

Abstract
Background
The origin of novel traits and their subsequent diversification represent central themes in evo-devo and evolutionary ecology. Here we explore the genetic and genomic basis of a class of traits that is both novel and highly diverse, in a group of organisms that is ecologically complex and experimentally tractable: horned beetles.
Results
We developed two high quality, normalized cDNA libraries for larval and pupal Onthophagus taurus and sequenced 3,488 ESTs that assembled into 451 contigs and 2,330 singletons. We present the annotation and a comparative analysis of the conservation of the sequences. Microarrays developed from the combined libraries were then used to contrast the transcriptome of developing primordia of head horns, prothoracic horns, and legs. Our experiments identify a first comprehensive list of candidate genes for the evolution and diversification of beetle horns. We find that developing horns and legs show many similarities as well as important differences in their transcription profiles, suggesting that the origin of horns was mediated partly, but not entirely, by the recruitment of genes involved in the formation of more traditional appendages such as legs. Furthermore, we find that horns developing from the head and prothorax differ in their transcription profiles to a degree that suggests that head and prothoracic horns are not serial homologs, but instead may have evolved independently from each other.
Conclusion
We have laid the foundation for a systematic analysis of the genetic basis of horned beetle development and diversification with the potential to contribute significantly to several major frontiers in evolutionary developmental biology.
Background
The origin of novel traits and their subsequent diversification have been central themes in evolutionary biology ever since the discipline's inception over 150 years ago [1,2]. Specifically, the genetic, developmental, and ecological mechanisms, and the interactions between them, that allow novel phenotypes and functions to arise from pre-existing variation, continue to represent major frontiers in our understanding of phenotypic diversity. With the advent of modern -omics approaches, researchers have increasingly departed from a candidate gene or pathway approach and begun to explore organismal development and evolution from a genome, transcriptome, or proteome perspective, focusing in large part on existing genetic model systems such as Drosophila or Caenorhabditis. However, many key questions in evolutionary biology, including the mechanisms underlying organismal innovation, the role of plasticity in diversification, and the interplay between ecology and developmental evolution, are often difficult to address solely within the confines of classic model systems. Recent efforts have therefore begun to generate genomic and developmental genetic resources for organisms with promise as future model systems in evolutionary developmental biology and ecological genetics (e.g. butterflies: [3,4]; honey bees: reviewed in [5]; red flour beetle: [6]). Here we present and apply the first genomic resources to advance the study of a class of traits that is both novel and highly diverse in a group of organisms that is ecologically complex and experimentally tractable: beetle horns and horned beetles.
Beetle horns possess many characteristics that make them interesting models for integrating genetic, developmental, and environmental perspectives on the development and evolution of complex, novel traits (reviewed in [7]). First, beetle horns are major structures, often dominating the phenotype of their bearers. Second, beetle horns function as weapons of sexual selection, thus playing a major role in the behavioral ecology of individuals and populations. Third, beetle horns are inordinately variable, both within and between species, including differences in number, size, shape, and location. Moreover, diversity in horn expression is paralleled by amazing species richness. For instance, the genus Onthophagus currently contains over 2,400 extant species, making it the most speciose genus in the animal kingdom [8,9]. Forth, beetle horns are influenced in their expression by both genetic and environmental factors, ranging from absence of environmental sensitivity to complete determination by nutritional condition. In some cases, both extremes of environmental sensitivity can be found in different horn types expressed by the same individual [10]. Finally, beetle horns lack any obvious homology to structures in other insects or non-insect arthropods. Beetle horns are not modified antennae or mouthparts, but instead horns were "invented" by beetles in addition to their traditional appendages [11], and now provide their bearers with an important new function: a weapon used in male-male competition. Beetle horns and horned beetles therefore offer rich opportunities to explore the mechanisms of organismal innovation and diversification.
Beetle horns are rigid outgrowths of the exoskeleton that originate as epidermal outbuddings of the head or prothoracic epithelium. Horns lack joints, muscles, and nervous tissue. Several recent studies have begun to shed light on how beetle horns develop and differentiate during ontogeny [11-15] and showed that two developmentally dissociated processes contribute to the final degree of horn expression seen in adults: a prepupal growth phase late in larval development followed by a pupal remodeling phase just prior to the final adult molt (reviewed in [10]). As such, the development of horns shows many qualitative similarities to the development of traditional appendages, but also exhibits important differences. For instance, prothoracic horn primordia are frequently resorbed during the pupal stage in a sex- and species-specific manner, a phenomenon not usually associated with regular appendages [14]. Furthermore, earlier studies have also begun to question whether horns that develop in different body regions, such as the head vs. prothorax, constitute serial homologs, or instead may have evolved and diversified independently of each other [13,16].
Here we present the first steps toward a systematic analysis of the genetic and genomic basis of horn development and diversification in the genus Onthophagus. We first present the results of a comprehensive EST analysis of two normalized cDNA libraries obtained from two disparate developmental stages of Onthophagus taurus: larva and pupa. Second, using microarrays developed from our EST libraries we contrast the transcription profiles of the primordia of developing prothoracic horns, head horns, and legs right after the transition from larva to pupa. We then use these contrasts to identify candidate genes involved in the development and diversification of beetle horns. Furthermore, we examine two basic questions regarding the origin and diversification of horns. (a) Are horns highly simplified versions of more traditional appendages such as legs? If so, transcription profiles of developing horn primordia should largely match those of developing legs. If not, transcription profiles of developing horn primordia should only partly match those of developing legs and also include horn specific transcription signatures. (b) Are different horn types produced in different body regions homonomous, i.e. serial homologs of the same ancestral structure? If so, different horn types should exhibit highly similar transcription profiles. However, if different horn types originated and diversified independently of each other, transcription profiles may be predicted to exhibit important horn-type specific differences. We discuss the significance of our findings in the context of the biology of horned beetles in particular, and the origins and diversification of novel traits in general.
Results
Production and analysis of EST sequences
We constructed two normalized, size selected, and directionally cloned cDNA libraries from (i) heads and thoraces dissected from larvae and prepupae (referred to as OtL [larval]) and (ii) whole pupae (referred to as OtP [pupal]). Individual 5'- sequencing reads were generated from 3,756 randomly selected cDNA clones (1,874 larval and 1,882 pupal). The set of EST sequences were trimmed of vector sequence, adaptor sequence, and poly(A) tails, and filtered to remove sequences that were either low quality, chimeric, or shorter than 100 nucleotides (Methods). This yielded 3,488 high-quality sequences (1,783 larval and 1,705 pupal, Table 1) that are available at GenBank (accession numbers FG539013-FG542500). We then used ESTPiper [17] to assemble these sequences into contigs (see Methods). A total of 1,158 of these sequences were assembled into 451 contigs with an average of 2.6 clones per contig and a maximum of 11 clones per contig. The remaining 2,330 sequences did not assemble into contigs and are referred to as singletons. Thus, the 3,488 sequences collapse into 2,781 distinct sequences (451 contigs and 2,330 singletons) that we refer to as "non-redundant" sequences.
Table 1.
Summary of cDNA libraries and EST sequence analysis
|
Larvae (OtL) |
Pupae (OtP) |
Mixed (OtL & OtP) |
|
| median cDNA fragment size (nt) | 715 | 975 | - |
| average read length (nt) | 624 | 667 | 645 |
| raw reads | 1,874 | 1,882 | 3,756 |
| cleaned reads | 1,783 | 1,705 | 3,488 |
| assembled contigs | 217 | 171 | 451 |
| singletons | 1,252 | 1,284 | 2,330 |
| independent sequences | 1,469 | 1,455 | 2,781 |
| annotated | 1,104 | 998 | 1,984 |
It is likely that some of the non-redundant sequences derive from the same transcript but do not overlap, possibly due to 5'-truncated cDNA clones. In order to estimate the magnitude of this redundancy, we aligned the Onthophagus non-redundant sequences to Drosophila proteins, filtered the alignments for highly similar matches (BLASTx, E-value < 10-60), and then manually examined the alignments for separate Onthophagus sequences that align to distinct regions of the same Drosophila protein. Among 534 non-redundant Onthophagus sequences we found 35 pairs of sequences that aligned to the same Drosophila protein. Of these, 19 pairs aligned with highly similar matches to different regions of the same protein, indicating they derive from non-overlapping regions of the same transcript; 12 pairs had co-linear alignments with 95-98% sequence identities, suggesting that they either derive from the same gene with polymorphisms and/or sequencing errors, or derive from highly similar duplicate genes; and 4 pairs appear to be splice variants. Thus, this sample of 534 Onthophagus non-redundant sequences represents approximately 499 distinct genes (93% unique). While this is not a random sample and thus can't be extrapolated to full set of non-redundant sequences, it does however indicate that false-negative assemblies are not a pervasive problem among the non-redundant sequences.
