

Abstract
This review gives a brief description of the theory and application of X-ray absorption spectroscopy, both X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS), especially, pertaining to photosynthesis. The advantages and limitations of the methods are discussed. Recent advances in extended EXAFS and polarized EXAFS using oriented membranes and single crystals are explained. Developments in theory in understanding the XANES spectra are described. The application of X-ray absorption spectroscopy to the study of the Mn4Ca cluster in Photosystem II is presented.
Keywords: Photosystem II, Water oxidation, Oxygen evolution, Manganese cluster, X-ray spectroscopy, EXAFS, XANES, X-ray dichroism
Introduction
During the past 30 years, X-ray absorption spectroscopy (XAS) has made major contributions to a wide variety of biochemical research topics. It has been raising important questions of correlation between structure and function of the metal sites in metallo-proteins, including the photosynthetic oxygen-evolving complex (OEC; Yano and Yachandra 2008). The history of the development of the experimental method of XAS (Sayers et al. 1971; Eisenberger and Kincaid 1978) overlaps the history of the structural research on the OEC in photosystem II (PS II). The historical background of the XAS study on PS II, especially the early work, has been reviewed in some detail (Yachandra et al. 1996; Penner-Hahn 1998; Yachandra 2005; Yano and Yachandra 2007; Sauer et al. 2008).
In X-ray spectroscopy, transitions are involved in absorption (XAS, X-ray absorption spectroscopy) or emission (XES, X-ray emission spectroscopy) of X-rays, where the former probes the ground state to the excited state transitions, while the latter probes the decay process from the excited state. Both methods characterize the chemical nature and environment of atoms in molecules, and synchrotron sources provide a range of X-ray energies that are applicable to most elements in the periodic table, in particular, those present in redox-active metallo-enzymes. The choice of the energy of the X-rays used, in most cases, determines the specific element being probed. This is quite a contrast with other methods, such as optical or UV absorption, fluorescence, magnetic susceptibility, electrochemistry etc., which have been applied to study biological redox systems. The results from infrared and Raman spectroscopy can be related to specific elements through isotopic substitution, but the analysis of such spectra for metal clusters is complicated when the structure is not known. In this article, we focus on XAS methods which have been used in the field of photosynthesis. The XES methods are discussed in the paper by Bergmann and Glatzel (this issue).
X-ray absorption spectroscopy (XAS) is the measurement of transitions from core electronic states of the metal to the excited electronic states (LUMO) and the continuum; the former is known as X-ray absorption near-edge structure (XANES), and the latter as extended X-ray absorption fine structure (EXAFS) which studies the fine structure in the absorption at energies greater than the threshold for electron release. These two methods give complementary structural information, the XANES spectra reporting electronic structure and symmetry of the metal site, and the EXAFS reporting numbers, types, and distances to ligands and neighboring atoms from the absorbing element (Koningsberger and Prins 1988).
X-ray absorption spectroscopy (XAS) allows us to study the local structure of the element of interest without interference from absorption by the protein matrix, water or air. Yet, X-ray spectroscopy of metallo-enzymes has been a challenge due to the small relative concentration of the element of interest in the sample. In the PS II, for example, Mn may be at the level of 10 parts per million or less. In such a case, the use of X-ray fluorescence for the detection of the absorption spectra, instead of using the transmission detection mode, has been the standard approach. The development of intense third generation synchrotron radiation X-ray sources has also permitted the study of dilute samples.
Metal complexes, as models with known structures, have been essential in order to understand the XAS of metallo-proteins. These complexes provide a basis for evaluating the influence of the coordination environment (coordination charge) on the absorption edge energy (Cinco et al. 1999; Pizarro et al. 2004). Study of structurally well-characterized model complexes also provides a benchmark for understanding the EXAFS from metal systems of unknown structure.
