

Criteria for the interpretability of coordinate differences and a new method for identifying rigid-body motions and nonrigid deformations in protein conformational changes are developed and applied to functionally induced and crystallization-induced conformational changes.
Keywords: bioinformatics, computation, validation, molecular machines, rigid parts
Abstract
Valid interpretations of conformational movements in protein structures determined by X-ray crystallography require that the movement magnitudes exceed their uncertainty threshold. Here, it is shown that such thresholds can be obtained from the distance difference matrices (DDMs) of 1014 pairs of independently determined structures of bovine ribonuclease A and sperm whale myoglobin, with no explanations provided for reportedly minor coordinate differences. The smallest magnitudes of reportedly functional motions are just above these thresholds. Uncertainty thresholds can provide objective criteria that distinguish between true conformational changes and apparent ‘noise’, showing that some previous interpretations of protein coordinate changes attributed to external conditions or mutations may be doubtful or erroneous. The use of uncertainty thresholds, DDMs, the newly introduced CDDMs (contact distance difference matrices) and a novel simple rotation algorithm allows a more meaningful classification and description of protein motions, distinguishing between various rigid-fragment motions and nonrigid conformational deformations. It is also shown that half of 75 pairs of identical molecules, each from the same asymmetric crystallographic cell, exhibit coordinate differences that range from just outside the coordinate uncertainty threshold to the full magnitude of large functional movements. Thus, crystallization might often induce protein conformational changes that are comparable to those related to or induced by the protein function.
1. Introduction
‘Protein flexibility’ is a widely used umbrella term denoting a broad variety of phenomena. At its extremes it is taken to mean either disorder (denaturation) or motions of rigid fragments, but it can also refer to everything in between. However, it is well understood that static protein models are a useful artifice and that protein structures always fluctuate to some extent owing to thermal motion or can be deformed under the influence of external factors. Experimentally, a disordered case would mean that it is not possible to determine the structure of the specific ‘flexible’ part of the protein (Uversky et al., 2008 ▶). In many cases, this disorder may mean that the structure cannot even be crystallized (Price et al., 2009 ▶; Tang & Gallagher, 2009 ▶). All these cases correspond to intrinsically disordered proteins or parts of proteins. On the other hand, specific crystallization conditions might artificially stabilize some regions of proteins which might otherwise be disordered. In some medium-resolution X-ray structures in the PDB (Berman et al., 2000 ▶), fragments of the chain remain unresolved or are represented by multiple conformations at ultrahigh resolution (e.g. Howard et al., 2004 ▶; Wang et al., 2007 ▶), suggesting a dynamic flexibility. Many coordinate differences of 1 Å or larger are found in the PDB between structures of the same protein from independent research groups or from crystals grown under different conditions. Such coordinate differences might reflect limitations of the crystallographic method or actual plasticity of proteins and can be considered as ‘positional uncertainties’ or ‘coordinate uncertainties’ as long as no clear functional or physical meaning can be associated with them. It might be useful to distinguish such ‘positional uncertainties’ from coordinate accuracy, coordinate errors or standard uncertainty as usually referred to in the literature (Richardson, 2007 ▶; Moss et al., 1998 ▶; Brown & Ramaswamy, 2007 ▶). The situation is even more complicated for NMR structures (Snyder et al., 2005 ▶) and will be discussed elsewhere.
Studies of protein flexibility from comparisons of two or more structural states of the same protein were pioneered by Chothia, Lesk, Gerstein and coworkers (Chothia et al., 1983 ▶; Chothia & Lesk, 1985 ▶; Lesk & Chothia, 1984 ▶; Gerstein & Chothia, 1991 ▶; Gerstein et al., 1994 ▶) and led to the creation of a database of significant protein motions (Gerstein et al., 1994 ▶; Gerstein & Krebs, 1998 ▶; Krebs & Gerstein, 2000 ▶; Krebs et al., 2003 ▶). R.m.s. fitting and finding screw transformations by solving matrix equations or by using singular value decomposition (Kabsch, 1976 ▶; McLachlan, 1979 ▶; Challis, 1995 ▶) were used to find and characterize the motions.
More recently, it was realised that the use of quaternions allows a more compact and convenient r.m.s. fitting (Horn, 1986 ▶; Bagci et al., 2003 ▶; Coutsias et al., 2004 ▶; Maiti et al., 2004 ▶; Kavraki, 2006 ▶) and that distance difference matrices (DDMs) might provide a more convenient and accurate measurement of structural dissimilarities than standard r.m.s. fitting (Keller et al., 2000 ▶; Maiti et al., 2004 ▶; Schneider, 2000 ▶, 2004 ▶). However, these newer ideas have not been systematically applied to a broad range of flexibility phenomena.
Currently, increasingly large numbers of protein structures are being determined in large-scale high-throughput research centers organized under the umbrella of the Protein Structure Initiative (PSI–Nature Structural Genomics Knowledgebase, 2009 ▶). Reviews have been published cautioning against the overinterpretation of the results of crystallographic analyses of proteins (Wlodawer et al., 2008 ▶) and pointing out a number of pitfalls and uncertainties, the lack of understanding of the roles of ions and of what constitutes proper model substrates for studies of protein functions and the role of luck in the crystallographic studies of proteins (Chruszcz, Wlodawer et al., 2008 ▶). A very recent paper stressing the necessity of validation of crystallographic protein models notes that in addition to possible errors
Given the same data, no two crystallographers will ever produce identical final models. Their different biases and skill and experience levels will manifest themselves especially during manual model building but also during model refinement (e.g. different ways to parameterize a model and the use of different refinement programs and protocols).
(Kleywegt, 2009 ▶). It has been found that bond lengths and angles depend on the refinement protocols used (Jaskolski et al., 2007 ▶), which might lead to an accumulation of small coordinate differences.
In this and subsequent papers we pursue a closely related aim: a validation of interpretations of coordinate differences between independently determined structures of the same protein.
The pioneering work of Gerstein & Chothia (1991 ▶) introduced a simple classification of the major types of protein motions (Gerstein & Krebs, 1998 ▶). They were characterized by three extents of magnitude (no motion, minor movers and major movers), three sizes (fragment, domain and subunit) and three mechanisms (hinge, shear and other).
The lower threshold for an interpretable change between coordinates from two studies of the same protein has not been consistently defined in the literature and different authors have chosen it to be between 0.1 and 0.4 Å (Sadasivan et al., 1998 ▶; Sinha & Nussinov, 2001 ▶; Gerstein & Chothia, 1991 ▶). While the r.m.s. difference between Cα coordinates of functionally different conformations of the same protein can be as small as 0.6 Å (Hausrath & Matthews, 2002 ▶), often only much larger differences were considered to be significant in the literature. All of this has also been confounded by the use of different alignment procedures.
The terms ‘fragment’ and ‘domain’ are still used very loosely in the literature. What constitutes a domain remains poorly defined according to recent reviews (Wernisch & Wodak, 2003 ▶; Veretnik et al., 2004 ▶), making many structural interpretations unclear.
The ‘shear’ mechanism describes a special kind of sliding motion that maintains a well packed interface, constraining individual shear motions to have very small magnitudes, while their added effect can move protein fragments by tens of angstroms. The mechanism of motion was classified as ‘hinge motion’ when no sliding of fragments on the surface of the protein was involved. The latter term is somewhat misleading, because any movement of protein fragments arises from rotations around one or more single bonds, all of which can be considered to be hinges. Except for the immunoglobulin ‘ball-and-socket joint’, which corresponds to a sliding of smooth surfaces with no packing constraints (Lesk & Chothia, 1988 ▶), other mechanisms or their combinations were neither clearly defined nor studied. In particular, functional conformational changes involving extensive refolding of proteins were also mentioned but were not discussed or studied in detail (Gerstein & Echols, 2004 ▶).
It can be noted that conformational changes with short lifetimes that cannot actually be observed in the ensemble-averaged X-ray structure have been considered to be involved in hydrogen exchange and satisfactorily explained either by local unfolding (Rashin, 1987 ▶) or domain breathing motions (Bahar et al., 1998 ▶).
It appears that one of the major contributions to doubts in the validity of interpretations of protein X-ray structures is a lack of understanding of the role of crystallization itself in the formation of the protein structure and in the utilization of its flexibility.
Initially, we thought that copies of the same molecule comprising an asymmetric crystallographic unit cell would be structurally nearly identical. However, in about half of the dozens of cases that we considered, pairs of structures of the same molecule from the same unit cell exhibited structural differences that were comparable to those derived from pairs of structures corresponding to different functional states of the same molecule.
Because crystals are not a natural medium for proteins, this raised questions of the possible functional or physical reasons for significant conformational differences within unit cells. Could specific reasons, clearly beyond any ‘handwaving’, be found? This might lead to a crucial question: if crystallization can lead to significant relative distortion of protein structures within a single unit cell, what could the role of crystallization be in selecting or even forming almost any structure determined with its help?
Overinterpretation and misinterpretation of structural differences in phenomena involving protein flexibility are a subset of problems faced in the structural studies of proteins. Here, we initiate studies on moderately sized sets of protein structures that are well suited for pinpointing problems, developing methods for their analysis and formulating further questions. We anticipate that studies of different groups of proteins might raise different types of questions. Attempting to find common answers for a very large pool of proteins from different groups is likely to fail since the answers might be group-specific. We plan to subsequently extend our analyses to a larger part of the PDB.
In this paper, we focus on assessing which coordinate differences observed in X-ray structures of the same protein are within the range of currently unexplained uncertainties and thus render such structures identical within the ‘coordinate uncertainty’ and which can be meaningfully assigned to functional changes, crystallization effects or other identifiable reasons. We systematically use distance difference matrices and novel simple quaternion rotations in our analysis (see §2). We show examples of how X-ray coordinate uncertainties and analysis of DDMs might affect previous interpretations of some conformational differences. We also demonstrate how ‘coordinate uncertainty’ thresholds and a simple fragment-superposition procedure allow distinction between ‘rigid-body’ fragment movements and nonrigid deformations in protein conformational changes. This should help to clarify our ideas about the structures and mechanisms of molecular machines, develop a detailed classification of the motions employed and identify and understand particular causes of the currently unexplained motions. All this becomes increasingly important with the publication of a rapidly growing number of structures with higher resolution.
2. Methods
2.1. Distance difference matrices (DDMs)
For a protein of N residues, the distance matrix (DM) is a square N × N matrix in which element ij represents either in a numerical or other way (e.g. by symbols or colors) the distance between residues i and j. A DM is symmetric (the distances i to j and j to i being equal) and therefore usually only half of the matrix is considered (Nishikawa et al., 1972 ▶). If the same protein chain is observed in two different conformations, then DMs can be computed for the two conformations and a distance difference matrix, DDM, can be constructed as a two-dimensional N × N matrix of differences (DDs) between the corresponding elements of the two DMs. In this study, we use distances between all Cα atoms in both the DM and the DDM. This differs significantly from the usual RMSD for two structures of the same molecule (or of its fragment with k residues), which is calculated from only the CA αi—CB αi distances (here, the superscript i denotes a position along the chain and the subscripts A and B denote the two structures being compared),
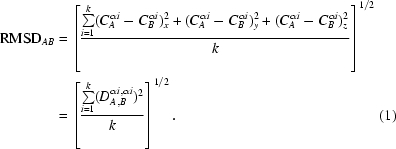 |
We evaluate the RMSDD for any protein fragment of k ≥ 3 residues from all values of DDij (i = 1, k; j = i + 2, k) in the DDM. We exclude DDii and DDi,i+1 because these are either zero or nearly constant. The total number, M, of DDs included is thus M = (k 2/2) − (k/2) − k + 1 (the first term is half of all the elements in the k × k square DDM, the second term excludes half of the DDM diagonal made of DDii and −k + 1 excludes all DDi,i+1). We treat RMSDD as the commonly used ‘sample standard deviation’ and in the denominator under the square root use M − 1 = k(k − 3)/2 for k > 3 and just 1 for k = 3,
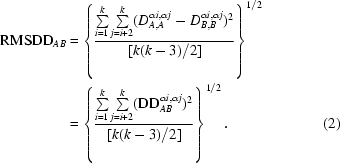 |
Some DDMs are presented in §3. We have chosen to represent these in three shades only (black, grey and white) based on the ranges of the absolute DD values. After various trials and analyses of previous work, we have concluded that additional gradations in shades, colors or symbols can actually serve to obscure the visual analysis of the DDMs.
The coordinate files of individual molecules were edited to contain only residues (or at least their main chains) that were present in the PDB in both molecules of the pair. For asymmetric units with more than two molecules, this could lead to comparisons of pairs of molecules with different numbers of residues in the different pairs. Furthermore, a few residues at the termini or around a crystallographically unresolved segment were edited out if this reduced the RMSDD. The number of residues included in calculations for each particular pair as well as their RMSDDs are given in §3.
2.2. DDM and B factors
A legitimate question arises of whether larger DDs can be rationalized in terms of B factors or of estimated standard deviations derived from the positional errors and B factors usually listed in the PDB. One method we used was to visually estimate the degree of correlation between the peaks in the DDs and the corresponding B factors, as performed by Daopin et al. (1994 ▶).
For estimated standard deviations, we used the expression
where
These expressions are similar to those suggested by Schneider (2000 ▶) and, following that work, a and b denote molecules while i and j denote residues.
For B ave we used the ‘mean B value’ from the PDB and for σave we used the average errors available in the PDB or their estimates (see supplementary material, hereafter referred to as SM1; Cruickshank, 1999 ▶; Read, 2005 ▶).
2.3. DD histograms
We represent DDs not only by a DDM but also as a histogram of the percentage of DDs. We found it useful to derive from the histograms of DDs another characteristic of the DDMs in addition to the RMSDD. The percentage of DDs lying outside the range −1 to 1 Å is denoted by Δ. While RMSDD shows the r.m.s. average of all DDs, Δ shows the percentage of DDs that are ‘large’. Let P(q) be the number of DDs equal to q in angstroms and M be the total number of DDs; then
2.4. Contact distance difference matrices (CDDMs)
It is often of interest to find out whether atoms distant from one another in one structure of a protein come into contact in another structure or how much contacting atoms shift relative to one another. To evaluate such changes, we have constructed contact distance difference matrices (CDDMs). Contact distance matrices (CDMs) for each structure (indexed by m = 1, 2) contain only |Cm αi—Cm αj| distances shorter than a ‘contact’ cutoff, chosen here as 8 Å based on tabulated distances between contacting helices and β-strands in proteins (Chothia & Janin, 1978 ▶; Chothia et al., 1981 ▶). For each ij marked as a ‘contact’ in at least one of two CDMs, a distance difference (the same as in the corresponding DDM) is calculated and marked on the CDDM. All ij positions that were not marked as a ‘contact’ on both CDMs remain blank in the CDDM (see SM11 for an example). The percentage of contact distance changes within any range can be computed from the CDDM. We only calculate such percentages for j > i + 4 in order to avoid domination of our contact statistics by contacts within α-helices and turns.
2.5. Fitting of ‘nearly rigid’ fragments
It is well known that superposition of three non-collinear points of a rigid body superimposes all points of the body. Any such superposition can be represented as an initial superposition of a single point followed by rotations around an axis passing through this point. It has been shown that using centers of mass in the initial superposition of the single point improves the fit (Horn, 1986 ▶; Kavraki, 2006 ▶). Therefore, we used the centers of mass of all Cα atoms in each of the two fragments being fitted as the pair of points to determine the translation of coordinates of the fragments in this initial fitting step. We decided to avoid possible complications in the usual RMSD-based fitting (Kabsch, 1976 ▶; McLachlan, 1979 ▶) by following a simple procedure (shown below) used for the fitting of two slightly distorted three-atom molecules (Rashin et al., 2001 ▶). Because of intrinsic uncertainties in atomic coordinates, this simplification should not introduce significant inaccuracies into the results of fitting. Nevertheless, a few choices were tried for the ends of the fragments being fitted as well as for reference Cα atoms, which must have a row of small DDs in the fragment of the DDM to ensure that the reference atoms move minimally relative to the atoms of the other fragment. Thus, a choice of only two reference Cα atoms in the DDM was required to determine the orientation of the axis of rotation passing through the center of mass and the angle of rotation. Our simple procedure allows us to avoid cumbersome and computationally intensive (Kabsch, 1976 ▶; Horn, 1986 ▶; Kavraki, 2006 ▶) matrix operations. We use a quaternion description of rotations (Kuipers, 1998 ▶) because of its transparency and simplicity for extracting rotational parameters. While quaternions have been known for 160 years, their rigorous use in rotational transformations dates back to 1986 (Horn, 1986 ▶) and is currently preferred in computer applications (Kavraki, 2006 ▶). Widespread application of quaternions to protein RMS fitting seems to have begun in 2003 (Bagci et al., 2003 ▶).
A single rotational transformation is comprised of the following two rotational transformations. Consider two nearly identical sets of points, a, b, c and a′, b′, c′, from structures A and B, with points b and b′ already superimposed (Fig. 1 ▶). Points b and b′ are the centers of mass of A and B. Points a, a′ and c, c′ are centers of ‘reference’ Cα atoms at the same positions along the identical sequences of structures A and B. To simplify the explanation, points b, c and c′ in Fig. 1 ▶ are shown to lie in the plane of the page, while points a and a′ are out of the plane. Atoms c and c′ can then be superimposed (or nearly superimposed if vectors bc and b′c′ have slightly different lengths) by rotating by the angle cbc′ around an axis v 1 passing through atom b and perpendicular to the plane of the page. This leads us from the top to the bottom configuration of points in Fig. 1 ▶. In actual calculations, the axis of this rotation v 1 (for presentation purposes, shown to be perpendicular to the plane of Fig. 1 ▶) is determined from the cross-product of vectors bc and b′c′,
where |v 1| is the length of vector v 1, u 1 = v 1/|v 1| is the unit vector along v 1 and the angle of rotation α1 is obtained from the dot product of the same vectors (the usual controls, which we do not discuss, of the sign of the angle may be required),
where |bc| is the length of vector bc. u 2 = bc/|bc| is the unit vector along bc.
Figure 1.