Functional annotation of assembled sequences
Given that insects express a broad diversity of genes during metamorphosis [18], we expected that the larval and pupal ESTs would be a rich source of gene discovery. In order to provide a first pass annotation for the putative function of the Onthophagus gene sequences, we annotated the non-redundant sequences using the UniProtKB/TrEMBL protein sequence database (E-value < 10-5). This successfully annotated 71.3% of the non-redundant sequences. As expected, these annotations covered a wide diversity of biological and molecular functions including the major expected categories such as cellular processes, metabolic processes, biological regulation, multicellular organismal processes, and developmental processes (see Additional files 1 and 2). This, coupled with the low redundancy within the Onthophagus libraries, indicates the set of ESTs as a rich source for gene discovery.
Given that the cDNA libraries derive from animals undergoing metamorphosis, which involves dramatic remodeling of the insect body accompanied by complex patterns of gene expression, it was expected that the EST libraries would include genes involved in a wide range of developmental processes. Indeed the assembled sequences included over 75 genes with close sequence similarity to genes with important functions in the development of other arthropods (Table 2). Briefly, these included the following major groups: 1) genes involved in axis-specification, patterning and morphogenesis, including many transcription factors (homothorax, extradenticle, spalt-related, bicaudal, prothoraxless, teashirt-like, Sex comb on midleg, Cephalothorax, Ultrabithorax, cut, tailup, pointed, Abdominal B, hairy, bab2, Additional sex combs); 2) proteins involved in several signaling pathways, including MAPK pathways (Epidermal growth factor-like protein, Star, MAP kinase-interacting serine/threonine kinase, licorne, puckered, DRas2, misshapen, discs large 1) the Wnt receptor signaling pathway (frizzled 4, shaggy, armadillo, hyrax, Wnt oncogene anolog 2), the Notch signaling pathway (Notch, fringe, dishevelled, kuzbanian, Enhancer of split, strawberry notch), the Hedgehog signaling pathway (hedgehog), the TGF-beta related pathway (bambi, cornichon), and the Toll signaling pathway (Spatzle-Processing Enzyme, pipe); 3) genes involved in endocrine regulation of development including ecdysone signaling (ultraspiracle, Ecdysone receptor, shade, disembodied, broad, Ecdysone-induced protein 78C, Ecdysone-induced protein 75B) and juvenile hormone signaling (Juvenile hormone epoxide hydrolase 3, Juvenile hormone acid methyl transferase). These clones represent a rich set of annotated genes for future studies investigating the function of the respective pathways in Onthophagus development and evolution.
Table 2.
Putative Onthophagus taurus orthologs with known functions in insect development and physiology
| Ot library ID | UniProtKB/FlyBase ID | Gene description | Identity (%) | E-value |
| contig201 | FBpp0089324 | shade | 47.8 | 2.00E-44 |
| contig473 | FBpp0077925 | fringe | 45.4 | 9.00E-60 |
| contig541 | FBpp0088364 | Autophagy-specific gene 12 | 53.2 | 7.00E-21 |
| contig1071 | FBpp0089159 | shaggy | 83.9 | 2.00E-49 |
| contig1223 | Q70WC9_TRICA | Homothorax | 93.3 | 6.00E-10 |
| contig1461 | FBpp0076186 | SHC-adaptor protein | 48.4 | 4.00E-55 |
| contig1583 | FBpp0088946 | transformer2 | 51.9 | 5.00E-22 |
| contig1873 | FBpp0075238 | PDCD-5 | 57.8 | 1.00E-37 |
| contig2291 | Q17P53_AEDAE | Sptzle 1B (Spz1B) | 28.5 | 2.00E-07 |
| contig2542 | Q70WD0_TRICA | Extradenticle | 95.7 | 4.00E-91 |
| contig2552 | FBpp0081258 | doublesex | 81.8 | 5.00E-23 |
| contig2972 | Q1HTM7_9MYRI | Daughterless (Fragment) | 32.9 | 2.00E-16 |
| contig3052 | FBpp0077659 | Star | 34.6 | 3.00E-47 |
| contig3563 | FBpp0089196 | chickadee | 81.7 | 4.00E-58 |
| contig3732 | FBpp0099954 | held out wings | 88.7 | 2.00E-78 |
| contig4043 | Q0IFK2_AEDAE | Map kinase-interacting serine/threonine kinase | 87.3 | 8.00E-48 |
| contig4483 | Q176R2_AEDAE | PIWI | 35.2 | 1.00E-57 |
| OtL001A07 | Q6RG14_AEDAE | Broad complex isoform Z1 | 95.1 | 4.00E-62 |
| OtL001C11 | Q178N5_AEDAE | Programmed cell death | 59.6 | 8.00E-10 |
| OtL003H10 | Q68QF3_LITFO | Notch (Fragment) | 61.8 | 5.00E-20 |
| OtL004F07 | FBpp0075677 | Autophagy-specific gene 1 | 70.6 | 5.00E-09 |
| OtL004G01 | FBpp0089035 | armadillo | 87.7 | 1.00E-124 |
| OtL005C09 | FBpp0079823 | spalt-related | 54.3 | 3.00E-14 |
| OtL005D10 | Q206L4_AEDAE | Juvenile hormone acid methyl transferase | 28.5 | 2.00E-13 |
| OtL006H06 | Q1HAY7_HOLDI | Epidermal growth factor-like protein | 47.0 | 1.00E-64 |
| OtL007E09 | FBpp0073551 | licorne | 70.7 | 4.00E-76 |
| OtL008H03 | FBpp0070977 | frizzled 4 | 50.3 | 7.00E-50 |
| OtL011A08 | HYEP1_CTEFE | Juvenile hormone epoxide hydrolase 1 (EC 3329) | 48.5 | 2.00E-39 |
| OtL011C06 | CNI_DROME | Protein cornichon | 75.9 | 3.00E-50 |
| OtL011G12 | IRS1_RAT | Insulin receptor substrate 1 (IRS-1) (pp185) | 32.1 | 9.00E-17 |
| OtL012D01 | FBpp0081288 | puckered | 39.7 | 1.00E-28 |
| OtL012F02 | Q1HEQ6_TRICA | Hedgehog (Fragment) | 43.7 | 7.00E-40 |
| OtL015G10 | FBpp0074770 | pipe | 56.7 | 4.00E-48 |
| OtL017B11 | Q3LFR2_BOMMO | Ecdysone 20-hydroxylase (EC 1149922) | 47.2 | 3.00E-42 |
| OtL017F12 | FBpp0081448 | hyrax | 67.4 | 1.00E-81 |
| OtL017H05 | FBpp0086896 | bicaudal | 69.3 | 7.00E-46 |
| OtL017H12 | FBpp0087596 | Wnt oncogene analog 2 | 51.6 | 5.00E-52 |
| OtL019E08 | Q16PS8_AEDAE | Mago nashi, putative | 95.2 | 7.00E-78 |
| OtL020B06 | Q17DN9_AEDAE | Enhancer of split protein, putative | 71.5 | 4.00E-43 |
| OtL020E02 | FBpp0071427 | Autophagy-specific gene 8a | 93.2 | 8.00E-60 |
| OtL020F12 | FBpp0089344 | forkhead box, sub-group O | 63.7 | 5.00E-33 |
| OtP001A11 | FBpp0089363 | bancal | 57.7 | 1.00E-15 |
| OtP001B03 | FBpp0073061 | disembodied | 53.6 | 3.00E-28 |
| OtP001C01 | FBpp0099532 | enabled | 79.1 | 3.00E-47 |
| OtP001F04 | FBpp0083832 | Spatzle-Processing Enzyme | 42.9 | 4.00E-29 |
| OtP003A12 | Q2F5M0_BOMMO | Ras-related protein 2 | 87.1 | 4.00E-75 |
| OtP004B11 | Q8MYD0_APICA | Creb protein (Fragment) | 75.8 | 5.00E-27 |
| OtP005F11 | FBpp0082472 | bitesize | 46.4 | 2.00E-31 |
| OtP006B04 | Q95UR2_TRICA | Homeodomain transcription factor Prothoraxless | 82.4 | 9.00E-54 |
| OtP006B10 | FBpp0080555 | Bicaudal D | 61.9 | 1.00E-67 |
| OtP007F08 | Q967X9_TRICA | Teashirt-like protein | 80.3 | 5.00E-92 |
| OtP008D04 | FBpp0081580 | Sex comb on midleg | 61.8 | 8.00E-61 |