The significant advantage of XAS over the X-ray crystallography is that the local structural information around the element of interest can be obtained even from disordered samples, such as powders and solution. However, ordered samples, such as membranes and single crystals, often increases the information obtained from XAS. For oriented single crystals or ordered membranes, the interatomic vector orientations can be deduced from dichroism measurements. These techniques are especially useful for determining the structures of multi-nuclear metal clusters, such as the Mn4Ca cluster associated with water oxidation in the photosynthetic oxygen-evolving complex (OEC). Moreover, quite small changes in geometry/structure associated with transitions between the intermediate states, known as the S-states, in the cycle of the water-oxidation reaction can be readily detected using XAS. Another useful approach has been to collect complementary EXAFS measurements, for example, at both the Mn and Ca K-edges for the OEC cluster (Cinco et al. 2002), or following a Sr → Ca replacement measuring data at the Mn and Sr K-edges (Latimer et al. 1995; Cinco et al. 1998; Pushkar et al. 2008). Such measurements greatly improve the information that can be obtained for multi-nuclear metal clusters, such as the Mn4Ca cluster in PS II, as the precision of the fits can be improved by such complementary data.
X-ray absorption spectroscopy (XAS) theory has been developed to an extent that it can be applied to complicated molecules of known structure (Teo 1986; Rehr and Albers 2000). Although it is less straightforward to apply it to the OEC, where its molecular environment is not yet precisely defined, the basic XAS equation allows us to interpret EXAFS spectra to considerable advantage. X-ray spectral properties to be expected from specified cluster geometries can be calculated and compared with experimental measurements. Density-functional theory (DFT) can be applied to issues like the stability of a proposed cluster arrangement or the likelihood of postulated reaction paths. Moreover, the time-dependent DFT calculations provide an important insight into the electronic structure of the metal site combined with the analysis of the XANES pre-edge region.
In the current review, we summarize the basics of XAS, and also discuss some techniques which have been applied to study the OEC of PS II.
Theory of XAS
X-ray absorption spectra of any material be it atomic or molecular in nature are characterized by sharp increases in absorption at specific X-ray photon energies, which are characteristic of the absorbing element. These sudden increases in absorption are called absorption edges, and correspond to the energy required to eject a core electron into the LUMO or to the continuum thus producing a photoelectron. The absorption discontinuity is known as the K-edge, when the photoelectron originates from a 1s core level, and an L-edge when the ionization is from a 2s or 2p electron. Figure 1 shows a typical energy level diagram. L-edge spectroscopy is, in general, more sensitive to the electronic, structural, and the spin state changes of the metal cluster compared to the K-edge spectroscopy, however, there are experimental difficulties in applying this technique to biological samples. We will focus on K-edge spectroscopy in the current review.
Fig. 1.

The energy level diagram for L-edge (LI, LII, and LIII) transitions (2s and 2p to 3d) and K-edge transitions (1s to 3d and 4p) for Mn(II). The energy levels are not drawn to scale. For example, the K-edge is at 6,539 eV and the L edges are at 769, 650, and 639 eV, respectively
XANES
X-ray absorption near-edge structure (XANES) spectra provide detailed information about the oxidation state and coordination environment of the metal atoms (Fig. 2). The K-edge absorption edge energy increases with increasing oxidation state. In general, the rising edge position shifts when the effective number of positive charges (in a simplified view, oxidation state) changes resulting from 1s core hole shielding effects (Shulman et al. 1976). In an atom with one electron, for example, the electron experiences the full charge of the positive nucleus. However, in an atom with many electrons, the outer electrons are simultaneously attracted to the positive nucleus and repelled by the negatively charged electrons. The higher the oxidation state of the metal, the more positive the overall charge of the atom, and therefore more energy is required to excite an electron from an orbital. Conversely, the XANES spectrum shifts to a lower energy when there is more negative charge on the metal.
Fig. 2.