An illustration of simple rigid-body movement transformations. There are two sets of three points: a, b, c and a′, b′, c′. Within each set the three points are rigidly fixed relative to each other in a rigid body. The triangles formed by the two sets are similar but not identical. Points b and b′ are superimposed. Points b (b′), c and c′ are in the plane of the figure. The bottom configuration of the points is obtained by rotating the rigid set a′, b′, c′ in the top configuration by the angle cbc′ around an axis v 1 passing through superimposed atoms b, b′ and perpendicular to the plane of the page. This superimposes points c and c′ in the bottom configuration (see text for further details).
Vectors ab and a′b can be superimposed (or nearly superimposed if the angles abc and a′b′c′ are slightly different) by rotation around the already superimposed vector (bc). In actual calculations, the angle of rotation around bc = v 2 (bottom image in Fig. 1 ▶) equals the angle α2 between the normal vectors p 1 and p 2 to two planes, one passing through points a, b, c and the other through points a′, b, c′,
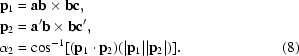 |
These two simply defined rotations with known axes and angles can be combined into a single rotation around a new axis using quaternions (Kuipers, 1998 ▶).
Quaternions a + b i + c j + d k can be viewed as the sum of a real number a and a three-dimensional vector u = b i + c j + d k. An addition of two quaternions yields a new quaternion,
Multiplication of quaternions also yields a quaternion through dot and cross products,
The absolute value of a quaternion, z = a + v, is defined as |z| = (a 2 + |v|2)1/2. The conjugate z* of the quaternion z = a + v is z* = a − v and for a unit quaternion its multiplicative inverse is z −1 = z*.
Rotation of a vector p counterclockwise by angle α around an axis g passing through the origin can be conveniently represented as conjugation by a unit quaternion z,
where
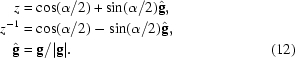 |
Two rotations by quaternions v 1 and v 2 correspond to a rotation by their product v 2 v 1. Our two rotations, first by an angle α1 around u 1 and then by an angle α2 around u 2, can be performed by one rotation around an axis t by an angle θ with a unit quaternion z 21,
Equating the scalar [cos(θ/2)] and vector [sin(θ/2)t] to the scalar and vector components of the quaternion product (see equation 10) on the right-hand side we can obtain the angle θ and the vector of the rotation axis t,
Fragment coordinates relative to the center of mass of the fragment are transformed by rotation by quaternion z 21 (11) and translation by the coordinates of this center of mass.
This is the simplest and fastest algorithm to code for the superimposition of nearly rigid protein fragments. To check its performance, we applied this algorithm and SUPERPOSE (Maiti et al., 2004 ▶) to 32 fragments from three protein pairs: 6ldh–1ldm, 1akz–1ssp and 1lfh–1lfg. For 18 fragments our RMSD was either better or less than 0.05 Å worse compared with that from SUPERPOSE, for six fragments our RMSD was 0.07–0.10 Å worse than that from SUPERPOSE, for three fragments it was 0.1–0.13 Å worse than that from SUPERPOSE and for five (three-residue or four-residue) fragments the SUPERPOSE RMSD was worse than ours by 0.33–1.5 Å (see SM21 for details). In comparisons of SUPERPOSE with MOLMOL (Maiti et al., 2004 ▶) for chains with 100% sequence identity three out of seven SUPERPOSE RMSDs were worse by about 0.1 Å. Thus, our algorithm performs well. Comparisons with several other available superposition algorithms will be reported elsewhere.
We use fragments of no fewer than three residues as rigid bodies (three points determine a rigid body), with the vast majority of all DDs within the black area of a DDM and a small minority in the gray area. Sometimes, we allow a few DDs within a moved fragment to exceed the 1 Å limit if this reduces the RMSDD of the entire pair of molecules. In the first step, the transformation parameters for fitting of the largest rigid fragment are calculated and applied to the coordinates of the protein atoms of the entire second molecule (this does not change the RMSDD). The coordinates corresponding to atoms of bound substrate (or cofactor) are not included in the calculations.
In the following fitting steps, the structures of all fragments of the entire sequence of the second molecule should be fitted to the structure of the corresponding fragments of the first molecule to verify whether the functional movement is (within the coordinate uncertainty) a result of a series of rigid-body movements of protein fragments. If the RMSDD and DD distribution after rigid-body fitting of a second structure to the first lie outside the uncertainty limits, it means that the functional movement involves significant nonrigid deformation of the main chain. The particular order of the fitting steps is arbitrary. Note that by fitting fragments it allows actual breaks (within the coordinate uncertainty limits) between their ends in the complete fitted structure. Before fitting of the entire structure is completed, the ends of consecutive fragments, one of which is already fitted and the other is not, can be distant from one another (a broken chain) because these fragments and their ends might have moved/rotated by large distances between two functional states of the protein.
2.6. Conformational differences between identical molecules in the same unit cell
We have randomly chosen 52 asymmetric units containing more than one molecule and studied 75 structural pairs from the same asymmetric unit. The coordinate files of individual molecules were edited as described in §2.1 above. The number of residues included in calculations for each particular pair together with their RMSDDs and Δs are shown in §3. Because the molecules are from the same PDB file, their pairs are denoted by a single PDB code followed by the chain identifiers in parentheses. In some cases we also cross-compared structures of the same protein from different PDB entries and the corresponding pairs are denoted by both PDB codes.
3. Results
3.1. Estimation of positional uncertainties
To determine the range of coordinate uncertainties, we calculated and analyzed the DDMs of 1014 pairs of structures of bovine ribonuclease A and of some whale myoglobin structures for which the authors of the X-ray studies did not report any significant structural movements (see SM31 for the list of structures used). To avoid subjective judgments in individual cases, our set does not contain proteins complexed with protein inhibitors, structures with low water content, structures at low temperature or structures of mutants. Any of these factors might lead to significant local or global conformational changes (Kishnan et al., 1995 ▶; Frauenfelder et al., 1987 ▶; Sinha & Nussinov, 2001 ▶; Chatani et al., 2002 ▶). Each pair was characterized by its DDM and RMSDD, by a histogram of numbers of DDs of different magnitudes and by the number of DDs outside the range −1 to +1 Å, termed Δ.
We also considered including hen egg-white lysozyme (HEWL) structures in our set; however, HEWL has often been reported to have flexible regions that coincide with contact areas in various crystal forms. The crystallographic unit cell is often comprised of several copies of the same protein. We found that about half of 75 pairs of structures from the same unit cell displayed significant structural differences (see below). Therefore, neither these structures nor HEWL structures have been included in the set for our evaluation of positional uncertainties and will be further analyzed in subsequent studies.
Figs. 2 ▶(a) and 2 ▶(b) give examples of DDMs with unexplained coordinate uncertainties. In Fig. 2 ▶(a), the Cα atom near the C-terminus of the S-peptide (residues 1–21) of one structure (1fs3) of RNase A is shifted by 1 Å more than in another form (1xpt). Similar shifts of residue 21 can be seen, for example, in the DDM 1bel–1xpt (not shown), suggesting that the shift of residue 21 occurs in the 1xpt structure. However, new white and light-gray areas appear in the DDM for the pair 1bel–1xpt compared with that of 1fs3–1xpt. White or gray strips or areas appear in clearly different positions in Fig. 2 ▶(b) compared with Fig. 2 ▶(a). The RMSDD average in Fig. 2 ▶(a) is 0.35 Å and in Fig. 2 ▶(b) it is 0.27 Å.
Figure 2.