| OtP008D05 | Q95UA8_TRICA | Cephalothorax | 77.6 | 4.00E-51 |
| OtP009C08 | Q1RP84_BLAGE | Ecdysone inducible protein 75 isoform B | 58.2 | 2.00E-61 |
| OtP009H01 | Q172A2_AEDAE | Staufen | 50.2 | 1.00E-43 |
| OtP010C08 | FBpp0074738 | absent, small, or homeotic discs 1 | 47.6 | 4.00E-45 |
| OtP011F05 | Q8T939_TRICA | Ultrabithorax | 74.1 | 1.00E-32 |
| OtP011F12 | FBpp0082957 | Autophagy-specific gene 8b | 36.4 | 7.00E-16 |
| OtP012A03 | Q176U2_AEDAE | Insulin receptor tyrosine kinase substrate | 58.2 | 6.00E-29 |
| OtP012E04 | FBpp0071026 | cut | 93.0 | 1.00E-40 |
| OtP012H05 | FBpp0080662 | tailup | 63.3 | 6.00E-25 |
| OtP012H08 | PNT2_DROME | ETS-like protein pointed, isoform P2 (D-ETS-2) | 46.0 | 4.00E-39 |
| OtP013E09 | FBpp0073311 | dishevelled | 39.0 | 6.00E-08 |
| OtP013G10 | FBpp0089312 | misshapen | 89.5 | 1.00E-126 |
| OtP014A10 | Q1KY82_9MYRI | Abdominal-B | 96.3 | 3.00E-07 |
| OtP014B07 | FBpp0079676 | basket | 81.8 | 6.00E-09 |
| OtP014B10 | DLG1_DROME | Discs large 1 tumor suppressor protein | 52.1 | 9.00E-19 |
| OtP015B03 | FBpp0074588 | gigas | 33.2 | 5.00E-36 |
| OtP015D05 | Q9U7D9_LOCMI | RXR | 71.8 | 2.00E-50 |
| OtP015H03 | FBpp0099504 | hairy | 56.8 | 6.00E-36 |
| OtP015H08 | Q17HJ1_AEDAE | Kuzbanian | 89.0 | 1.00E-119 |
| OtL016D12 | Q6B0K6_9CUCU | LIM protein | 88.8 | 4.00E-36 |
| OtP016E12 | FBpp0088965 | cheerio | 78.7 | 1.00E-108 |
| OtP017C12 | FBpp0089115 | groucho | 68.8 | 4.00E-47 |
| OtP017F02 | FBpp0072535 | bab2 | 56.3 | 7.00E-26 |
| OtP017F03 | FBpp0111762 | strawberry notch | 86.3 | 6.00E-34 |
| OtP018C02 | FBpp0086622 | Additional sex combs | 70.0 | 8.00E-08 |
| OtP018E03 | Q17J62_AEDAE | Ras | 83.7 | 9.00E-78 |
| OtP019D06 | O02035_TENMO | Ecdysone receptor | 72.3 | 3.00E-40 |
| OtP019F10 | ILPR_BRALA | Insulin-like peptide receptor precursor | 28.6 | 2.00E-07 |
| OtP019G07 | Q6F2E0_XENTR | Bambi (BMP and activin membrane-bound inhibitor) | 36.3 | 1.00E-09 |
| OtP020A12 | FBpp0077963 | Ecdysone-induced protein 78C | 67.6 | 2.00E-10 |
1 = contigs consisting of larval ESTs only; 2 = contigs consisting of pupal ESTs only; and 3 = contigs consisting of larval and pupal ESTs.
Comparative analysis of the Onthophagus transcriptome
While the beetle order is incredibly species rich and diverse, this study represents only the second systematic study of beetle genes [6]. Comparative analyses of gene content in Tribolium castaneum revealed that the proportion of universal and insect specific genes is similar to that in other insects [6]. However, the proportion of genes without similarity to other organisms is higher in red flour beetles than in other insects [6]. In order to explore the conservation of Onthophagus genes across metazoans, we aligned the 2,781 non-redundant Onthophagus sequences to the protein sequences derived from the annotated genomes of Tribolium castaneum (NCBI GenBank), Drosophila melanogaster (FlyBase), Caenorhabditis elegans (Ensembl), human (Ensembl), as well as non-redundant protein dataset (nr) from GenBank (Table 3). We also aligned our translated sequences with combined "invertebrate protein datasets" from NCBI ftp://ftp.ncbi.nih.gov/refseq/release/invertebrate.
Table 3.
Datasets used in this study.
| Genome | Database | Version (date uploaded, YYMMDD) |
| Drosophila melanogaster | FlyBase | 5.2 (070725) |
| Tribolium castaneum | NCBI | (060410) |
| Homo sapiens | Ensembl | NCBI 36 release 46 (070803) |
| Caenorhabditis elegans | Ensembl | Wormpep 180 (070819) |
| Invertebrate | NCBI | (080508) |
| non-redundant | NCBI | (080514) |
In order to group the Onthophagus sequences according to patterns of conservation and divergence across these datasets we filtered them for those with BLASTx sequence matches with proteins in the various datasets (E-value < 1 × 10-5), and then clustered them according to the bit scores (Figure 1 and Additional file 3). A total of 1,086 non-redundant Onthophagus sequences (39%) had sequence matches to proteins in all the datasets searched (Figure 1, Group 1). A further 868 of the non-redundant sequences (31%) had matches to proteins in the Tribolium dataset, as well as to proteins in one or more of the other datasets (Figure 1, Group 2). Within group 2 there are two prominent sub-groups. First, there were 300 non-redundant (10.8%) with matches to fly, "invertebrate", and nr proteins, but no matches to worms and humans, and are thus putatively restricted to insects. We manually inspected the matched sequences and confirmed that there are no protein matches from non-insect species. While Gene Ontology annotations of these sequences show no striking enrichment for specific biological processes, they do include proteins with functions specific to insects, such as cuticle proteins (data not shown). Second, there were 212 (7.6%) non-redundant sequences with matches to proteins in either only Tribolium, or Tribolium and either/both of "invertebrate" and nr proteins. In order to test if these sequences are indeed restricted to beetles we filtered them to meet the following criteria: (i) the Onthophagus sequences did not have a sequence match (BLASTx, E-value < 10-20) with proteins from non-beetle species in the nr dataset; (ii) the Tribolium protein to which the Onthophagus sequence had the best alignment did not have a sequence match (BLASTx, E-value < 10-20) with proteins from non-beetle species in the nr dataset. In total, 44 of the non-redundant sequences (1.6%) met these criteria and thus are restricted to beetle species among the currently available protein sequence information. This suggests that these genes may have arisen de novo, or may be fast evolving, in beetles. A total of 194 translated non-redundant sequences (7%) did not match proteins from Tribolium, but did have matches in one or more of flies, worms, humans, invertebrates or nr (Figure 1, group 3). Among these, 43 sequences had matches in all other protein datasets, and 33 additional sequences had matches to Drosophila, "invertebrate", and nr. Combined, these data raise the possibility that at least some of these 76 sequences may either have been lost from the Tribolium lineage, or alternatively, may be conserved but simply not yet annotated in Tribolium. Indeed, we found 22 cases where the Onthophagus sequences matched against regions of the Tribolium genome (BLASTn E-value < 1 × 10-5, data not shown) with no gene annotations. These may represent genes that are unannotated in the Tribolium genome. Thus, the Onthophagus expressed sequences reported here would be useful in refining the annotation of the Tribolium genome.
Figure 1.
