DH, et al. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12(15):2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmitt TM, Zuniga-Pflucker JC. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity. 2002;17(6):749–756. doi: 10.1016/s1074-7613(02)00474-0. [DOI] [PubMed] [Google Scholar]
- 33.Schmitt TM, Zuniga-Pflucker JC. T-cell development, doing it in a dish. Immunol Rev. 2006;209:95–102. doi: 10.1111/j.0105-2896.2006.00353.x. [DOI] [PubMed] [Google Scholar]
- 34.Bertwistle D, Zindy F, Sherr CJ, Roussel MF. Monoclonal antibodies to the mouse p19Arf tumor suppressor protein. Hybrid Hybridomics. 2004;23(5):293–300. doi: 10.1089/hyb.2004.23.293. [DOI] [PubMed] [Google Scholar]
- 35.Malissen M, Minard K, Mjolsness S, et al. Mouse T cell antigen receptor: structure and organization of constant and joining gene segments encoding the beta polypeptide. Cell. 1984;37(3):1101–1110. doi: 10.1016/0092-8674(84)90444-6. [DOI] [PubMed] [Google Scholar]
- 36.Williams RT, Roussel MF, Sherr CJ. Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2006;103(17):6688–6693. doi: 10.1073/pnas.0602030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams RT, den Besten W, Sherr CJ. Cytokine-dependent imatinib resistance in mouse BCR-ABL+, Arf-null lymphoblastic leukemia. Genes Dev. 2007;21(18):2283–2287. doi: 10.1101/gad.1588607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maillard I, Schwarz BA, Sambandam A, et al. Notch-dependent T-lineage commitment occurs at extrathymic sites following bone marrow transplantation. Blood. 2006;107(9):3511–3519. doi: 10.1182/blood-2005-08-3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zindy F, Williams RT, Baudino TA, et al. Arf tumor suppressor promoter monitors latent oncogenic signals in vivo. Proc Natl Acad Sci U S A. 2003;100(26):15930–15935. doi: 10.1073/pnas.2536808100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van der Lugt NMT, Domen J, Linders K, et al. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 1994;8(7):757–769. doi: 10.1101/gad.8.7.757. [DOI] [PubMed] [Google Scholar]
- 41.Miyazaki M, Kawamoto H, Kato Y, et al. Polycomb group gene mel-18 regulates early T progenitor expansion by maintaining the expression of Hes-1, a target of the Notch pathway. J Immunol. 2005;174(5):2507–2516. doi: 10.4049/jimmunol.174.5.2507. [DOI] [PubMed] [Google Scholar]
- 42.Palomero T, Lim WK, Odom DT, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103(48):18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weng AP, Millholland JM, Yashiro-Ohtani Y, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20(15):2096–2109. doi: 10.1101/gad.1450406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shank-Calvo JA, Draheim K, Bhasin M, Kelliher MA. p16Ink4a or p19Arf loss contributes to Tal1-induced leukomogenesis in mice. Oncogene. 2006;25(21):3023–3031. doi: 10.1038/sj.onc.1209326. [DOI] [PubMed] [Google Scholar]
- 45.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4(2):199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 46.Beverly LJ, Felsher DW, Copobianco AJ. Suppression of p53 by Notch in lymphomagenesis: implications for initiation and regression. Cancer Res. 2005;65(16):7159–7168. doi: 10.1158/0008-5472.CAN-05-1664. [DOI] [PubMed] [Google Scholar]
- 47.Cullion K, Draheim KM, Hermance N, et al. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood. 2009;113(24):6172–6181. doi: 10.1182/blood-2008-02-136762. [DOI] [PMC free article] [PubMed] [Google Scholar]