DDMs for pairs of structures of bovine ribonuclease A. White space in the DDM means that the absolute value of the distance difference (DD) between the corresponding pair of Cα atoms in the two structures (e.g. PDB entries) is greater than 1 Å, black areas mean that the DD is below 0.5 Å and gray areas indicate DDs between 0.5 and 1 Å. Short thick bars or segments of thin double lines along the tops and sides of the triangular matrices denote the positions of helices or β-strands (taken from the PDB file). Distances between neighboring tick marks on the top and left are at intervals of 20 residues. If the DDM name does not show the chain identifier in parentheses after the PDB name then this indicates that either there is only one chain in the unit cell or the first (usually denoted A) chain is used. (a) 1fs3 (wild-type trigonal crystal) versus 1xpt (monomer A of phosphate-free monoclinic crystal) structures (the DDM is denoted 1fs3–1xpt). (b) DDM 1afu(A)–1afu(B): two monomers from the unit cell of 1afu (monoclinic crystal).
To probe whether the variation in the DDs might be explained by the more direct crystallographic data from the PDB, in Fig. S2(a) in SM41 we compared DDs (Cα21–Cαi)1fs3—(Cα21—Cαi)1xpt, i ≥ 21, which correspond to the brightest line in Fig. 1 ▶(a), directly with the B factors of the Cα atoms of 1fs3 and 1xpt and in Fig. S2(b) in SM41 with σ(DD21,i 1fs3–1xpt) calculated according to (3) and (4). In Figs. S2(c) and S2(d) in SM41 we perform analogous comparisons for DDs (Cα38–Cαi)1fs3–(Cα38–Cαi)1xpt, i ≥ 38, which showed up as the second brightest set of spots in Fig. 2 ▶(a).
We found (see SM41 for details) that the B factors do not explain high/low values of DDs in the 1fs3–1xpt pair. We came to the same conclusion from studying a few more structure pairs. A similar conclusion was reached in another investigation (Sinha & Nussinov, 2001 ▶): the largest B factors do not systematically correspond to the largest DDs in structural pairs of a variety of proteins. A recent investigation of ensemble refinement also suggests that B factors systematically underestimate RMS deviations from the average coordinates (Levin et al., 2007 ▶).
We also checked whether the mean values of B factors of Cα atoms, B α ave, might explain the RMSDD values for six structures of ribonuclease A (1fs3, 1xpt, 1qhc, 1rbx, 1bel and 1jvu) with B α ave between 12.62 and 29.59 Å2 and RMSDD between 0.21 and 0.40. The highest RMSDD was for the pair 1jvu–1bel, with B α ave values of 22.43 and 15.05 Å2, and the lowest RMSDD was for the pair 1jvu–1qhc, with B α ave values of 22.43 and 29.59 Å2. The highest average RMSDD of 0.344 among these six structures was for 1bel, with a B α ave of only 15.05 Å2, followed by the average RMSDD of 0.33 for 1fs3 with a B α ave of 13.88 Å2, while the lowest average RMSDD of 0.282 was for 1qhc with the highest B α ave of 29.59 Å2. Thus, there seems to be no apparent correlation between RMSDDs and B factors.
Fig. 3 ▶ shows the distribution of distance differences in the DDMs for the pairs 1fs3–1xpt and 1afu(A)–1afu(B) (for the A and B chains in the 1afu structure), which are depicted in a simplified smoothed way in three shades in Figs. 2 ▶(a) and 2 ▶(b). Note that a DD distribution can be strongly asymmetric relative to zero DD, as seen in the curve for 1fs3–1xpt. The percentage of DDs within the 0–0.25 Å range is more than twice as small as in the −0.25–0 Å range. On the other hand, the curve for 1afu(A)–1afu(B) is almost symmetric. Such asymmetry or near-symmetry often occurs for pairs of independently determined structures. The sign of the shift of the curve depends on which of the structures is arbitrarily chosen as the reference. However, the presence of a significant shift itself has been shown (or suggested) to be important (see below) in some previous publications (Frauenfelder et al., 1987 ▶; Kundrot & Richards, 1987 ▶; Tilton et al., 1992 ▶). We will re-examine the results from some of these publications below.
Figure 3.

Distribution of the distance differences in the DDMs for ribonuclease A. DD ranges are in increments of ±0.25 Å around 0. The y coordinate shows the percentage of DDs in the range noted on the x axis of all DDs in the corresponding DDM (see §2). A filled triangle for 1fs3–1xpt and a filled square for 1afu(A)–1afu(B) in the column marked by 0.25 on the x axis show the percentage of DDs in the range 0–0.25 Å, those in the column marked by −0.25 show the percentage of DDs in the range −0.25–0 Å and those in the column marked by 0.75 show the percentage of DDs in the range 0.5–0.75 Å, with the corresponding negative DD range marked by −0.75.
The distribution of the RMSDDs in 1014 structural pairs is shown in Fig. 4 ▶.
Figure 4.

Numbers of occurrences of RMSDD magnitudes in 0.01 Å steps in 1014 structural pairs of ribonuclease A and residues 1–151 of myoglobin (see list in SM31). Black bars represent the contributions of RMSDDs from only the ribonuclease pairs and gray bars those of RMSDDs from both ribonuclease and whale myoglobin.
The distribution is asymmetric and bimodal, with the maximum at an RMSDD of 0.29–0.31 Å. We currently do not understand why it is bimodal. The first hump (at lower RMSDD) is more pronounced in the histogram for all 1014 pairs of RNases and myoglobins, but remains in place for 861 RNase pairs. It is possible that in the myoglobin subset the same structural models were used more often as a source of phasing and introduced more artificial similarities and thus smaller RMSDDs. However, it remains unclear why any such similarities would form a hump instead of a monotonically dropping tail in both distributions. The calculated mean for the entire distribution is at an RMSDD of 0.28 Å with a dispersion σ of 0.08 Å. Thus, an RMSDD of 0.44 Å is within 2σ of the entire distribution. For the RNases-only distribution the mean RMSDD is at 0.29 Å and σ = 0.07 Å. If we repeat the calculations for the more symmetric part of the distribution of Fig. 4 ▶ within about σ from its mean (RMSDDs between 0.19 and 0.43 Å), we obtain a new mean for this part of RMSDD = 0.31 Å and σ = 0.05 for both full and RNase distributions. Thus, the high end of the RMSDD distribution at 0.44 Å is now within 2σ or very close to it. In fact, a 2σ cutoff is a completely arbitrary choice for identifying outliers. The coordinate-uncertainty cutoff suggested by Fig. 4 ▶ has a very simple empirical basis. Ribonuclease A and sperm whale myoglobin are rather rigid molecules. If no well justified explanation could be found for an RMSDD value for a pair of structures of a rigid molecule, then the same (or a smaller) unexplained RMSDD value has uncertain causes for any pair of structures of another molecule (which could be softer). Two such structures thus might be considered to be identical within the current coordinate-uncertainty threshold, unless new justified reasons are found to explain a particular case and/or possibly to change the ‘structural identity’ or ‘uncertainty’ thresholds: e.g. we excluded dehydrated proteins from our uncertainty set because they systematically show significant structural changes.
From the distributions of DDs, we derived another characteristic, Δ, of the DDMs (see equation 5). While the RMSDD gives the RMS of all DDs, Δ gives the percentage of ‘large’ DDs (|DD| > 1 Å). For example, for the DDM 1afu(A)–1afu(B) with an RMSDD of 0.27 Å Δ = 0.65%, while for 1fs3–1xpt with an RMSDD of 0.35 Å Δ = 1.22% (see Figs. 2 ▶ and 3 ▶). The DDM of 1fs3–1rca (RNase A, not shown) has an RMSDD of 0.37 Å and Δ = 2.06%.
Fig. 5 ▶ shows the relationship between Δ and RMSDD for 1014 DDMs with coordinate uncertainties. It also includes Δ and RMSDD for two DDMs (endothiapepsin, 4ape–5er2, and thermolysin, 1l3f–3tmn) corresponding to functionally significant motions (Krebs & Gerstein, 2000 ▶) and having the lowest RMSDDs (0.45 and 0.46 Å) among the 20 DDMs of functional motions that we have studied.
Figure 5.

The relationship between Δ and RMSDD for 1014 DDMs. Δ (the percentage of DDs outside the range −1 to 1 Å in DDM) is plotted against the corresponding RMSDD. The two rightmost points in the scatter plot correspond to significant motions in 4ape–5er2 and 1l3f–3tmn (see text).
Note that in the scatter plot in Fig. 5 ▶ the points corresponding to coordinate uncertainty have either smaller RMSDDs or both smaller RMSDD and smaller Δ than the two rightmost points (RMSDD = 0.45 Å, Δ = 5.21% and RMSDD = 0.46 Å, Δ = 3.35%), corresponding to significant functional movements. Therefore, in this paper we will use the criteria that a DDM does not indicate a significant motion but only a coordinate uncertainty when the RMSDD is below 0.46 Å and its Δ is less than 5%. Further accumulation and analysis of data might change these criteria somewhat. However, we find them to be useful working values. Fig. 5 ▶ shows that only a few DDMs have no DDs above ±1 Å. An examination of DDMs show that each one has gray areas corresponding to DDs between ±0.5 and 1 Å.
3.2. Coordinate uncertainty, asymmetry of DD distribution and some previous applications of DDM
3.2.1. Using a single reference structure and ignoring the coordinate uncertainty in multiple pairwise structure comparisons can lead to rather serious misinterpretations of structural differences
DDMs have been used (Sinha & Nussinov, 2001 ▶) to identify structure perturbations caused by point mutations in a few proteins. It was concluded that
regardless of the location of a mutation in the protein structure and of its type, the observed movements of the backbone recur largely at the same positions in the structures regardless of the distance from the mutation.
All mutant structures of a given protein were compared with the same reference structure of that protein. Using the same reference structure for all mutants suggests that the observed ‘recurrence’ of movements may be caused by some peculiarity in the reference structure. The most significant recurrent movement with DDs in the range 2.75–7.17 Å was reported between the mutant and wild-type structure of myoglobin. Of 54 mutants, 49 contained the mutation D122N. As a reference structure, the authors used PDB structure 105m (sperm whale myoglobin at pH 9 with bound N-butyl isocyanide).
Fig. 6 ▶(a) shows the DDM 105m–109m between the reference N-butyl isocyanide structure and the ethyl isocyanide mutant D122N. Large white areas (DDs larger than 1 Å) align with the GH loop and the adjacent terminus of the G helix. In Fig. 6 ▶(b) we show the DDM of 105m compared with the high-resolution structure 1bz6 of aquomet myoglobin at neutral pH. DDM 105m–1bz6 (Fig. 6 ▶ b) has practically the same large white areas aligned with the GH loop and the C-terminus of the G helix as the DDM 105m–109m. However, while Figs. 6 ▶(a) and 6 ▶(b) show the same major large movements, Fig. 6 ▶(a) (Sinha & Nussinov, 2001 ▶) compares the ‘wild type’ with a mutant whereas Fig. 6 ▶(b) (105m–1bz6) involves no mutations.
Figure 6.