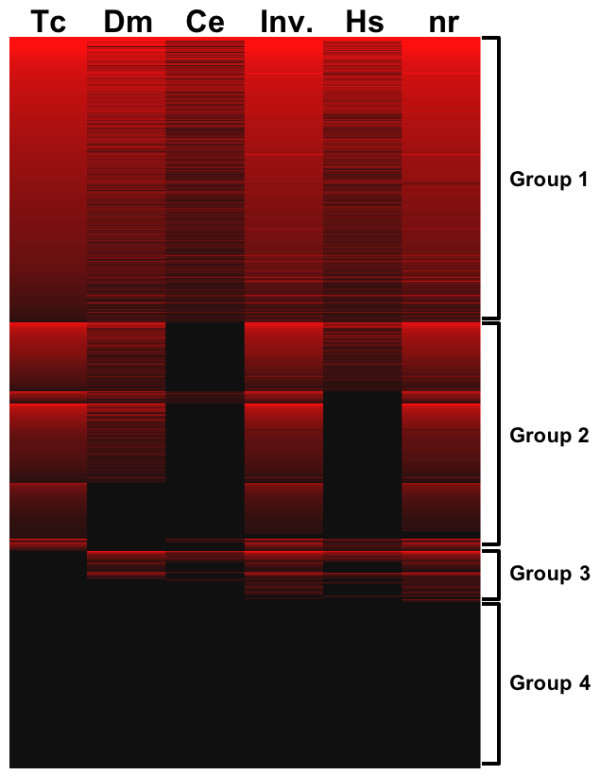
Overall comparison of O. taurus sequences with other protein datasets. Filtering and clustering analysis of assembled O. taurus ESTs based on BLASTx. Shown are bit scores against protein sequences from Tribolium castaneum (Tc, NCBI), Drosophila melanogaster (Dm, FlyBase), Caenorhabditis elegans (Ce, Sanger), invertebrate proteins (inv., NCBI), Homo sapiens (Hs, Ensembl), and non-redundant protein dataset (nr, NCBI). Each row represents a single Onthophagus sequence, and each column represents sequence matches to proteins from the indicated datasets, where the color intensity is proportional to the bit score (0 = black to 789 = brightest red). The Onthophagus sequences are grouped (Groups 1-4) according to the patterns of BLASTx sequence matches with proteins in the various datasets (E-value cut-off = 1 × 10-5), and clustered according to the bit scores. There are 1,086, 868, 194, and 633 sequences in groups 1-4, respectively. The complete dataset for this figure is available as Additional file 3.
Our analysis also identified 633 of the non-redundant sequences (23.2%) to have "no-hit" (Figure 1 Group 4) to any of the proteomes. This is consistent with the finding that approximately 23% of genes annotated in the Tribolium genome lack sequence matches in a wide range of other species [6]. However, our estimate of Onthophagus specific sequences is likely to be inflated by (i) sequences that are largely, or entirely, within the UTRs of protein coding transcripts, or (ii) sequences that may be non-coding transcripts. Resolving the question of whether these sequences do in fact represent genes that are unique to Onthophagus must await large-scale sequencing of the transcriptomes and/or genomes of Onthophagus and related species. However, the observation that 44% of theses sequences include ORFs of greater than 300 nucleotides (data not shown), suggests that at least some of these represent protein-coding genes that have not yet been identified in the species sequenced to date.
Gene expression profiles in pupal appendage primordia
While our EST analysis identified many genes homologous to interesting Drosophila developmental genes, and such an approach to identify candidate genes has been successful in beetles [11,15,19,20], this approach is limited to identifying obvious candidates. Given that Onthophagus horns appear to be novel structures invented in beetles, it is highly likely that unexpected, or indeed previously uncharacterized genes may be important in their development. We therefore developed a custom microarray spotted with the 3,756 cDNA clones from which the ESTs were derived (Methods), undertook gene expression profiling of developing horns (early pupal stage) as an unbiased means of identifying such candidates. Since there is evidence that head horns and prothoracic horn are quite distinct structures (not simply serial homologs; [13,16]), we analyzed gene expression in each of these organs separately. Since there is evidence that some, but not all, appendage patterning genes play a role in horn development [15], we included legs in our analysis in order to distinguish similarities and differences between horns and a canonical appendage. Finally, since beetle horns and legs both develop by out-budding of the epithelium, we use non-appendage bearing epithelium (dorsal abdomen) as a common reference sample.
The design of the expression profiling experiments included three comparisons, each done with five independent biological replicates (Figure 2A). The complete microarray data are available at GEO http://www.ncbi.nlm.nih.gov/geo/, accession number GPL7555). The percentage of array elements that detected signal (where feature intensity > average + 2 SD background intensity) was uniformly high across the four tissues interrogated: an average of 78.5% in head horns, 82.7% in prothoracic horns, 84.3% in legs, and 83.7% in abdominal epithelium. Signal intensities were also reproducible across both technical replicates hybridized on the same microarray (average correlation coefficient = 0.94, n = 2, SD = 0.04), and independent biological replicates hybridized on different microarrays (average correlation coefficient = 0.901, n = 60, SD = 0.062). These data indicate the microarrays were sensitive and the experiments were reproducible. A total of 1,542 of the 3,756 cDNA array elements detected statistically significant differential expression (adjusted p-value < 0.05) in one or more of the three comparisons - head horns, prothoracic horns and legs all compared to abdomen. In order to examine the overall similarities and differences in the patterns of these differentially expressed genes we used two-dimensional hierarchical clustering (175 array elements with some missing data-points were excluded from the clustering). This revealed that expression patterns of head horns, prothoracic horns, and legs are remarkably similar (Figure 2B). Similar results were obtained when the data were collapsed into non-redundant sequences (data not shown). 83% of the array elements (1,135 out of 1,367) detected enriched or depleted expression in all three tissues compared to abdominal epithelium. Despite the high degree of similarity, the expression patterns in head horns and prothoracic horns are still significantly closer to each other than they are to those in legs (reflected in the branch lengths in the sample tree in Figure 2B). 11% of the array elements (150 out of 1,367) detected enriched or depleted expression in both head and prothoracic horns and not legs. Thus, in terms of overall patterns of gene expression, head and prothoracic pupal horn primordia are similar to but distinct from pupal leg primordia.
Figure 2.
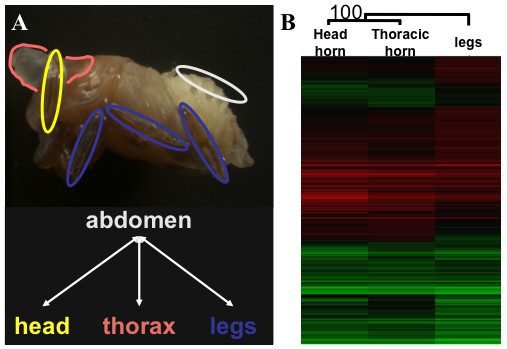
Experimental design and clustering analysis of the gene expression pattern in O. taurus day 1 pupa. A. Microarray experimental design. The pupal tissues used are indicated in the upper panel and the microarray hybridizations are illustrated in the lower panel. Head horn (head) is labeled yellow, thoracic horn (thorax) is labeled pink, legs are labeled blue, and abdominal epithelium (abdomen) is labeled white. B. Hierarchical clustering of differentially expressed genes. 1,367 spots were clustered based on their M-values when compared to abdominal epithelium. Each row represents a single spot and each column represents the sample. Relative magnitude of gene expression level is indicated by color brightness; red indicates enriched compared to abdominal epithelium whereas green indicates depleted relative to abdominal epithelium. M-values ranged from -4.85 to 4.12. Bootstrap values were obtained after 5000 trials. Branch lengths represent relative distances between the samples.
Identifying candidate genes based on expression in horn primordia
We expected that at least some genes involved in horn development would show differential expression in pupal horn primordia. In order to focus on these genes, we collapsed the data from array elements into non-redundant sequences (contigs and singletons), and then filtered the non-redundant sequences for those that were both statistically differentially expressed and showed at least two-fold changes in either head horns, prothoracic horn or both types of horns. A total of 306 non-redundant sequences met these criteria (adjusted p-value < 0.05 and fold-change > 2); 73 in head horns only, 38 in prothoracic horns only, and 195 in both head and prothoracic horns (Figure 3). These 306 non-redundant sequences included 74 with no BLASTx matches (E-value < 10-5) to protein sequences in UniProtKB, Tribolium castaneum (NCBI GenBank), Drosophila melanogaster (FlyBase), Caenorhabditis elegans (Ensembl), human (Ensembl), or non-redundant protein dataset (nr) from GenBank. The remaining 232 were putatively annotated based on matches to proteins from other species (Additional file 4). The candidate genes for the development of beetle horns can be grouped into three conceptual categories (expected, unexpected, and unknown genes).
Figure 3.
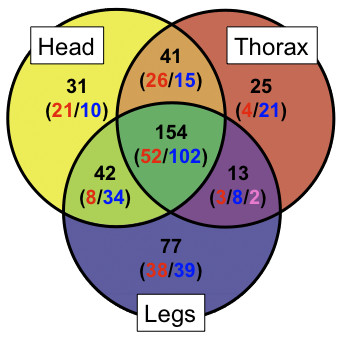
Expression differences between horn and leg primordia relative to abdominal epithelium. Categorization of genes that exhibited significantly differential (p value < 0.05 and > 2-fold difference). The labels on each category represent the tissue types (head = head horns, thorax = prothoracic horns, and legs = legs). Numbers indicated in the Venn diagram represent the counts of non-redundant sequences in each category. The numbers in parenthesis indicate the counts of sequences that showed enriched or depleted expression relative to abdominal epithelium, where: red = enriched, blue = depleted, and pink = mixed (i.e. enriched in thoracic horns and depleted in legs).
First, we identified genes whose expression in the context of horn development could be expected given existing insights into the developmental biology of horns, and knowledge about the function of these genes in other organisms. For instance, the Hox gene Sex combs reduced (Scr) is enriched in the prothoracic horn (19.2 fold) and legs (7.9 fold) relative to abdominal epithelium (Additional file 4). In Drosophila and Tribolium Scr patterns the identity of the labial and first thoracic segment [21-23]. Preliminary results showed that Onthophagus Scr executes similar functions during labial and thoracic development in addition to playing a major role in the regulation of prothoracic horn development (Wasik, Rose, and Moczek, unpublished data).
Secondly, we identified genes that although functionally well characterized in Drosophila or elsewhere, would not readily be expected to be expressed in the context of horn development. Genes in this category include the putative ortholog of Drosophila doublesex (dsx), enriched more than 2-fold in the head and prothoracic horns relative to abdominal epithelium (Additional file 4). In Drosophila the expression of sex-specific DSX isoforms regulate somatic sex-determination sexually dimorphic differentiation [24,25]. While Onthophagus horns are sexually dimorphic, our observation that the putative dsx ortholog is expressed preferentially in the male horn tissue when compared to male abdominal tissue was unexpected. Expression and functional studies are now under way to identify the role of dsx in the development and diversification of horns.
Similarly, we found that the putative Onthophagus orthologs of yellow-c, -e, and -f were enriched more than 2-fold in head and/or prothoracic horns relative to abdominal epithelium (Additional file 4). The functions of yellow family genes are remarkably diverse and include the regulation of pigmentation [26,27], the production of a major component of royal jelly in the honeybee [28] as well as expression of normal male courtship behavior in Drosophila [29]. Combined, these observations suggest that yellow genes may be involved in the regulation of a wide array of sex- or caste-specific functions, at least among insects, though it remains to be determined, what, if any, function the gene family may be executing in Onthophagus beetles.
Lastly, we identified 74 genes that were significantly differentially expressed in either head horns or prothoracic horns, or both, that lack obvious homology to proteins in any of the datasets used in this study. Of those 74, at least 29 (39%) contained predicted ORFs with longer than 300 nucleotides (100 codons).
Discussion
Horned beetles, most notably in the genus Onthophagus, are increasingly being recognized as an emerging model system in evo-devo and eco-devo studies [13,30-34]. Below we discuss the major findings of our study and their applicability to ongoing and future research efforts in horned beetles and beyond.
Onthophagus taurus expressed sequences as a resource
The expressed sequences and the corresponding cDNAs presented here provide a valuable entry point for studies of gene function in Onthophagus taurus. The sequences derived from normalized larval and pupal cDNA libraries had a low level of redundancy. The 3,488 high quality EST sequences from both libraries assembled into 2,781 non-redundant sequences (contigs and singletons). The low level of redundancy resulted in a sample of sequences derived from a wide range of biological functions.
The Onthophagus transcriptome
This study provides a first pass survey of genes found in Onthophagus. Prior to this study, Tribolium castaneum was the only species of beetle for which comprehensive sequence information was available [6,35]. Comparative analyses indicate that the gene repertoire of Tribolium is consistent with the general trends seen across sequenced insects and vertebrates [6,36]. Our estimates of the proportions of Onthophagus sequences that are common to other species are consistent with those in Tribolium [6]. For instance, we found that 39% of Onthophagus sequences had sequence matches to proteins in all the datasets searched, which is consistent with the Tribolium genome in which ~35% of genes have orthologs in all species examined [6]. Of particular interest are the 23% of Onthophagus sequences that lack orthology (Group 4 in Figure 1) to proteins from six proteomes including the non-redundant dataset which is very close to the corresponding estimate of 23% of annotated Tribolium genes [6]. About 40% of these Group 4 Onthophagus sequences exhibited appreciable putative open reading frames and thus need to be considered potentially protein-coding. This group of genes likely contains genes unique to, or fast evolving in, Onthophagus beetles, and studies are under way to further characterize and analyze the significance of these genes for the evolution, diversification, and radiation of horned beetles.
From ESTs to candidate genes for the evolutionary biology of beetle horns and horned beetles
Beetle horns and horned beetles are attractive system to address several current frontiers in evolutionary biology. The EST resources and array results presented here provide the first genomic resources to identify candidate genes, pathways, and networks underlying morphological, behavioral, and developmental aspects of the biology of horned beetles, as well as providing insights into their respective evolutionary histories. Below we briefly highlight two broad categories of current research efforts and how they are being advanced by the results presented here.
The origins of horns
Beetle horns have attracted attention because they lack obvious homology to other appendages or outgrowths in the insects. Horns therefore constitute an evolutionary novelty. Understanding how novel traits arise from pre-existing variation remains one of the most challenging and poorly understood questions in evolutionary biology.
One hypothesis that has been proposed toward explaining the origin of horns is based on the observation that horns share many morphological and developmental features with traditional appendages (e.g. epithelial origin, prepupal growth, dorso-ventral axis formation, or pupal remodeling presumably via programmed cell death; [10]). Furthermore, in several other respects horns are much simpler than legs or mouthparts (e.g. they lack nerves, muscles, or joints). Horns may therefore have evolved via the large-scale co-option of genes ancestrally used to instruct appendage development. Our microarray results suggest that horns and legs are indeed highly similar in gene expression profiles and support the hypothesis that many genes involved in leg formation may also play a role in horn development. Earlier research has begun to implicate a small subset of appendage patterning genes in horn development (Distal-less, dachshund, extradenticle, homothorax, [11,13,15]). The results presented here add a substantial list of gene candidates (Additional file 4) that may have mediated the origin of horns via co-option from traditional appendage development.
At the same time, horn-specific transcription profiles also included genes not represented in developing legs, suggesting that horns should not be viewed solely as being simplified appendages. While this fraction of genes was small in comparison, it nevertheless highlights a possible class of genes involved in developmental processes of horn formation that are not represented, or at least not to the same degree, during the development of traditional appendages. If correct, this would suggest that the origin of horns may have been mediated by the co-option of appendage patterning genes alongside integration of genes and pathways unrelated to appendage formation. Clearly, additional contrasts including the sampling of other developmental time points, as well as gene function studies, are needed to establish the general validity of these conclusions.
The diversification of beetle horns and horned beetles
Beetle horns and horned beetles are attractive study organisms because they permit investigation of the mechanisms underlying phenotypic diversification on many interesting levels. First, species differ in the body region involved in horn expression: horns may extend from the head, prothorax, or both, and while their function as weapons in male combat appears to be conserved across species, recent studies suggest that different horn types may have originated and diversified at least in part independently of one another [13]. Our results support this scenario by identifying a list of genes whose expression differs significantly across horn types such as yellow-e (head horns), tailup (encodes a LIM-homeodomain protein; prothoracic horns), or Scr (prothoracic horns and legs). While the function, if any, of these candidate genes in the context of horn development remains to be explored our results presented here provide an important starting point toward untangling shared, independent, and convergent aspects in the evolution of different horn types across horned beetles.
Substantial diversity in horn expression also exists within species in the form of sexual and male dimorphisms. Sexual dimorphisms are brought about via sex-specific regulation of horn expression whereas male dimorphisms are predominantly the product of nutritional differences experienced during larval life (reviewed in [37]). Endocrine factors such as juvenile hormone (JH) are likely to play important roles in the regulation of both types of diversity [38-40]. Furthermore, the same nutritional or hormonal manipulations affect sexual and male dimorphisms differently in different species and populations, suggesting that evolutionary changes in the interplay between endocrine factors, nutrition, and sexual differentiation have contributed to the diversification of horned beetles [40,41]. Our EST resources and microarray results provide an important starting point to begin exploring putative candidate genes that may be associated with sex-specific (such as doublesex, transformer-2 or members of the yellow gene family) or nutrition-dependent (e.g. foxo) expression of horns. Moreover, the resources presented here should support the development of experiments towards characterizing sex- and morph-specific transcriptomes in O. taurus and closely related species in the genus (Snell-Rood, Cash, Kijimoto, Andrews, Moczek; unpublished data).
Conclusion
In conclusion, the EST resources and microarray results present here provide a first step toward a systematic analysis of the molecular basis of horn development and diversification in beetles with the potential to inform several major frontiers in evolutionary developmental biology.