DDMs for pairs of myoglobin (residues 1–153) structures (symbols are the same as in Fig. 2 ▶). (a) 105m–109m, reference N-butyl isocyanide structure and ethyl isocyanide mutant D122N; (b) 105m–1bz6, reference N-butyl isocyanide structure and high-resolution aquomet myoglobin; (c) 1bz6–109m, reference high-resolution aquomet myoglobin and ethyl isocyanide mutant.
Thus, it is possible that mutations might have no role in the major movements reported for myoglobin. This is confirmed by DDM 1bz6–109m (Fig. 6 ▶ c), which compares the mutant D122N (structure 109m) with the high-resolution 1bz6 structure. All major movements present in DDM 105m–109m are absent in 1bz6–109m. RMSDDs for DDMs involving ‘wild-type’ structure 105m shown in Figs. 6 ▶(a) and 6 ▶(b) are 0.55 and 0.57 Å, respectively. Δ values for the same two DDMs are 5.99 and 5.51%. These RMSDDs and Δ values indicate significant motions beyond the coordinate uncertainty. In contrast, the DDM of Fig. 6 ▶(c) has an RMSDD of 0.28 and Δ = 0.73%, both of which are characteristic of only a coordinate uncertainty.
Comparisons of structures 105m and 109m to a variety of independently determined whale myoglobin structures (1bzr, 5mbn, 1l2k and 1mbo; not shown) further confirm the conclusion that the reported large movements in myoglobin mutants are likely to arise from peculiarities of structure 105m used as a reference. Practically all other movements in other proteins ascribed to mutations (Sinha & Nussinov, 2001 ▶) seem to have small DDs that are characteristic of coordinate uncertainties and are not necessarily related to mutations. Some reported differences (Sinha & Nussinov, 2001 ▶) might also arise from comparisons of proteins from different species (e.g. 105m and 1mdn).
3.2.2. Can small coordinate shifts within the uncertainty threshold reliably be interpreted using a careful refinement?
Expansion/contraction effects were expected to be within 0.1 Å in a comparison of hen egg-white lysozyme (HEWL) structures at pressures of 101 MPa (3lym) and 101 kPa (2lym) (Kundrot & Richards, 1987 ▶). The refinement of the high-pressure data was started from a partial refinement of the low-pressure structure 2lym. The study can be viewed as a carefully controlled structure-perturbation refinement. The contraction was reported to be non-uniformly distributed, with residues 40–88 being essentially incompressible. The authors state that they
consider changes in structure to be more accurate than the absolute structure.
The DD histogram (0.6% between −0.5 and −0.25 Å, 84.2% between −0.25 and 0 Å, 15.1% between 0 and 0.25 Å) representing the original structures 2lym and 3lym shows a narrow peak of DD distribution that agrees with the expectations of the authors of the original study (Kundrot & Richards, 1987 ▶). However, there are quite a few pairs of HEWL in the same crystal form P43212 which exhibit very similar narrow high peaks shifted in the negative direction in their DD histograms and quite similar DDMs, while they do not differ in pressure. For example, 193l–1bvx(A) has 10, 81.6 and 6.9% of all DDs in the corresponding DD histogram regions (−0.5 to −0.25 Å, −0.25 to 0 Å and 0 to 0.25 Å, respectively). Interestingly, 1bvx was refined starting from the refined structure of 193l, which had the highest resolution (1.33 Å). Could such refinement sufficiently restrict differences between these two structures, which were otherwise studied under rather similar conditions? Could the refinement of 3lym starting from a partial refinement of 2lym (Kundrot & Richards, 1987 ▶) impose a similar restraint? According to some opinions, comparison of structures when the source of phasing was the same structural model may show artificial similarity. Returning to Fig. 3 ▶ for the 1fs3–1xpt ribonuclease pair, would it be possible to take a well refined 1xpt structure as a starting structure for refinement of 1fs3 and obtain a narrower and about twice higher peak for the newly produced 1fs3–1xpt pair? Similar high negatively shifted peaks and DDMs are observed for 193l–1azf or 194l–1azf, where the 1azf crystals were grown in a bromide solution. There are more pairs that exhibit these characteristics. Therefore, we can conclude that an apparent small contraction of the HEWL structure might be caused by a variety of chemical or computational factors. Before all the possibilities have been checked, we might be better off erring on the side of caution and considering the small coordinate changes in the HEWL pressure experiment (Kundrot & Richards, 1987 ▶) as possibly ‘having uncertain causes’.
3.3. Functional motions, their evaluation and classification
Functional (or protein association-induced) motions may or may not involve an actual hinge. A deformation in the main chain ‘allowing’ a significant motion far from this deformation may be a continuous deformation of a flexible fragment of a chain (like a bent spring) as well as a series of hinges. Thus, we suggest that we distinguish an ‘allowing’ deformation in the main chain from a motion remote from this main-chain deformation. There may be a chain of smaller motions that allow a remote motion (as, for example, in citrate synthase, where small shifts and deformations of contacting helices accumulate, leading to a large remote conformational movement; Lesk & Chothia, 1984 ▶).
We retain the name hinge motion for motions allowed by hinges in the main chain but neither forming new long-range contacts nor grabbing a target, e.g. as in molecules of the viral capsid of 2tbv (Gerstein et al., 1994 ▶; Harrison, 1980 ▶). If a motion grabs a target (e.g. substrate) and brings remote protein parts into contact, we shall liken it to closing of the tips of tweezers and call it a tweezers motion. A motion in which remote protein parts lock onto a target but their tips do not form a close contact we shall call a pliers motion. If transforming one functional conformation into another (within coordinate uncertainty) can only be achieved by a large number (over a dozen) of rigid-body motions of its fragments, we shall call the entire motion a glove tweezers/pliers motion. More (and alternatively named) types can be suggested as detailed analysis of motions in proteins progresses. It should be noted that we often cannot tell which of a number of rigid-body transformations between two conformational states are required by the function of a protein and which might be caused by independent factors, e.g. crystal forces (see below).
Any mostly black right-angle triangle on the diagonal side of a DDM can be considered to be a rigid body within the coordinate uncertainty. (This can be verified by directly calculating the RMSDD for the part of the DDM represented by the triangle.) A rigid-body motion of any such protein substructure can be fully described by the translation of its center of mass between its position in one conformation to its position in another, the directional angles of the axis of rotation passing through the center of mass and the angle of rotation around this axis (see §2). The borders of the triangular rigid-body parts of DDM often are clearly delineated in the DDM. However, they might require a trial-and-error adjustment of its ends to achieve the largest reduction in RMSDD upon the rigid-body movement (see §2).
3.3.1. Pliers
DDM 1hrd–1bgv and the superimposed wire images for glutamate dehydrogenase apo to holo structures are shown in Figs. 7 ▶(a) and 7 ▶(b). The DDM is characterized by RMSDD = 1.89 Å, Δ = 32.85%. CDDM does not show any newly formed long-range contacts with large DDs (pliers). The boundaries of all rigid fragments are almost delineated by the white rectangular or stripe-like areas of the DDM. The marking of the secondary structure on the borders of the DDM allows an easy description of the movements of the fragments in terms of the movements of the secondary-structure elements. Therefore, we do not specifically focus here on the movements of individual secondary-structure elements or their groups.
Figure 7.

An example of pliers motion for glutamate dehydrogenase (1hrd–1bgv). (a) The DDM with a rather clear delineation of rigid fragments: four essential rigid-body motions (two major, two small) after the initial superposition; notation is the same as in Fig. 2 ▶. (b) A superposition of fragment 1–203 of wire images of 1hrd (black) and 1bgv (gray, where it does not practically coincide with 1hrd); all wire images in this work were produced with the MOLE package (Kurochkina & Privalov, 1998 ▶; MOLE CD and manual available from G. P. Privalov, gpriv@axonx.com).
The following five rigid-body transformations led to a DDM with RMSDD = 0.22 Å and Δ = 0.01%, which are both well within the characteristics of coordinate uncertainties alone. Two molecules were superimposed using the fragments corresponding to the top dark triangle in the DDM (1–203). This was followed by fitting fragments 204–372, 373–393, 394–431 and 432–449. Rigid-body movement of 204–372 reduced the RMSDD by 1.29 Å and that of 394–431 reduced it by 0.32 Å, with the other two motions contributing 0.06 Å to RMSDD reduction. Note that the CATH domains (Orengo et al., 1997 ▶) for 1hrd are (1–51) + (425–449), 52–187 and 297–373. We find that fragments 1–51 and 52–187 move together as one rigid body within fragment 1–203, with fragment 425–449 also practically not moving relative to them. If we limited the fitting to only two rigid-body movements (initial 1–203 followed by 204–431), this would result in an RMSDD of 0.35 Å and Δ = 1.84%. Thus, either five or two rigid-body movements can lead to structures that are identical within the coordinate-uncertainty thresholds.
3.3.2. Tweezers
Fig. 8 ▶ shows the DDMs 9aat–1ama (mitochondrial aspartate aminotransferase) with corresponding superimposed wire images of 9aat and 1ama before and after a series of transformations of 1ama: 228–319 (initial superposition; second largest uninterrupted dark triangle), 382–401, 350–381 and 14–33. Rigid-body fittings of fragments 320–349, 2–13 and 34–36 produced only very small changes and are not shown separately. Initially, 9aat–1ama was characterized by an RMSDD of 1.2 Å and Δ = 23%. The DDM after transformations has the characteristics of only a coordinate uncertainty, with an RMSDD of 0.36 Å and Δ = 2.52%. Thus, structures 9aat and 1ama are obtained from one another by a short series of rigid-body motions.
Figure 8.

Comparison of DDMs and wire superpositions in a sequence of rigid-body transformations for tweezers functional motion of aspartate aminotransferase (9aat–1ama). (a) DDM of the initial two structures; (d) final DDM after a sequence of rigid-body transformations (see text); (e–h) wire-frame superpositions of the same structures depicted by DDMs in (a–d): the black wire shows the unchanging reference conformation of 9aat(A) and the gray wire shows the 1ama structure changing on rigid-body movement (see text). Notation is the same as in Fig. 2 ▶, except that only residues present in the PDB files of both molecules are included in the DDM calculation and these residues are numbered sequentially.
It is interesting to see how the RMSD of fragments and corresponding RMSDD of the entire structure change with individual rigid-body transformations. Moving the C-terminal fragment 382–401 reduces its RMSD from 4.36 to 0.56 Å; however, the RMSDD of the entire pair of structures remains at 1.19 Å. Comparison of Figs. 8 ▶(a) and 8 ▶(b) shows that fragment 382–401 improved its fit to a large portion of the rest of the structure of 9aat but the fit becomes worse for two neighboring fragments 350–381 and 319–349. The RMSD of 350–381 drops from 3.73 to 0.62 Å and the corresponding RMSDD is reduced from 1.19 to 1.10 Å. For 320–349 the RMSD falls from 3.74 to 0.4 Å and the RMSDD of the entire pair of structures falls to 1 Å. The result of these two rigid-body transformations (Fig. 8 ▶ c) shows that all the vertical white strips corresponding to DDs larger than 1 Å disappear and that all remaining white space is horizontal and associated with the N-terminal fragments. After the N-terminal fragments are moved as rigid bodies most of the white space disappears, as shown in Fig. 8 ▶(d). The resultant DDM of 9aat–1ama (Fig. 8 ▶ d) has characteristics that indicate only coordinate uncertainties. In this work, we concentrate on the characteristics of the entire pair of structures. It may be noted that some individual fragments of 1ama remain distorted beyond the uncertainty threshold (e.g. 14–33). However, the rigid-body transformation of this fragment reduces the RMSDD of the entire 9aat–1ama from 1.0 to 0.4 Å. One might suggest moving the rigid fragments according to the CATH domain assignment: 47–319, 13–46, 320–401 (note that 13–46 is shorter than the usually accepted domain size). In fact it does lead to a final structure within the uncertainty threshold with an RMSDD of 0.44 Å and Δ = 4.34% (see SM5 for a comparison with Fig. 8 ▶ d). In particular, fitting of the C-terminal CATH domains as one unit leads to an RMSDD of 0.42 Å and Δ = 4.2% for this domain, while fitting of three separate fragments yields an RMSDD of 0.37 Å and Δ = 2.38%. (Note that fragment 2–12 was not moved individually but only with the initial fitting of 47–319; also, attempts to move fragments 13–46 and 320–401 together as a rigid discontinuous domain were not successful). Thus, both transformations (with either seven or three rigid-body movements; see DDMs in SM5) yielded structures that were identical within the uncertainty threshold, while the structural papers reported small movements and distortions within the domains. It was not tested whether the more accurate fitting could be required in a fitting of the biological dimer, in which actual movement does occur. Comparing the 9aat–1ama DDMs with the corresponding superimposed wire pairs (Fig. 8 ▶), it is easy to see that DDMs show movements, as well as their location, very clearly, while these are more difficult to see and position in the sequence in the wire pairs, requiring finding an advantageous orientation of the wire pair. However, the two representations might be complementary. For example, wire frames (Fig. 8 ▶ e–8h) as well as CDMs clearly show that the ‘hanging’ N-terminal tail does not interact with the main body of its monomer and is kept ‘rigid’ by interactions in the dimer.
3.3.3. Glove tweezers
The DDM 4ake–1ank for adenylate kinase apo-to-holo motion and the superimposed wire representation are shown in Figs. 9 ▶(a) and 9 ▶(b). The DDM is characterized by an RMSDD of 6.45 Å, Δ = 59.62%. The motion is described as ‘tweezers’ because CDDM (see SM1) shows newly formed long-range contacts with large DDs (tweezers). Note that practically the entire adenylate kinase DDM looks like the area of a conformational change, with much white space close to the diagonal indicating nonrigid deformations. The largest rigid fragment (black triangle 120–156) possesses almost no secondary structure (Figs. 9 ▶ a and 9 ▶ b). Attempts to transform the holo structure to the apo structure by a sequence of rigid-body motions did not succeed despite attempts to move many sets of differing rigid fragments. The resultant RMSDDs were around 1 Å, obviously indicating highly flexible structures undergoing glove movements with tweezers closure.
Figure 9.

An example of glove tweezers motion for adenylate kinase (4ake–1ank). (a) DDM showing a flexible structure with many fragments not having rigid conformations and thus not reducible to rigid-body motions. Notation is the same as in Figs. 7 ▶ and 8 ▶. (b) Wire image superposition of fragment 120–156 (the largest black triangle in the DDM); 4ake, black wire; 1ank, grey wire (the fitted region is almost completely covered by black wire).
3.4. Conformational differences between identical chains in the same unit cell: general statistics
Table 1 ▶ presents a compilation of the results of our analysis together with other available data for 52 asymmetric unit cells with more than one molecule, providing 75 structural pairs each from the same asymmetric unit, and for over two dozen test cross-comparisons of structures.
Table 1. Characteristics of pairs of molecules in unit cells and of their flexibilities.
| No.† | Pairs‡ | Protein name | Residues§ | Resolution (Å) | In cell | Biomol.¶ | Space group | RMSDD (Å) | Δ (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1kv3 (A:F) | Transglutaminase | 651 | 2.8 | 6 | d | P212121 | 0.04 | 0.00 |
| 2 | 1b8j (AB) | Phosphatase | 449 | 1.9 | 2 | d | I222 | 0.06 | 0.00 |
| 3 | 4pbg (AB) | β-Galactosidase | 468 | 2.5 | 2 | c | P21212 | 0.09 | 0.00 |
| 4 | 1cle (AB) | Cholesterol esterase | 534 | 2.0 | 2 | m | P1 | 0.10 | 0.00 |
| 5 | 4cha (AB) | α-Chymotrypsin | 238 | 1.68 | 2 | d | P21 (P1211) | 0.14 | 0.02 |
| 6 | 1gtm (AC) | Glu dehydrogenase | 417 | 2.2 | 3 | h | P42212 | 0.16 | 0.01 |
| 7 | 1dq4 (AB) | Concanavalin | 223 | 2.9 | 2 | te | P21212 | 0.17 | 0.53 |
| 8 | 9aat (AB) | Asp aminotransferase | 401 | 2.2 | 2 | d | P1 | 0.18 | 0.01 |
| 9 | 1ajr (AB) | Asp aminotransferase | 412 | 1.74 | 2 | d | P212121 | 0.18 | 0.01 |
| 10 | 1bjw (AB) | Asp aminotransferase | 382 | 1.8 | 2 | d | P212121 | 0.19 | 0.27 |
| 11 | 1gtm (AB) | Glu dehydrogenase | 417 | 2.2 | 3 | h | P42212 | 0.19 | 0.34 |
| 12 | 2gd1 (O:R) | Glyceraldehyde dehydrogenase | 334 | 2.5 | 4 | te | P21 (P1211) | 0.20 | 0:0.11 |
| 13 | 13pk (BC) | Phosphoglycerate kinase | 415 | 2.5 | 4 | d | P212121 | 0.20 | 0.25 |
| 14 | 1bmd (AB) | Malate dehydrogenase | 327 | 1.9 | 2 | d | P212121 | 0.20 | 0.54 |
| 15 | 1gtm (BC) | Glu dehydrogenase | 417 | 2.2 | 3 | h | P42212 | 0.21 | 0.37 |
| 16 | 3tim (AB) | Triosephosphate isomerase | 249 | 2.8 | 2 | d | P212121 | 0.26 | 0.08 |
| 17 | 2ccy (AB) | Cytochrome c | 127 | 1.67 | 2 | d | P212121 | 0.27 | 1.47 |
| 18 | 4cts (AB) | Citrate synthase | 437 | 2.9 | 2 | d | P43212 | 0.28 | 0.13 |
| 19 | 1b47 (AB) | CBL/ZAP-70 N-domain | 304 | 2.2 | 3 | h | C2 (C121) | 0.29 | 0.65 |
| 20 | 9wga (AB) | Lectin | 171 | 1.8 | 2 | d | C2 (C121) | 0.30 | 0.83 |
| 21 | 1gam (AB) | γB crystallin C-domain | 86 | 2.6 | 2 | d | P3221 | 0.31 | 1.34 |
| 22 | 1b47 (AC) | CBL/ZAP-70 N-domain | 304 | 2.2 | 3 | h | C2 (C121) | 0.32 | 0.92 |
| 23 | 1cbu (AB) | Cobinamide kinase | 180 | 2.3 | 3 | h | C2221 | 0.34 | 1.57 |
| 24 | 1beb (AB) | β-Lactoglobulin | 156 | 1.8 | 2 | d | P1 | 0.35 | 1.14 |
| 25 | 1ggu (AB) | Blood coagulation factor XIII | 701 | 2.1 | 2 | d | P21 (P1211) | 0.36 | 2.56 |
| 26 | 4lyt (AB) | HEW lysozyme | 129 | 1.9 | 2 | c | P21 (P1211) | 0.36 | 1.48 |
| 27 | 1b47 (BC) | CBL/ZAP-70 N-domain | 304 | 2.2 | 3 | h | C2 (C121) | 0.37 | 1.02 |
| 28 | 1cbu (BC) | Cobinamide kinase | 180 | 2.3 | 3 | h | C2221 | 0.38 | 1.87 |
| 29 | 1cdl (CB) | Calmodulin | 138 | 2.2 | 4 | d | P21212 | 0.39 | 2.43 |
| 30 | 1cbu (AC) | Cobinamide kinase | 180 | 2.3 | 3 | h | C2221 | 0.40 | 3.05 |
| 31 | 13pk (AB) | Phosphoglycerate kinase | 415 | 2.5 | 4 | d | P212121 | 0.42 | 4.06 |
| 32 | 1oxt (AB) | ABC ATPase | 352 | 2.1 | 3 | m | P212121 | 0.43 | 3.58 |
| 33 | 1pp2 (LR) | Phospholipase A | 122 | 2.5 | 2 | d | P212121 | 0.44 | 4.39 |
| 34 | 1oxt (BD) | ABC ATPase | 352 | 2.1 | 3 | m | P212121 | 0.44 | 3.92 |
| 35 | 1a5d (AB) | γE crystallin | 173 | 2.3 | 2 | c | P21 (P1211) | 0.45 | 3.40 |
| 36 | 13pk (CD) | Phosphoglycerate kinase | 415 | 2.5 | 4 | d | P212121 | 0.46 | 6.18 |
| 37 | 1cdl (BD) | Calmodulin | 138 | 2.2 | 4 | d | P21212 | 0.47 | 3.99 |
| 38 | 1cdl (AB) | Calmodulin | 138 | 2.2 | 4 | d | P21212 | 0.47 | 4.52 |
| 39 | 1g51 (AB) | Asp tRNA synthase | 580 | 2.4 | 2 | d | P212121 | 0.48 | 2.92 |
| 40 | 1cdl (AC) | Calmodulin | 138 | 2.2 | 4 | d | P21212 | 0.49 | 4.71 |
| 41 | 1aa7 (AB) | Flu virus protein M1 | 157 | 2.08 | 2 | d | P3121 | 0.49 | 5.29 |
| 42 | 13pk (AC) | Phosphoglycerate kinase | 415 | 2.5 | 4 | d | P212121 | 0.49 | 6.17 |
| 43 | 1b3a (AB) | Anti-HIV protein | 67 | 1.6 | 2 | d | P212121 | 0.49 | 7.41 |
| 44 | 6adh (AB) | Alcohol dehydrogenase | 374 | 2.9 | 2 | d | P1 | 0.50 | 4.66 |
| FM | 8adh→6adh | ADH functional motion | 374 | 2.4/2.9 | 1 | d | C2221/P1 | 1.05 | 21.90 |
| 45 | 13pk (BD) | Phosphoglycerate kinase | 415 | 2.5 | 4 | d | P212121 | 0.50 | 7.75 |
| 46 | 1oxt (AD) | ABC ATPase | 352 | 2.1 | 3 | m | P212121 | 0.53 | 6.04 |
| 47 | 1g59 (AC) | Glu tRNA synthase | 468 | 2.4 | 2 | d | C2221 | 0.54 | 6.64 |
| 48 | 1njg (AB) | E. coli polymer clamp loader | 239 | 2.2 | 2 | m | P21 (P1211) | 0.55 | 5.56 |
| 49 | 1cdl (AD) | Calmodulin | 138 | 2.2 | 4 | d | P21212 | 0.57 | 7.22 |
| 50 | 4dfr (AB) | Dihydrofolate reductase | 159 | 1.7 | 2 | d | P61 | 0.58 | 4.45 |
| 51 | 1cdl (CD) | Calmodulin | 138 | 2.2 | 4 | d | P21212 | 0.63 | 10.72 |
| FM | 1cll→1ctr | Calmodulin functional motion | 138 | 1.7/2.45 | 1 | m | P1/P3221 | 12.83 | 54.92 |
| 52 | 13pk (AD) | Phosphoglycerate kinase (PGK) | 415 | 2.5 | 4 | d | P212121 | 0.65 | 7.75 |
| FM | 16pk→13pk | PGK functional motion | 415 | 1.6/2.5 | 1 | m | P212121 | 3.07 | 45.61 |
| 53 | 1j7n (AB) | Anthrax toxin | 725 | 2.3 | 2 | m | P21 (P1211) | 0.67 | 12.60 |
| 54 | 2ak3 (AB) | Adenylate kinase | 215 | 1.85 | 2 | m | P212121 | 0.68 | 10.54 |
| FM | 4ake→1ank | Adenylate kinase functional motion | 214 | 2.2/2.0 | 2→1 | d→m | P1/C2 (C121) | 6.45 | 59.62 |
| 55 | 1gyr (AB) | rRNA A dimethyltransferase | 252 | 2.1 | 2 | m | C2 (C121) | 0.68 | 13.48 |
| 56 | 1dbw (AB) | Transcriptional protein FIXJ-N | 123 | 1.6 | 2 | d | P1 | 0.69 | 10.31 |
| 57 | 2tbv (AB) | Tomato bushy stunt virus | 287 | 2.9 | 3 | h | I23 | 0.75 | 6.63 |
| 58 | 6tim (AB) | Triosephosphate isomerase | 249 | 2.2 | 2 | d | P21212 | 0.83 | 6.90 |
| Test 1a | 3tim–6tim (AA) | Cross-test | 249 | 0.20 | 0.00 | ||||
| Test 1b | 3tim–6tim (BA) | Cross-test | 249 | 0.27 | 0.50 | ||||
| 59 | 1ivy (AB) | Carboxypeptidase | 450 | 2.2 | 2 | d | P21212 | 0.87 | 8.31 |
| 60 | 2j1p (AB) | Diphosphate synthase | 271 | 1.8 | 2 | d | P21 (P1211) | 0.94 | 14.08 |
| 61 | 1ajs (AB) | Asp aminotransferase | 412 | 1.6 | 2 | d | P212121 | 0.97 | 18.96 |
| Test 2a | 1ajr (AB) | Asp aminotransferase | 412 | 1.74 | 2 | d | P212121 | 0.18 | 0.01 |
| Test 2b | 1ajs–1ajr (BB) | Cross-test | 0.39 | 0.54 | |||||
| Test 2c | 1ajs–1ajr (AA) | Cross-test | 0.94 | 17.31 | |||||
| Test 2d | 1ajs–1ajr (AB) | Cross-test | 1.00 | 18.52 | |||||
| FM | 9aat→1ama | Asp aminotransferase functional motion | 401 | 2.3/2.3 | 2 | d | P1/C2221 | 1.20 | 22.96 |
| 62 | 1ex6 (AB) | Apo guanylate kinase | 186 | 2.3 | 2 | d | P31 | 0.99 | 23.86 |
| FM | 1ex6→1ex7 | Guanylate kinase functional motion | 186 | 2.3/1.9 | 2→1 | d→m | P31/P43212 | 2.99 | 36.89 |
| 63 | 2eia (AB) | Anemia virus capsid | 204 | 2.7 | 2 | te | P6122 | 2.86 | 39.94 |
| 64 | 2tbv (AC) | Tomato bushy stunt virus | 287 | 2.9 | 3 | h | I23 | 1.37 | 32.22 |
| 65 | 1jkt (AB) | Death-associated kinase CD | 276 | 3.49 | 2 | d | P41 | 1.06 | 26.58 |
| Test 3a | 1jks–1jkt (A) | 276 | 1.5/3.49 | 1/2 | P21212/P41 | 1.10 | 24.30 | ||
| Test 3b | 1jks–1jkt (B) | 276 | 1.5/3.49 | 1/2 | P21212/P41 | 1.02 | 21.13 | ||
| 66α test | 1xz2 (αα) | Hemoglobin dimer in tetramer | 141 | 1.9 | 4 | te | P212121 | 0.16 | 0.00 |
| 67β test | 1xz2 (ββ) | Hemoglobin dimer in tetramer | 146 | 1.9 | 4 | te | P212121 | 0.21 | 0.19 |
| 68 | 2bj1 (AB) | NikR + 4 Ni, 1 Cl | 133 | 3.0 | 2 | te | P41212 | 2.52 | 40.27 |
| 69 | 2bj3 (AB) | NikR + 2 Cl, 2 Mg = apo | 134 | 2.2 | 4 | te | P21 (P1211) | 2.71 | 49.02 |
| 70 | 2bj3 (AC) | Same as above | 135 | 2.2 | 4 | te | P21 (P1211) | 2.76 | 51.33 |
| 71 | 2bj3 (AD) | Same as above | 128 | 2.2 | 4 | te | P21 (P1211) | 0.40 | 3.35 |
| 72 | 2bj3 (BC) | Same as above | 137 | 2.2 | 4 | te | P21 (P1211) | 0.42 | 3.55 |
| 73 | 2bj7 (AB) | NikR + 4 Ni, 1 Cl, 2 PG4, 2 EDO | 137 | 2.1 | 2 | te | P3221 | 7.87 | 51.93 |
| 74 | 2bj8 (AB) | NikR + 6 Ni, 1 Cl, 1 PG4, 1 EDO | 136 | 2.1 | 2 | te | P3221 | 7.79 | 51.80 |
| 75 | 2bj9 (AB) | NikR + 5 Ni, 2 PO4, PG4 | 133 | 3.0 | 2 | te | P3221 | 7.77 | 50.79 |
| Test 4a | 2bj3–2bj1 (AA) | Cross-test | 132 | 0.89 | 14.87 | ||||
| Test 4b | 2bj3–2bj1 (BB) | Cross-test | 131 | 1.33 | 36.94 | ||||
| Test 4c | 2bj3–2bj7 (AA) | Cross-test | 135 | 4.63 | 47.66 | ||||
| Test 4d | 2bj3–2bj7 (BB) | Cross-test | 137 | 4.24 | 55.12 | ||||
| Test 4e | 2bj3–2bj8 (AA) | Cross-test | 135 | 4.55 | 47.69 | ||||
| Test 4f | 2bj3–2bj8 (BB) | Cross-test | 137 | 4.22 | 55.12 | ||||
| Test 4g | 2bj3–2bj9 (AA) | Cross-test | 133 | 4.68 | 46.10 | ||||
| Test 4h | 2bj3–2bj9 (BB) | Cross-test | 137 | 4.25 | 55.33 | ||||
| Test 4i | 2bj7–2bj8 (AA) | Cross-test | 136 | 0.11 | 0.00 | ||||
| Test 4j | 2bj7–2bj9 (AA) | Cross-test | 133 | 0.28 | 0.28 | ||||
| Test 4k | 2bj8–2bj9 (AA) | Cross-test | 133 | 0.32 | 0.62 | ||||
| Test 4l | 2bj7–2bj8 (BB) | Cross-test | 138 | 0.13 | 0.01 | ||||
| Test 4m | 2bj7–2bj9 (BB) | Cross-test | 138 | 0.40 | 1.31 | ||||
| Test 4n | 2bj8–2bj9 (BB) | Cross-test | 138 | 0.41 | 1.33 | ||||
| Test 4o | 2bj1–2bj9 (AA) | Cross-test | 132 | 5.16 | 46.46 | ||||
| Test 4p | 2bj1–2bj9 (BB) | Cross-test | 132 | 4.05 | 45.52 | ||||
| Test 4q | 2bj1–2bj9 (AB) | Cross-test | 132 | 3.29 | 45.19 | ||||
| Test 4r | 2bj1–2bj9 (BA) | Cross-test | 132 | 4.33 | 44.08 | ||||
| Test 4s | 2bj3–2bj9 (AB) | Cross-test | 135 | 3.97 | 49.40 | ||||
| Test 4t | 2bj3–2bj9 (BA) | Cross-test | 132 | 4.44 | 52.71 |
Each numbered entry denotes a pair of identical chains in the same unit cell; FM denotes a functional motion entered for comparison purposes; test entries (with the exception of hemoglobin) compare structures from different PDB files.
PDB file names for pairs of chains with chain identifiers shown in parentheses (for compactness, without the dash used in the text); in 1kv3 all chains from A to F have practically identical conformations and therefore all pairs are represented by a single entry.
Number of residues used in pairwise comparison (see §2).
Number of chains in biomolecule according to PQS: m, monomer; d, dimer; c, complex; te, tetramer; h, hexamer.
39 of 75 structural pairs showed differences in RMSDD and Δ that were within coordinate uncertainty thresholds. 19 of these 39 pairs were from cells with two chains each. Their RMSDD ranged from 0.04 to 0.45 Å and Δ ranged between 0.00 and 4.39%. Of four cells with three chains each, three cells had all three pairs of structures identical within the uncertainty threshold. In one cell, 1oxt, one pair of structures differed beyond the uncertainty limits. One cell with four chains, 2gd1, and one cell with six chains, 1kv3, had all pairs of chains with a practically identical RMSDD and Δ within the uncertainty threshold and are listed only once in Table 1 ▶. One cell, 1cdl, with four chains had only one pair of six within the uncertainty threshold and another cell, 13pk, had two pairs within this threshold. Four pairs (66α, 67β, 71, 72) from two cells with four chains in each differed within the uncertainty threshold but were placed among pairs of structures with much larger structural differences for comparison and discussion purposes.
22 of 36 pairs of structures differing by more than the uncertainty limits came from cells with two chains each, three pairs came from cells with three chains each and 12 pairs from cells with four chains each. RMSDD and Δ in this group were in ranges corresponding to smaller functionally significant movements (RMSDD = 0.46–0.48 Å, Δ = 3–7%) up to major functional movements (RMSDD = 1–2.8–7.8 Å, Δ = 24–51%).
Thus, cells with two chains contribute comparable percentages to the set of structural pairs differing only within the coordinate uncertainty threshold (50%) and to the set of pairs differing significantly beyond this threshold (61%). A predominance of P212121 crystal symmetry in our sample agrees with the distributions established in previous large-scale surveys (Chruszcz, Potrzebowski et al., 2008 ▶). The predominance of similar or dissimilar pairs in unit cells with more than two chains is likely to be statistically unreliable in our small set.
In a vast majority of cases, larger differences between pairs of structures from the same unit cell are quantified by the RMSD by the authors of crystallographic papers. However, no factual explanations for the origins of the differences are usually provided.
3.5. Conformational differences between identical chains in the same unit cell: some specific results
3.5.1. Triosephosphate isomerase
The DDM comparing two structures in the unit cell of 3tim (the PDB file shows no ions or substrates and is an ‘open’ apo form) is shown in Fig. 10 ▶(a). The corresponding DDM for 6tim, which has substrate bound to one of two subunits in the unit cell, is shown in Fig. 10 ▶(b). Tests 1a and 1b (in Table 1 ▶) show that subunit A of 6tim has a structure that is identical to the structures of both subunits (A and B) of 3tim within the coordinate uncertainty threshold. The difference between Figs. 10 ▶(a) and 10 ▶(b) mainly arises from the large movement of loop 169–178, reflected in the largest L-shaped white strip in Fig. 10 ▶(b). The black corner of the L-shaped strip means that the Cα—Cα distances within the loop itself mainly remain within 0.5 Å comparing 6tim(A) and 6tim(B). Thus, the loop moves as a rigid body (within the coordinate uncertainty). Original crystallographic papers stated that the loop and its movement are functionally important and the loop moves as a rigid body for up to 7 Å to its ‘closed’ position at the bound substrate or bound sulfate ion. The loop of subunit A makes contacts in the crystal which apparently prevent it from moving to the closed position (Noble et al., 1991 ▶). However, structure 1tsi with another substrate (Verlinde et al., 1992 ▶) shows the substrate binding to both subunits with open loop conformation, with better substrate occupancy observed in the subunit with the loop locked in the open conformation by the crystal contacts. Could this mean that the motion of the loop is not important for the function? We will return to this question in §4.
Figure 10.