Methods
cDNA library construction
Adult Onthophagus taurus were collected from pastures near Bloomington, IN and reared as described previously [15]. We constructed two separate libraries from larval and pupal stages. For the larval library we dissected heads and thoraces from mid third instar larvae, late third instar larvae, and early and late prepupal stages. For the pupal library tissues included whole individuals one, two, three and four days after pupation. For both libraries we harvested at least two individuals for each stage and sex, and all samples were frozen in liquid nitrogen, immediately transferred to -80°C for storage until RNA extraction. Total RNA was extracted using TRIreagent (Sigma, MO), precipitated with ethanol and stored at -80°C. The normalized cDNA libraries were each constructed from 1 μg of total RNA, using the TRIMMER-DIRECT cDNA normalization kit (Evrogen, Moscow, Russia) for the library normalization, followed by the Creator SMART cDNA library construction kit (Clontech, CA) for cDNA library construction, as described in Zhulidov et al. 2004 [42]. We followed the manufacturers protocols with the following modifications and specific conditions: (i) the cycle conditions for the PCR-based double-strand cDNA synthesis were 16 cycles of [95°C for 7 sec, 66°C for 30 sec, and 72°C for 6 min]; (ii) we used 2 μl of cDNA mixture for PCR during cDNA library construction and normalization; and (iii) the conditions for the two step amplification of the normalized cDNA were 18 cycles [95°C for 7 sec, 66°C for 30 sec, and 72°C for 6 min] for the first step, and the second amplification was cycled for 12 cycles using the same conditions. Normalized and amplified cDNA fragments were size-fractionated, digested by Sfi I, and ligated with the plasmid vector pDNR-LIB according to manufacturer's instruction. Electro-transformed E. coli cells were spread on LB plate containing chloramphenicol (final concentration of 30 μg/ml). The estimated titer of both of the libraries were ~1 × 10-8CFU. A total of 3,756 colonies were picked at random. Unless stated otherwise standard molecular procedures were used to execute basic molecular analyses [43].
EST sequencing
DNA samples were prepared for sequencing using a Beckman Coulter Biomek FX Laboratory Automation Workstation as described in Burr et al. 2006 [44]. Each picked clone was incubated overnight at 37°C in 96-well tissue culture plates with 100 μl of SOC medium with chloramphenicol (final concentration of 30 μg/ml), without rotation. 20 μl of the cultured cells were mixed with 80 μl of water and heat-punctured at 95°C for 10 min. Insert DNA was PCR-amplified using cell lysate (10 μl) as template, 0.1 μM M13fw primer (5'-GTG TAA AAC GAC GGC CAG TAG-3'), 0.1 μM M13rev primer (5'-AAA CAG CTA TGA CCA TGT TCA C-3'), 0.2 mM each dNTP, 0.5 U/20 μl reaction Taq polymerase (Bioline, MA), and 1× reaction buffer (Bioline, MA). The reaction was incubated at 95°C for 5 min then 35 cycles of [95°C for 1 min, 54°C for 1 min, and 2 min at 72°C]. The amplified DNA was purified using the Multiscreen-PCR 96-well purification system (Millipore, MA). The purified DNA was subjected to agarose gel electrophoresis against molecular weight standard and visualized using a Kodak 440cf imaging station. Sequencing reactions were performed with the primer pDNRlib30-50 (5'-TAT ACG AAG TTA TCA GTC GAC G-3') and ABI BigDye chemistry and ABI Prism 3730 sequencer (Applied BioSystems, CA).
EST processing, assembly, and annotation
ESTPiper [17] was used to analyze EST sequences including base calling, data cleaning, de novo assembly, and annotation. A total of 3,756 EST sequences were generated in FASTA format with quality scores after base calling. For data cleaning, ESTPiper first removed low quality and vector sequences using LUCY [45] program with the default parameter settings. PolyA/T tails were then trimmed, where within 50 bp searching range from both ends of the sequences, the minimum length of continuous polyA/T region was set to 9 bp and the maximum number of mismatches within the polyA/T region was set to 3. Potentially chimeric clones, which were defined as sequences with at least 30 bp continuous A/T or adaptors occurring in the middle of sequences, were removed. Finally, shorter sequences (< 100 bp) were also removed. A total of 3,488 high quality sequences passed data cleaning procedure. We then performed de novo assembly to assemble EST sequences into contigs and singletons. Parameters were set as follows: (i) overlap percent identity cutoff was 95%, (ii) overlap length cutoff was 49, and (iii) maximum number of word matches was 10,000 (this parameter defines the maximum number of matches that the program will consider for a given sequence, and was set high to improve accuracy [46]. For annotation, ESTPiper matched contigs/singletons to UniProt database [47] using BLASTX with an E-value cutoff of 1 × 10-5 and only the top match was taken.
Microarray printing
We developed the cDNA microarray using all the clones used for the EST analysis (3,756 clones) as well as GAPDH and actin-5c (internal positive controls). Insert DNA was PCR amplified and purified as described above in the EST sequencing section. We followed the protocol of Indiana University Drosophila Genomics Resource Center [48] to print microarrays with a minor revision to post-print washes. Purified insert DNA was dried completely, re-dissolved in DGRC spotting solution (1.5 M Betaine in 3 × SSC), and spotted to GAPSII Microarray Slides (Corning) using an OmniGrid 300 printing. The microarray design included 4,320 spots arranged in 48 blocks of 90. A total of 3,756 of these spots were cDNA fragments (each spotted once) and 564 of these consisted of control spots (GAPDH, actin-5c, and spotting buffer only). The gene list and platform description is available at Gene Expression Omnibus http://www.ncbi.nlm.nih.gov/geo/ accession number GPL7555. After printing, the microarrays were heated at 85°C for 3 hrs and rinsed with 5 × SSC/0.1%SDS (55°C), water (twice at RT, once at 95°C, and once again at RT) and then centrifuged to dry. All microarrays were kept dry at room temperature until they were used.
Target RNA preparation, hybridization and obtaining data sets
Tissues were dissected from 20 male O. taurus (day 1 pupae) that were collected from our laboratory colony. Dissections and RNA extractions (RNeasy Mini kit, Qiagen, CA) of head horn, prothoracic horn, leg, and abdominal epithelium were performed separately for each animal. Independent biological replicates of RNA samples were created by pooling an equal mass of RNA isolated from the same type of tissue from 4 individuals. For each RNA sample 1 μg of RNA was reverse transcribed using Oligo(dT)-T7 primer (Ambion, TX) and SuperScriptIII reverse transcriptase (Invitrogen, CA), and DNA polymerase and RNase H (Invitrogen, CA) were used for second strand synthesis. Amplified RNA (aRNA) was generated by in vitro transcribing the cDNA using the MEGAscript kit (Ambion, TX). The aRNA was directly labeled with Cy3 or Cy5 using the ULS aRNA Fluorescent Labeling Kit (KREATECH, Amsterdam, The Netherlands). Three sets of amplified RNA samples from head horns, prothoracic horns, and legs were labeled with Cy5, while abdominal epithelial tissue samples were labeled with Cy3. The remaining two sets of samples were labeled in the opposite way. After measuring the quantity and labeling efficiency, amplified and labeled RNA samples from test (head horns, prothoracic horns, and legs) samples and abdomen (reference sample) were mixed and hybridized onto arrays. aRNA with 50 pmol dye from the test sample and reference sample were mixed with KREAblock (ULS aRNA Fluorescent Labeling kit) and 2 × enhanced cDNA hybridization buffer (Genisphere, PA), then heated at 80°C for 10 min. Arrays were pre-treated for more than one hour at 55°C in pre-hybridization buffer (5 × SSC, 0.1%SDS, 1% I-block (Applied Biosystems, CA)). Both mixed sample and microarray were kept at 55°C until the hybridization step. Hybridization was performed in a dark humidified chamber at 55°C overnight. The microarray was rinsed in buffer A (2 × SSC/0.2%SDS) at 55°C then incubated in buffer A at 65°C for 10 min. The microarray was transferred to 2 × SSC (room temperature) for 10 min, followed by incubation in 0.2% SSC for 10 min at room temperature. The rinsed microarray was dried by centrifuging at 500rcf for 4 min.
The hybridized microarrays were scanned by GenePix scanner 4200 (Molecular Devices, CA) to obtain raw data sets. After initial quality check of results using OLIN in Bioconducter (Basic Hybridization Analysis, Costello et al. 2005, https://dgrc.cgb.indiana.edu/microarrays/support/bha.html), differential expression was assessed using Limma [49]. The values for each spot were shown as log2 ratios between the two signal intensities (M-values). The microarray data are available at Gene Expression Omnibus http://www.ncbi.nlm.nih.gov/geo/, accession number GPL7555.