Comparisons of the structures of two monomers of triosephosphate isomerase from the same asymmetric unit cell. Notation is the same as in Figs. 2 ▶ and 8 ▶. (a) DDM 3tim(A–B) for a pair of structures 3tim(A) and 3tim(B) from the same unit cell 3tim. (b) DDM 6tim(A–B) for a pair of structures 6tim(A) and 6tim(B) from the 6tim unit cell. (c) Wire representation of the pair of structures 6tim(A) and 6tim(B) superimposed using the rigid fragment 2–129. There are three significant differences between the two structures going from the left to the right of the wire picture: the left-most corresponds to the fragment 130–138 and forms a fading L-shaped band in the DDM in (b), the next and largest difference corresponds to the fragment 170–179 and the brightest L-shaped band in the same DDM, the rightmost difference is the smallest one around residue 211 and corresponds to the narrowest L-shaped band in the DDM in (b).
3.5.2. Calmodulin
Just one of six pairs of structures from the asymmetric unit cell of 1cdl differed only within the coordinate uncertainty limits. The other five pairs showed structurally significant differences beyond the coordinate uncertainty. We have selected the DDMs of two pairs (AD and CD) with the largest conformational differences and compared them with DDM 1cll–1ctr, representing a full functional motion of calmodulin from the apo to the holo form. The main feature of the apo–holo DDM (1cll–1ctr) is a large white rectangle flanked by two more or less solidly black triangles. A black triangle marks a continuous fragment (corresponding to the diagonal of the triangle) with minimal changes (<0.5 Å) in transition from the apo to the holo form between all pairs of Cα atoms within the structure of the fragment. It signifies that the fragment moves as a rigid body (within the coordinate uncertainty) in the transition. The white square shows that all Cα atoms in the two fragments change their pairwise distances by more than 1 Å. The white areas in Figs. 11 ▶(a) and 11 ▶(b) are in the same parts of the DDM where there are white areas in Fig. 11 ▶(c). Thus, the conformational differences between calmodulin monomers in the unit cell might represent a partial movement utilizing the same degrees of freedom as the full functional change reflected in Fig. 11 ▶(c). The original crystallographic paper on 1cdl does not offer any explanation for the intra-cell conformational differences.
Figure 11.

Calmodulin. (a) DDM 1cdl(A–D); (b) DDM 1cdl(C–D); (c) DDM 1cdl–1ctr, representing a full functional motion of calmodulin from the apo to holo form. DDM notation is the same as in Fig. 10 ▶.
3.5.3. Phosphoglycerate kinase (PGK)
Two of six pairs of structures from the asymmetric unit cell of 13pk only differed within the coordinate uncertainty limits. This is a holo form of this protein. Similarly to calmodulin (1cdl) above, we compared two DDMs 13pk(A–B) (Fig. 12 ▶ a) and 13pk(A–D) (Fig. 12 ▶ b) with the DDM 16pk–13pk (Fig. 12 ▶ c) of a functional movement. As in calmodulin, described above, practically all the white spots in DDMs of pairs from the same unit cell appeared within the larger white areas of the function-reflecting DDM. However, the functional DDM, 16pk–13pk (Fig. 12 ▶ c), has a significantly more complex structure than the DDM 1cll–1ctr (Fig. 11 ▶ c) for calmodulin and therefore it might be more difficult to clearly relate motions inside the cell to those involved in the function. We found that the conformational change 16pk→13pk involves over 12 rigid-body motions. The original crystallographic paper on 13pk does not offer any explanation of the intra-cell conformational differences.
Figure 12.

Phosphoglycerate kinase. (a) DDM 13pk(A–B); (b) DDM 13pk(A–D); (c) DDM 16pk–13pk representing a functional motion; DDM notations are as in Fig. 10 ▶.
3.5.4. Adenylate and guanylate kinases
In both cases there are two identical chains in the asymmetric unit cell. Also in both cases the white areas in the DDMs (Figs. 13 ▶ and 14 ▶ a) reflecting intra-cell conformational differences are mainly within the larger white areas of the corresponding functional DDMs (Figs. 9 ▶ and 14 ▶ b). The intra-cell change in 2ak3(A–B) (Fig. 13 ▶) has a smaller area than that in 1ex6(A–B) (Fig. 14 ▶ a). However, 1ex6 is an apo form while 2ak3 has AMP bound and substrate binding in the latter might increase its rigidity compared with the apo form, thus accounting for this difference. Functional movement in adenylate kinase 4ake→1ank (see Fig. 9 ▶) involves over 12 movements of rigid fragments (see §3.3.3). The original paper on 2ak3 only mentions the movement of three domains and we did not find any explanation for the origins of the intra-cell conformational differences. The original paper on 1ex6 states nonspecifically that ‘the differences are partially due to crystal packing’.
Figure 13.

Adenylate kinase. DDM 2ak3(A–B); this should be compared with DDM 4ake–1ank (Fig. 9 ▶) representing a full functional motion. DDM notation is the same as in Fig. 10 ▶.
Figure 14.

Guanylate kinase. (a) DDM 1ex6(A–B); (b) DDM 1ex6–1ex6 representing a full functional motion. DDM notation is the same as in Fig. 10 ▶.
3.5.5. Asp aminotransferase
It was reported in the original crystallographic paper (Rhee et al., 1997 ▶) on 1ajs that
In the presence of 2-methylaspartate, one of the subunits (subunit A) shows a ligand-induced conformational change that involves a large movement to produce a ‘closed’ conformation. No such transition is observed in the other subunit (subunit B), because crystal lattice contacts lock it in an ‘open’ conformation.
DDM 1ajs(A–B) (Fig. 15 ▶ a) clearly shows a large conformational difference between molecules A and B (entry 61 in Table 1 ▶). DDM 1ajr(A–B) shows differences within coordinate uncertainty limits (test 2a in Table 1 ▶) in agreement with PDB file 1ajr, which presents the apo form of the same protein. Structures 1ajs(A) and 1ajs(B) are compared with structures 1ajr(A) and 1ajr(B) (tests 2b–2d in Table 1 ▶). Only structure 1ajs(A) produces DDMs that show large conformational differences with both 1ajr(A) and 1ajr(B). DDM 1ajs(A)–1ajr(B) (Fig. 15 ▶ b) is practically indistinguishable from DDM 1ajs(A–B) (Fig. 15 ▶ a), in agreement with the crystallographic paper. However, to further confirm that DDM 1ajs(A–B) represents a functional motion, one can compare it with DDM 9aat–1ama (Fig. 8 ▶ a), which reportedly represents a functional movement in the same protein from a different species. The two DDMs are very similar, both visually and in their RMSDD and Δ. This confirms that the conformational change in molecule A of the unit cell of 1ajs is a function-induced (or related) change. Note, however, that the holo form 1ama from chicken crystallizes with a different symmetry and does not lock one of the molecules in the asymmetric unit cell in an open conformation. These consequential crystallographic differences were not adequately commented on in the original papers.
Figure 15.