Clustering analysis
We performed clustering analysis and support tree construction using TIGR MultiExperiment Viewer of the TM4 system [50]. We performed hierarchical clustering by using Cosine Correlation with average linkage to obtain the cluster and tree.
Authors' contributions
TK, APM, and JA designed the study. TK developed cDNA libraries and microarrays. TK, JC, ZT and JA analyzed libraries. TK performed microarray experiments. TK, APM, and JA analyzed the microarray results. TK, JC, ZT, APM and JA wrote the paper. All authors read and approved the final manuscript.
Supplementary Material
Summary of BLASTx results of Onthophagus taurus ESTs against UniProtKB. Summary of BLASTx sequence alignments of non-redundant sequences from Ot libraries (refer Results section) against UniProtKB protein dataset. The top BLASTx hits with E-values < 10-5 are shown.
Summary of BLASTx results of Onthophagus taurus ESTs against FlyBase. Summary of BLASTx sequence alignments of non-redundant sequences from Ot libraries (refer Results section) against FlyBase protein dataset. The top BLASTx hits with the E-values < 10-5 are shown.
Summary of BLASTx results of Onthophagus ESTs against all the datasets used in this study. Summary of BLASTx sequence alignments of Onthophagus ESTs against all the datasets used in this study. The bit score, E-value, and percent identity as well as the gene names from Tribolium, Drosophila, C. elegans, invertebrate, human, and nr dataset are shown for of each Ot non-redundant sequences.
List of genes exhibiting differential expression in horn- or leg primordia compared to abdominal epithelium. Listed are genes exhibiting significant (p < 0.05) and at least 2-fold difference in expression intensity in primordial head horns and/or prothoracic horns and/or legs. Shown are spot- or contig IDs, the tissue(s) in which differential expression was detected (H: head horns, T: prothoracic horns, L: legs), fold-differences in expression relative to abdominal epithelium (positive values indicate enrichment relative to abdominal epithelium), sequence match descriptions (UniProtKB, FlyBase, Triboium, invertebrate, or nr), percent amino acid sequence identity, E-values, and bit score.
Acknowledgments
Acknowledgements
This manuscript benefited greatly from comments by Amy Cash, Emilie Snell-Rood, and two anonymous reviewers. We thank the Center for Genomics and Bioinformatics at Indiana University and its staff, especially John Colborne, Zhao Lai, Zach Smith, Jade Buchanan-Carter, and Heejung Yang for their advice and expertise in executing this study. Erin Yoder and Sarah Jones provided expert beetle care. Funding for this study, as well as for work in the Center for Genomics and Bioinformatics, was provided in part by the METACyt Initiative of Indiana University, funded in part through a major grant from the Lilly Endowment. Additional support was provided by National Science Foundation grants IOS 0820411 to JA and APM.
Contributor Information
Teiya Kijimoto, Email: tkijimot@indiana.edu.
James Costello, Email: jccostel@indiana.edu.
Zuojian Tang, Email: tangz@cgb.indiana.edu.
Armin P Moczek, Email: armin@indiana.edu.
Justen Andrews, Email: jandrew@bio.indiana.edu.
References
- Raff RA. The Shape of Life: Genes, Development, and the Evolution of Animal Form. Chicago: University of Chicago Press; 1996. [Google Scholar]
- West-Eberhard MJ. Developmental Plasticity and Evolution. Oxford, UK: Oxford University Press; 2003. [Google Scholar]
- Beldade P, Rudd S, Gruber JD, Long AD. A wing expressed sequence tag resource for Bicyclus anynana butterflies, an evo-devo model. BMC genomics. 2006;7:130. doi: 10.1186/1471-2164-7-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapan DD, Flanagan NS, Tobler A, Papa R, Reed RD, Gonzalez JA, Restrepo MR, Martinez L, Maldonado K, Ritschoff C, et al. Localization of Mullerian mimicry genes on a dense linkage map of Heliconius erato. Genetics. 2006;173:735–757. doi: 10.1534/genetics.106.057166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson GE, Fernald RD, Clayton DF. Genes and social behavior. Science. 2008;322:896–900. doi: 10.1126/science.1159277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribolium Genome Sequencing Consortium The genome of the model beetle and pest Tribolium castaneum. Nature. 2008;452:949–955. doi: 10.1038/nature06784. [DOI] [PubMed] [Google Scholar]
- Moczek AP. The Evolution and Development of Novel Traits, or How Beetles Got Their Horns. BioScience. 2005;55:937–951. doi: 10.1641/0006-3568(2005)055[0937:TEADON]2.0.CO;2. [DOI] [Google Scholar]
- Balthasar V. Monographie der Scarabaeidae und Aphodiidae der palaearktischen und orientalischen Region (Coleoptera: Lamellicornia. Band 2, Coprinae. Prag: Verlag der tschechoslowakischen Akademie der Wissenschaften; 1963. [Google Scholar]
- Matthews E. A revision of the Scarabaeinae dung beetles of Australia. I. Tribe Onthophagini. Aus J Zool. 1972. pp. 1–330.
- Moczek AP. Integrating micro- and macroevolution of development through the study of horned beetles. Heredity. 2006;97:168–178. doi: 10.1038/sj.hdy.6800871. [DOI] [PubMed] [Google Scholar]
- Moczek AP, Rose D, Sewell W, Kesselring BR. Conservation, innovation, and the evolution of horned beetle diversity. Dev Genes Evol. 2006;216:655–665. doi: 10.1007/s00427-006-0087-2. [DOI] [PubMed] [Google Scholar]
- Emlen DJ, Corley Lavine L, Ewen-Campen B. On the origin and evolutionary diversification of beetle horns. Proc Natl Acad Sci USA. 2007;104:8661–8668. doi: 10.1073/pnas.0701209104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emlen DJ, Szafran Q, Corley LS, Dworkin I. Insulin signaling and limb-patterning: candidate pathways for the origin and evolutionary diversification of beetle 'horns'. Heredity. 2006;97:179–191. doi: 10.1038/sj.hdy.6800868. [DOI] [PubMed] [Google Scholar]
- Moczek AP. Pupal remodeling and the development and evolution of sexual dimorphism in horned beetles. Am Nat. 2006;168:711–729. doi: 10.1086/509051. [DOI] [PubMed] [Google Scholar]
- Moczek AP, Nagy LM. Diverse developmental mechanisms contribute to different levels of diversity in horned beetles. Evol Dev. 2005;7:175–185. doi: 10.1111/j.1525-142X.2005.05020.x. [DOI] [PubMed] [Google Scholar]
- Moczek AP, Cruickshank TE, Shelby A. When ontogeny reveals what phylogeny hides: gain and loss of horns during development and evolution of horned beetles. Evolution. 2006;60:2329–2341. [PubMed] [Google Scholar]
- Tang Z, Choi J, Hemmerich C, Sarangi A, Colbourne JK, Dong Q. ESTPiper - a web-based analysis pipeline for expressed sequence tags. BMC genomics. 2009;10:174. doi: 10.1186/1471-2164-10-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbeitman MN, Furlong EE, Imam F, Johnson E, Null BH, Baker BS, Krasnow MA, Scott MP, Davis RW, White KP. Gene expression during the life cycle of Drosophila melanogaster. Science. 2002;297:2270–2275. doi: 10.1126/science.1072152. [DOI] [PubMed] [Google Scholar]
- Beermann A, Schroder R. Functional stability of the aristaless gene in appendage tip formation during evolution. Dev Genes Evol. 2004;214:303–308. doi: 10.1007/s00427-004-0411-7. [DOI] [PubMed] [Google Scholar]
- Tomoyasu Y, Wheeler SR, Denell RE. Ultrabithorax is required for membranous wing identity in the beetle Tribolium castaneum. Nature. 2005;433:643–647. doi: 10.1038/nature03272. [DOI] [PubMed] [Google Scholar]