Asp aminotransferase. (a) DDM 1ajs(A–B); (b) cross-test DDM 1ajs(A)–1ajr(B); these should be compared with DDM 9aat–1ama (Fig. 8 ▶ a) representing a full functional motion in another species. DDM notation is the same as in Fig. 10 ▶.
3.5.6. Death-associated kinase (DAPK)
Two monomers in the unit cell of 1jkt (entry 65 in Table 1 ▶) exhibit significant differences beyond the coordinate uncertainty threshold which are uncommented on in the original paper and in a subsequent review (Bialik & Kimchi, 2006 ▶), while even the numbers of α and β fragments in the two monomers differ in the PDB file. For this pair RMSDD is 1.06 Å and Δ is 26.58% and the pair forms only an approximately symmetric dimer. These two apo monomers differ from one another to practically the same extent as each of them differs from the single apo monomer comprising the unit cell 1jks (tests 3a and 3b in Table 1 ▶). Such a large asymmetric distortion of monomers upon dimerization seems rather unusual. Commonly, either both monomers are distorted similarly by a dimerization or one monomer retains the monomeric conformation. It has recently been suggested that EGFR kinase is activated by asymmetric dimerization (Zhang et al., 2006 ▶; Ferguson, 2008 ▶). In DAPK, both dimeric and monomeric forms are reported to be activated. Thus, the role of the asymmetric monomer-distorting dimerization of DAPK remains unclear, in contrast to the case of EGFR.
3.5.7. NikR: a puzzling molecule
The structures of this nickel-responsive repressor (Chivers & Tahirov, 2005 ▶) are the most puzzling in our set. It is a homotetramer in which subunits with identical sequences adopt two drastically different conformations in all reported structural forms. Subunit A differs from subunits B and C, while subunits B and C have the same conformation within the coordinate uncertainty (if we ignore the somewhat different lengths of their disordered termini). Subunit D has the most unlocalized residues, including a few in the middle of the chain, but otherwise it has the same structure as subunit A within the coordinate uncertainty. Thus, the homotetramer exhibits some features resembling those of (αβ)2 hemoglobin, introduced into Table 1 ▶ for comparison (entries 66–67 in Table 1 ▶). Because the A and B subunits differ beyond the coordinate uncertainty in asymmetric unit cells with three different crystal symmetries, their conformational differences can hardly be explained in all cases by crystal-packing effects. However, the authors provided no structural or functional explanation of the A–B structural differences.
There are a few other examples of significant conformational differences between identical chains in homo-oligomers (Gerstein & Echols, 2004 ▶). However, in these examples the asymmetry is explained either by a difference in the bound ions (a change from Ni to Zn) or by a gating function. None of these explanations seemed to be applicable or were offered for NikR.
DDMs 2bj3(A–B) (apo form), 2bj1(A–B) (four Ni bound) and 2bj7(A–B) (four Ni bound plus PG4 and EDO) are shown in Figs. 16 ▶(a)–16 ▶(c). There are significant differences between these DDMs visually, numerically and in their crystal symmetries (Table 1 ▶).
Figure 16.

Nickel-responsive repressor (NikR). (a) DDM 2bj3(A–B) (apo form); (b) DDM 2bj1(A–B) (four Ni bound); (c) DDM 2bj8(A–B) (four Ni bound + PG4 and EDO); (d) DDMs 2bj8(A–B) (six Ni + PG4 and EDO); (e) DDM 2bj9(A–B) (five Ni + PG4 + EDO + PO4). DDM notations are the same as in Fig. 10 ▶.
However, there are no significant visual, numeric or crystal symmetry differences between DDMs 2bj7(A–B), 2bj8(A–B) and 2bj9(A–B) with four or more nickels plus EDO and PG4, regardless of whether phosphates are bound (Figs. 16c–e, Table 1 ▶). Comparisons of pairs of the same subunits from structures 2bj7, 2bj8 and 2bj9 show that they have the same structure within the coordinate uncertainty (tests 4i–4n in Table 1 ▶). Thus, after the binding of four Ni ions (with PG4 and EDO added) the sensitivity to further Ni binding levels off.
The very high degree of similarity of these DDMs and their numeric characteristics in Table 1 ▶ does not seem to support the original claims of high sensitivity of the NikR structure to phosphate binding claimed in the original structural paper, but shown there only at the level of a few changes in the side-chain conformations. The high sensitivity of the NikR structure to phosphate was only exhibited by the dissolution of 2bj8 crystals upon soaking for half an hour in high concentrations of sodium phosphate. Phosphate-containing 2bj9 crystals were obtained by soaking in low-concentration solutions of sodium phosphate with flash-freezing after 10 min of soaking. Thus, 2bj9 might be an artificial metastable form that is possibly unrelated to the in vivo binding mode of the phosphates.
4. Discussion and conclusions
We have shown that the coordinate uncertainty thresholds derived from comparing pairs of independently determined structures of the same protein allow the suggestion of objective limits to the interpretation of main-chain conformational changes in proteins. Because they are derived from RMSDDs of large sets of independently determined structures, which we screened for the absence of major biasing factors, these thresholds present the highest objectivity at this time. Further accumulation, analysis and re-evaluation (Kleywegt, 2009 ▶; Terwilliger et al., 2007 ▶; Levin et al., 2007 ▶; Chen & Brooks, 2007 ▶) of the structural data may lead to their modification.
In particular, it seems possible that uncertainty thresholds might be much lower for high-resolution structures. Most such structures are solved at cryogenic temperatures, which creates its own set of problems, and were intentionally excluded from this study. However, we calculated the uncertainty thresholds for higher resolution subsets of 1014 structural pairs studied here (see supplementary material for details). We found that for ribonuclease A structures with a resolution of 1.6 Å or better (28 pairs of structures) the highest RMSDD was 0.37 Å, which is significantly lower than the maximum RMSDD of 0.44 Å found for all 1014 pairs. However, for the subset with resolutions of 1.7 Å or better (120 pairs) the highest RMSDD rose to 0.41 Å. For the total of 153 pairs of myoglobin structures (at near room temperature) the highest RMSDD was 0.40 Å. The highest RMSDD was in the distribution tail and therefore it can be expected to increase with the size of the set. Thus, we currently have too few statistics for high-resolution structures to resolve the opposing effects of an increase in resolution and of the number of pairs on the value of the uncertainty threshold.
However, we have demonstrated that the use of the thresholds derived here together with DDMs and DD distributions could help to reduce the possibility of the misinterpretation of coordinate differences observed in particular studies. We also have shown that a combined use of uncertainty thresholds, various difference distance matrices and simple transformations opens up possibilities for a more precise and detailed classification and description of protein motions. In particular, this allows an easy distinction between conformational changes of proteins comprised of rigid-body movements of their fragments, also designated as ‘collective’ elsewhere (Yang et al., 2007 ▶), and changes which are dominated by continuous deformations of the polypeptide chain. Such a division of an entire conformational change into sets of rigidly and not rigidly moving fragments is novel and allows better understanding of the mechanics of the molecular machines.
We suggest distinguishing between allowing deformations in the main chain (which arise from hinge-like rotations around single bonds) and motions that are remote from this main-chain deformation. There may be a chain of motions that allow a remote motion. In the motion classification suggested here, we focus on remote motions. It is generally clear that for rigid-body motions the allowing changes in the main-chain ϕ, ψ dihedral angles occur mainly in chain regions between rigid fragments which are usually clearly seen in the DDMs and can be compared with plots of ϕ, ψ differences between two conformations.
Here, we also did not consider the details of shear motions, which have been introduced previously (Lesk & Chothia, 1984 ▶) as one of two major types of conformational motions. In a more detailed paper (Rashin et al., 2009 ▶) we use an approximated degree of shear, obtained from the CDDM, only as one of the characteristics of a remote motion. However, a detailed analysis of shear motions might be warranted.
It is known that in many cases protein subunits only acquire a stable structure upon association or binding of a cofactor or substrate. The same was suggested to be true for some protein domains (Petsko & Ringe, 2004 ▶) whose definition and location remain controversial (Wernisch & Wodak, 2003 ▶; Veretnik et al., 2004 ▶). Our preliminary calculations (Rashin et al., 2009 ▶) suggest that this might often be the case even for motions that are identified as rigid body. It may be that unaccounted-for cofactor binding, intersubunit or crystal contacts might stabilize fragments moving as a rigid unit.
We also find that conformational changes which are often thought to be required or caused by the protein function might be irrelevant to the function and be caused by the crystallization itself. Because we are mostly interested in the protein functions, we need to be able to diagnose reliably specific crystallization effects in protein conformational changes. This might require the analysis of many aspects of crystallization, some of which may be more tractable than others.
Here, we employ a useful ‘coordinate uncertainty’ threshold derived from a rather large set of independently solved X-ray structures with unexplained relatively small differences. This threshold (while possibly just a temporarily useful device) has an additional explanatory advantage. To the best of our knowledge, nobody has claimed to have found a function-triggered conformational change in a protein with a magnitude within this threshold. Alternatively, it has been suggested that using ‘single-conformer structures’ might underestimate uncertainties in protein structures and that multiple structures fitting electron densities should be constructed and considered for a more accurate evaluation of uncertainties (Levin et al., 2007 ▶; Knight et al., 2008 ▶). However, deriving uncertainty thresholds from actual differences in a large number of independently solved ‘single-conformer’ structures of the same protein seems to be at least as valid a procedure.
Outside the uncertainty threshold, we find presumably function-triggered as well as comparably large nonfunctional conformational changes that might be caused, for example, by the crystallization itself. This is a particular case of the old question: can an observation significantly perturb the object of observation? Unfortunately, in protein crystallography the causes of such perturbations and the limits at which they become possible have been insufficiently studied and documented. In particular, relatively rarely definitive perturbations by intruding crystal contacts, a specific intermolecular bond or a specific binding of an ion have been shown to cause a conformational change. More often, plausible but undemonstrated causes have been mentioned. One such suggestion (Andrec et al., 2007 ▶) refers to the possible effect of the crystal field on the choice of molecular conformation, which has been successfully used for small organic molecules (Pertsin & Kitaigorodsky, 1987 ▶).
In principle, crystallization might select a protein conformation that best fits a crystal (Tung & Gallagher, 2009 ▶). According to statistical mechanics, all conformations that might be selected by crystallization should be present in solution. However, some would have very high free energy and therefore would be poorly populated. If crystallization can provide sufficient energy to stabilize its preferred conformation then we will find it in the crystal, regardless of what is preferred in solution or what is really involved in solution biochemistry. Crystallization might start with a conformation in the neighborhood of its preferred conformation and then invest energy to further transform it towards a desirable state. (This may be the entire difference between ‘pre-existing equilibrium’ and ‘induced-fit’; Xu et al., 2008 ▶.) In every particular case the question would be only whether crystallization can afford it. If it cannot then a protein will not crystallize. Limited crystallization-energy resources would allow only limited deviations in the crystal from the conformation preferred in solution. How much energy is required is determined by the stability and rigidity of a particular protein in solution. However, a quantitative evaluation of these characteristics remains a difficult problem (Knight et al., 2008 ▶).
The binding of a substrate analog in an ‘open’ conformation of the enzyme and locking this conformation invokes another often-met problem: how do we know which analogs faithfully imitate the short-lived binding of a real substrate (Chruszcz, Wlodawer et al., 2008 ▶)? At present, good criteria for answering this question seem to be lacking.
Of many more possible questions we will mention only one; however, it is one that often arises. A change of the ions in the mother liquor often leads to a change in the crystal symmetry, in which an alternative protein conformation is often observed. This does not happen for bovine RNase A used here for derivation of the coordinate uncertainty thresholds. However, apo-form thermolysin 1l3f only crystallized with Zn ions (Hausrath & Matthews, 2002 ▶). Did they only change the crystal symmetry or did they directly stabilize the conformation observed in this form, or both? Note that 1l3f has high B factors and high solvent content ‘suggesting some hinge-bending motion within this crystal form.’ Ion regulation is apparently rather common and in our set is most pronounced for NikR. However, it is not clear how much is understood about the mechanisms by which ions change protein conformations, stabilities and crystal symmetries.
An understanding of the workings of molecular machines requires a clearer elucidation of their various motions in order to fully understand the designs, parts and their interconnections and to be able to predict possible movements. Such a deeper understanding of protein motions is also critical to enable the design of new proteins. The methods and approaches presented in this paper should lead to a more objective distinction between rigid-body motions, plastic deformation and their various combinations employed in molecular machines as well as to distinctions between functional and nonfunctional motions. We have only presented a few examples here and more are forthcoming (Rashin et al., 2009 ▶).
Supplementary Material
Supplementary material file. DOI: 10.1107/S090744490903145X/dz5165sup1.pdf
Acknowledgments
This work was supported by NIH grants R01GM072014, 1R01GM073095, R01GM081680 and NSF grant CNS-0521568.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: DZ5165). Services for accessing this material are described at the back of the journal.
References
- Andrec, M., Snyder, D. A., Zhou, Z., Young, J., Montelione, G. T. & Levy, R. M. (2007). Proteins, 69, 449–465. [DOI] [PubMed]
- Bagci, Z., Kloczkowski, A., Jernigan, R. L. & Bahar, I. (2003). Proteins, 53, 56–67. [DOI] [PubMed]
- Bahar, I., Wallqvist, A., Covell, D. G. & Jernigan, R. L. (1998). Biochemistry, 37, 1067–1075. [DOI] [PubMed]
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). Nucleic Acids Res.28, 235–242. [DOI] [PMC free article] [PubMed]
- Bialik, S. & Kimchi, A. (2006). Annu. Rev. Biochem.75, 189–210. [DOI] [PubMed]
- Brown, E. N. & Ramaswamy, S. (2007). Acta Cryst. D63, 941–950. [DOI] [PubMed]
- Challis, J. H. (1995). J. Biomech.28, 733–737. [DOI] [PubMed]
- Chatani, E., Hayashi, R., Moriyama, H. & Ueki, T. (2002). Protein Sci.11, 72–81. [DOI] [PMC free article] [PubMed]
- Chen, J. & Brooks, C. L. III (2007). Proteins, 67, 922–930. [DOI] [PubMed]
- Chivers, T. & Tahirov, T. H. (2005). J. Mol. Biol.348, 597–607. [DOI] [PubMed]
- Chothia, C. & Janin, J. (1978). Proc. FEBS Meet.52, 117–126.
- Chothia, C. & Lesk, A. M. (1985). J. Mol. Biol.182, 151–158. [DOI] [PubMed]
- Chothia, C., Lesk, A. M., Dodson, G. G. & Hodgkin, C. (1983). Nature (London), 302, 500–505. [DOI] [PubMed]
- Chothia, C., Levitt, M. & Richardson, D. (1981). J. Mol. Biol.145, 215–250. [DOI] [PubMed]
- Chruszcz, M., Potrzebowski, W., Zimmerman, M. D., Grabowski, M., Zheng, H., Lasota, P. & Minor, W. (2008). Protein Sci.17, 623–632. [DOI] [PMC free article] [PubMed]
- Chruszcz, M., Wlodawer, A. & Minor, W. (2008). Biophys. J.95, 1–9. [DOI] [PMC free article] [PubMed]
- Coutsias, E. A., Seok, C. & Dill, K. A. (2004). J. Comput. Chem.25, 1849–1857. [DOI] [PubMed]
- Cruickshank, D. W. J. (1999). Acta Cryst. D55, 583–601. [DOI] [PubMed]
- Daopin, S., Davies, D. R., Schlunegger, M. P. & Grütter, M. G. (1994). Acta Cryst. D50, 85–92. [DOI] [PubMed]
- Ferguson, K. M. (2008). Annu. Rev. Biophys.37, 353–373. [DOI] [PMC free article] [PubMed]
- Frauenfelder, H., Hartmann, H., Karplus, M., Kuntz, I. D., Kuriyan, J., Parak, F., Petsko, G. A., Ringe, D., Tilton, R. F., Connolly, M. L. & Max, N. (1987). Biochemistry, 26, 254–261. [DOI] [PubMed]
- Gerstein, M. & Chothia, C. (1991). J. Mol. Biol.220, 133–149. [DOI] [PubMed]
- Gerstein, M. & Echols, N. (2004). Curr. Opin. Chem. Biol.8, 14–19. [DOI] [PubMed]
- Gerstein, M. & Krebs, W. G. (1998). Nucleic Acids Res.26, 4280–4290. [DOI] [PMC free article] [PubMed]
- Gerstein, M., Lesk, A. & Chothia, C. (1994). Biochemistry, 33, 6739–6749. [DOI] [PubMed]
- Harrison, S. C. (1980). Biophys. J.32, 139–151. [DOI] [PMC free article] [PubMed]
- Hausrath, A. C. & Matthews, B. W. (2002). Acta Cryst. D58, 1002–1007. [DOI] [PubMed]
- Horn, B. K. P. (1986). J. Opt. Soc. Am.4, 629–642.
- Howard, E. I., Sanishvili, R., Cachau, R. E., Mitschler, A., Chevrier, B., Barth, P., Lamour, V., Van Zandt, M., Sibley, E., Bon, C., Moras, D., Schneider, T. R., Joachimiak, A. & Podjarny, A. (2004). Proteins, 55, 792–804. [DOI] [PubMed]
- Jaskolski, M., Gilski, M., Dauter, Z. & Wlodawer, A. (2007). Acta Cryst. D63, 611–620. [DOI] [PubMed]
- Kabsch, W. (1976). Acta Cryst. A32, 922–923.
- Kavraki, L. E. (2006). Molecular Distance Measures. http://cnx.org/content/m11608/latest.
- Keller, P. A., Leach, S. P., Luu, T. T. T., Titmuss, S. J. & Griffith, R. (2000). J. Mol. Graph. Model.18, 235–241. [DOI] [PubMed]
- Kishan, R. V. R., Chandra, N. R., Sudarsanakumar, C., Suguna, K. & Vijayan, M. (1995). Acta Cryst. D51, 703–710. [DOI] [PubMed]
- Kleywegt, G. J. (2009). Acta Cryst. D65, 134–139. [DOI] [PMC free article] [PubMed]
- Knight, J. L., Zhou, Z., Gallicchio, E., Himmel, D. M., Friesner, R. A., Arnold, E. & Levy, R. M. (2008). Acta Cryst. D64, 383–396. [DOI] [PMC free article] [PubMed]
- Krebs, W. G. & Gerstein, M. (2000). Nucleic Acids Res.28, 1665–1675. [DOI] [PMC free article] [PubMed]
- Krebs, W. G., Tsai, J., Alexandrov, V., Echols, N., Junker, J., Jansen, R. & Gerstein, M. (2003). Methods Enzymol.374, 544–584. [DOI] [PubMed]
- Kuipers, J. B. (1998). Quaternions and Rotation Sequences. Princeton University Press.
- Kundrot, C. E. & Richards, F. M. (1987). J. Mol. Biol.193, 157–170. [DOI] [PubMed]
- Kurochkina, N. & Privalov, G. (1998). Protein Sci.7, 897–905. [DOI] [PMC free article] [PubMed]
- Lesk, A. M. & Chothia, C. (1984). J. Mol. Biol.174, 175–191. [DOI] [PubMed]
- Lesk, A. M. & Chothia, C. (1988). Nature (London), 335, 188–190. [DOI] [PubMed]
- Levin, E. J., Kondrashov, D. A., Wesenberg, G. E. & Phillips, G. N. Jr (2007). Structure, 15, 1040–1052. [DOI] [PMC free article] [PubMed]
- Maiti, R., Van Domselaar, G. H., Zhang, H. & Wishard, D. S. (2004). Nucleic Acids Res.32, W590–W594. [DOI] [PMC free article] [PubMed]
- McLachlan, A. D. (1979). J. Mol. Biol.128, 49–79. [DOI] [PubMed]
- Moss, D. S., Tickle, I. J. & Laskowski, R. (1998). Estimation of Precision and Accuracy in Protein Structure Refinement from X-ray Data http://people.cryst.bbk.ac.uk/∼ubcg05m/precgrant.html.
- Nishikawa, K., Ooi, T., Isogai, Y. & Saito, N. (1972). J. Phys. Soc. Jpn, 32, 1331–1337.
- Noble, M. E., Wierenga, R. K., Lambeir, A. M., Opperdoes, F. R., Thunnissen, A. M., Kalk, K. H., Groendijk, H. & Hol, W. G. (1991). Proteins, 10, 50–69. [DOI] [PubMed]
- Orengo, C. A., Michie, A. D., Jones, S., Jones, D. T., Swindells, M. B. & Thornton, J. M. (1997). Structure, 5, 1093–1108. [DOI] [PubMed]
- Pertsin, A. J. & Kitaigorodsky, A. I. (1987). The Atom-Atom Potential Method. Application to Organic Molecular Solids. New York: Springer-Verlag.
- Petsko, G. A. & Ringe, D. (2004). Protein Structure and Function. London: New Science Press.
- Price, W. N. et al. (2009). Nature Biotechnol.27, 51–57. [DOI] [PMC free article] [PubMed]
- PSI–Nature Structural Genomics Knowledgebase (2009). http://kb.psi-structuralgenomics.org.
- Rashin, A. A. (1987). J. Mol. Biol.198, 339–349. [DOI] [PubMed]
- Rashin, A. A, Rashin, A. H. L. & Jernigan, R. L. (2009). Submitted.
- Rashin, A. A., Tawa, G., Topol, I. A. & Burt, S. K. (2001). Chem. Phys. Lett.335, 327–333.
- Read, R. J. (2005). Protein Crystallography Course. http://www-structmed.cimr.cam.ac.uk/Course/Statistics/statistics.html.
- Rhee, S., Silva, M. M., Hyde, C. C., Rogers, P. H., Metzler, C. M., Metzler, D. E. & Arnone, A. (1997). J. Biol. Chem.272, 17293–17302. [PubMed]
- Richardson, J. (2007). The Anatomy and Taxonomy of Protein Structure. C. Levels of Error. http://suna.biochem.duke.edu/teaching/anatax/html/anatax.1c.html [DOI] [PubMed]
- Sadasivan, C., Nagendra, H. G. & Vijayan, M. (1998). Acta Cryst. D54, 1343–1352. [DOI] [PubMed]
- Schneider, T. R. (2000). Acta Cryst. D56, 714–721. [DOI] [PubMed]
- Schneider, T. R. (2004). Acta Cryst. D60, 2269–2275. [DOI] [PubMed]
- Sinha, N. & Nussinov, R. (2001). Proc. Natl Acad. Sci. USA, 98, 3139–3144. [DOI] [PMC free article] [PubMed]
- Snyder, D. A., Bhattacharia, A., Huang, Y. J. & Montelione, G. T. (2005). Proteins, 59, 655–661. [DOI] [PubMed]
- Terwilliger, T. C., Grosse-Kunstleve, R. W., Afonine, P. V., Adams, P. D., Moriarty, N. W., Zwart, P., Read, R. J., Turk, D. & Hung, L.-W. (2007). Acta Cryst. D63, 597–610. [DOI] [PMC free article] [PubMed]
- Tilton, R. F. Jr, Dewan, J. C. & Petsko, G. A. (1992). Biochemistry, 31, 2469–2481. [DOI] [PubMed]
- Tung, M. & Gallagher, D. T. (2009). Acta Cryst. D65, 18–23. [DOI] [PubMed]
- Uversky, V. N., Oldfield, C. J. & Dunker, A. K. (2008). Annu. Rev. Biophys.37, 215–246. [DOI] [PubMed]
- Verlinde, C. L, Witmans, C. J., Pijning, T., Kalk, K. H., Hol, W. G., Callens, M. & Opperdoes, F. R. (1992). Protein Sci.1, 1578–1584. [DOI] [PMC free article] [PubMed]
- Veretnik, S., Bourne, P. E., Alexandrov, N. N. & Shindyalov, I. N. (2004). J. Mol. Biol.339, 647–678. [DOI] [PubMed]
- Wang, J., Dauter, M., Alkire, R., Joachimiak, A. & Dauter, Z. (2007). Acta Cryst. D63, 1254–1268. [DOI] [PubMed]
- Wernisch, L. & Wodak, S. J. (2003). Structural Bioinformatics, edited by P. E. Bourne, H. Weissig & M. A. Ariano, pp. 365–385. New York: Wiley–Liss.
- Wlodawer, A., Minor, W., Dauter, Z. & Jaskolski, M. (2008). FEBS J.275, 1–21. [DOI] [PMC free article] [PubMed]
- Xu, Y., Colletier, J. P. H., Jiang, H., Silman, I., Sussman, J. L. & Weik, M. (2008). Protein Sci.17, 601–605. [DOI] [PMC free article] [PubMed]
- Yang, L., Song, G. & Jernigan, R. L. (2007). Biophys. J.93, 920–929. [DOI] [PMC free article] [PubMed]
- Zhang, X., Gureasko, J., Shen, K., Cole, P. A. & Kuriyan, J. (2006). Cell, 125, 1137–1149. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material file. DOI: 10.1107/S090744490903145X/dz5165sup1.pdf
Supplementary material file. DOI: 10.1107/S090744490903145X/dz5165sup1.pdf