- Lewis RA, Kaufman TC, Denell RE, Tallerico P. Genetic Analysis of the Antennapedia Gene Complex (Ant-C) and Adjacent Chromosomal Regions of DROSOPHILA MELANOGASTER. I. Polytene Chromosome Segments 84b-D. Genetics. 1980;95:367–381. doi: 10.1093/genetics/95.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RA, Wakimoto BT, Denell RE, Kaufman TC. Genetic Analysis of the Antennapedia Gene Complex (Ant-C) and Adjacent Chromosomal Regions of DROSOPHILA MELANOGASTER. II. Polytene Chromosome Segments 84A-84B1,2. Genetics. 1980;95:383–397. doi: 10.1093/genetics/95.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeman RW, Stuart JJ, Brown SJ, Denell RE. Structure and function of the homeotic gene complex (HOM-C) in the beetle, Tribolium castaneum. Bioessays. 1993;15:439–444. doi: 10.1002/bies.950150702. [DOI] [PubMed] [Google Scholar]
- Saccone G, Salvemini M, Pane A, Polito LC. Masculinization of XX Drosophila transgenic flies expressing the Ceratitis capitata DoublesexM isoform. Int J Dev Biol. 2008;52:1051–1057. doi: 10.1387/ijdb.082657gs. [DOI] [PubMed] [Google Scholar]
- Sanchez L, Gorfinkiel N, Guerrero I. Sex determination genes control the development of the Drosophila genital disc, modulating the response to Hedgehog, Wingless and Decapentaplegic signals. Development. 2001;128:1033–1043. doi: 10.1242/dev.128.7.1033. [DOI] [PubMed] [Google Scholar]
- Han Q, Fang J, Ding H, Johnson JK, Christensen BM, Li J. Identification of Drosophila melanogaster yellow-f and yellow-f2 proteins as dopachrome-conversion enzymes. Biochem J. 2002;368:333–340. doi: 10.1042/BJ20020272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash WG, Yarkin RJ. Genetic regulation and pattern formation: a study of the yellow locus in Drosophila melanogaster. Genet Res. 1974;24:19–26. doi: 10.1017/S0016672300015044. [DOI] [PubMed] [Google Scholar]
- Malecova B, Ramser J, O'Brien JK, Janitz M, Judova J, Lehrach H, Simuth J. Honeybee (Apis mellifera L.) mrjp gene family: computational analysis of putative promoters and genomic structure of mrjp1, the gene coding for the most abundant protein of larval food. Gene. 2003;303:165–175. doi: 10.1016/S0378-1119(02)01174-5. [DOI] [PubMed] [Google Scholar]
- Drapeau MD, Radovic A, Wittkopp PJ, Long AD. A gene necessary for normal male courtship, yellow, acts downstream of fruitless in the Drosophila melanogaster larval brain. J Neurobiol. 2003;55:53–72. doi: 10.1002/neu.10196. [DOI] [PubMed] [Google Scholar]
- Brakefield PM, French V. Evo-devo focus issue. Heredity. 2006;97:137–138. doi: 10.1038/sj.hdy.6800878. [DOI] [Google Scholar]
- Emlen DJ. Integrating Development with Evolution:A Case Study with Beetle Horns. BioScience. 2000;50:403–418. doi: 10.1641/0006-3568(2000)050[0403:IDWEAC]2.0.CO;2. [DOI] [Google Scholar]
- Jenner RA, Wills MA. The choice of model organisms in evo-devo. Nat Rev Genet. 2007;8:311–319. doi: 10.1038/nrg2062. [DOI] [PubMed] [Google Scholar]
- Monteiro A, Podlaha O. Wings, horns, and butterfly eyespots: how do complex traits evolve? PLoS Biol. 2009;7:e37. doi: 10.1371/journal.pbio.1000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shubin N, Tabin C, Carroll S. Deep homology and the origins of evolutionary novelty. Nature. 2009;457:818–823. doi: 10.1038/nature07891. [DOI] [PubMed] [Google Scholar]
- Park Y, Aikins J, Wang LJ, Beeman RW, Oppert B, Lord JC, Brown SJ, Lorenzen MD, Richards S, Weinstock GM, et al. Analysis of transcriptome data in the red flour beetle, Tribolium castaneum. Insect Biochem Mol Biol. 2008;38:380–386. doi: 10.1016/j.ibmb.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyder S, Kriventseva EV, Schroder R, Kadowaki T, Zdobnov EM. Quantification of ortholog losses in insects and vertebrates. Genome Biol. 2007;8:R242. doi: 10.1186/gb-2007-8-11-r242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moczek A. Developmental plasticity and the origins of diversity: a case study on horned beetles. In: Whitman DW, Ananthakrishnan TN, editor. Phenotypic Plasticity of Insects: Mechanisms and Consequence. Plymouth, UK: Science Publishers Inc; 2009. pp. 81–134. [Google Scholar]
- Emlen DJ, Nijhout HF. Hormonal control of male horn length dimorphism in the dung beetle Onthophagus taurus (Coleoptera: Scarabaeidae) J Insect Physiol. 1999;45:45–53. doi: 10.1016/S0022-1910(98)00096-1. [DOI] [PubMed] [Google Scholar]
- Emlen DJ, Nijhout HF. Hormonal control of male horn length dimorphism in Onthophagus taurus (Coleoptera: Scarabaeidae): a second critical period of sensitivity to juvenile hormone. J Insect Physiol. 2001;47:1045–1054. doi: 10.1016/S0022-1910(01)00084-1. [DOI] [PubMed] [Google Scholar]
- Shelby JA, Madewell R, Moczek AP. Juvenile hormone mediates sexual dimorphism in horned beetles. J Exp Zool B Mol Dev Evol. 2007;308:417–427. doi: 10.1002/jez.b.21165. [DOI] [PubMed] [Google Scholar]
- Moczek AP, Nijhout HF. Developmental mechanisms of threshold evolution in a polyphenic beetle. Evol Dev. 2002;4:252–264. doi: 10.1046/j.1525-142X.2002.02014.x. [DOI] [PubMed] [Google Scholar]
- Zhulidov PA, Bogdanova EA, Shcheglov AS, Vagner LL, Khaspekov GL, Kozhemyako VB, Matz MV, Meleshkevitch E, Moroz LL, Lukyanov SA, et al. Simple cDNA normalization using kamchatka crab duplex-specific nuclease. Nucleic Acids Res. 2004;32:e37. doi: 10.1093/nar/gnh031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning. Third. Cold Spring Harbor: Cold Spring Harbor Press; 2001. [Google Scholar]
- Burr A, Bogart K, Conaty J, Andrews J. Automated liquid handling and high-throughput preparation of polymerase chain reaction-amplified DNA for microarray fabrication. Methods Enzymol. 2006;410:99–120. doi: 10.1016/S0076-6879(06)10005-1. [DOI] [PubMed] [Google Scholar]
- Chou HH, Holmes MH. DNA sequence quality trimming and vector removal. Bioinformatics. 2001;17:1093–1104. doi: 10.1093/bioinformatics/17.12.1093. [DOI] [PubMed] [Google Scholar]
- Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium The Universal Protein Resource (UniProt) Nucleic Acids Res. 2007:D193–197. doi: 10.1093/nar/gkl929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews J, Bogart K, Burr A, Conaty J. Fabrication of DGRC cDNA Microarrays. CGB Technical Report. 2006;2006-11 [Google Scholar]
- Smyth GK, Speed T. Normalization of cDNA microarray data. Methods. 2003;31:265–273. doi: 10.1016/S1046-2023(03)00155-5. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of BLASTx results of Onthophagus taurus ESTs against UniProtKB. Summary of BLASTx sequence alignments of non-redundant sequences from Ot libraries (refer Results section) against UniProtKB protein dataset. The top BLASTx hits with E-values < 10-5 are shown.
Summary of BLASTx results of Onthophagus taurus ESTs against FlyBase. Summary of BLASTx sequence alignments of non-redundant sequences from Ot libraries (refer Results section) against FlyBase protein dataset. The top BLASTx hits with the E-values < 10-5 are shown.
Summary of BLASTx results of Onthophagus ESTs against all the datasets used in this study. Summary of BLASTx sequence alignments of Onthophagus ESTs against all the datasets used in this study. The bit score, E-value, and percent identity as well as the gene names from Tribolium, Drosophila, C. elegans, invertebrate, human, and nr dataset are shown for of each Ot non-redundant sequences.
List of genes exhibiting differential expression in horn- or leg primordia compared to abdominal epithelium. Listed are genes exhibiting significant (p < 0.05) and at least 2-fold difference in expression intensity in primordial head horns and/or prothoracic horns and/or legs. Shown are spot- or contig IDs, the tissue(s) in which differential expression was detected (H: head horns, T: prothoracic horns, L: legs), fold-differences in expression relative to abdominal epithelium (positive values indicate enrichment relative to abdominal epithelium), sequence match descriptions (UniProtKB, FlyBase, Triboium, invertebrate, or nr), percent amino acid sequence identity, E-values, and bit score.