a The Mn K-edge XANES and EXAFS spectra. Top left: the X-ray absorption spectrum from a PS II sample showing the XANES and EXAFS regions of the spectrum. The energy levels are indicated on top of the panel. The enlargements show the Mn K-edge XANES and the k-space EXAFS spectrum. The Fourier transform of the k-space EXAFS data is shown on the right. b A schematic of the outgoing and backscattered photoelectron wave, which illustrates the concept of interference in EXAFS. Left: E 1 is the energy of the incident X-ray photon. The central atom (blue) is the absorbing atom and the photoelectron is backscattered from the surrounding atoms (red). The backscattered wave from the surrounding atoms (dashed blue circular lines) is in phase with the outgoing wave (solid blue circular lines). This leads to an increase in the absorption coefficient at E 1. Right: similarly, at energy E 2 > E 1 (notice that the wavelength of the photo-electron is shorter at E 2 compared to E 1), the backscattered wave can destructively interfere with the outgoing wave, which leads to a decrease in the cross section. The attenuation in the cross section in the absorption coefficient, called EXAFS, is a consequence of this phenomenon
The dominant contribution to the K-edge spectrum comes from 1s → np transitions, where np represents the lowest unoccupied p orbital of the absorbing atom. This transition, with ∆l = 1 (l is the orbital momentum quantum number), is quantum mechanically allowed and is typically intense. For transition metals with partially occupied d orbitals, additional insights can be gained by examination of pre-edge features that result from 1s to (n − 1)d transitions. These are relatively weak in intensity (∆l = 2; hence, formally forbidden or dipole-forbidden), but they can be detected as they occur at energies slightly less than that of the main absorption edge. The pre-edge peak intensity increases when the ligand environment is perturbed from octahedral symmetry (see “Mn K-edge pre-edge spectra and DFT calculations”).
EXAFS
At energies somewhat greater than the LUMO level, the absorption of an X-ray provides sufficient energy to cause the absorbing atom to release the electron (ionize). Any excess energy is carried off as translational kinetic energy, which is alternatively reflected in the wavelength associated with the electron treated as a wave phenomenon. The EXAFS modulations, shown in Fig. 2, are a direct consequence of the wave nature of the photoelectron with the velocity ν imparted to the photoelectron by the energy of the absorbed X-ray photon, which is in excess of the binding or threshold energy for the electron. The kinetic energy of the photoelectron is given by the following relation:
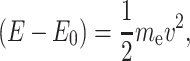 |
1 |
where E is the X-ray photon energy, E 0 is the ionization or threshold energy for the electron, and m e is the electron mass. The EXAFS modulations are better expressed as a function of the photoelectron wave vector k (k = 2π/λ, where λ is the wavelength given by the de Broglie relation, λ = h/m e v, h is Planck’s constant), which is expressed as follows:
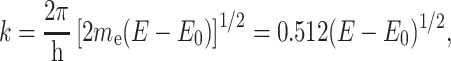 |
2 |
where E and E 0 are expressed in electron volts (eV) and k has the units of inverse angstroms (Å−1).
The wave nature of the departing electron results in interference owing to scattering off nearby atoms. Thus, the EXAFS oscillations result from the interference between the outgoing photoelectron wave and components of backscattered wave from neighboring atoms in the molecule, which start immediately past an absorption edge and extending to about 1 keV above the edge. The general definition for the EXAFS phenomenon χ(k), which is the oscillatory portion of the absorption coefficient, is the difference between the observed absorption coefficient μ(k) and the free-atom absorption coefficient μ 0(k), normalized by the free-atom contribution:
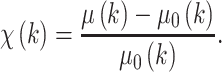 |
3 |
One can envision the EXAFS phenomena by the help of a schematic of the outgoing and backscattered waves as shown in Fig. 2b. As the energy of the photoelectron changes, so does the wavelength of the photoelectron. At a particular energy E 1, the outgoing and the backscattered waves are in phase and constructively interfere, thus increasing the probability of X-ray absorption or, in other words, increase the absorption coefficient. At a different energy E 2, the outgoing and backscattered waves are out-of-phase and destructively interfere, decreasing the absorption coefficient. This modulation of the absorption coefficient by the backscattered wave from neighboring atoms is essentially the basic phenomenon of EXAFS. And, Fourier transform (FT) of the modulation provides distance information describing the vector(s) between the absorbing atom and atoms to which it is bound—typically within a range limit of 4–5 Å.
A quantitative EXAFS modulation χ(k) can be expressed as follows:
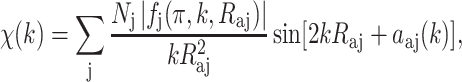 |
4 |
where N j is the number of equivalent backscattering atoms j at a distance R aj from the absorbing atom, f j(π, k) is the backscattering amplitude which is a function of the atomic number of the backscattering element j, and α aj(k) includes the phase shift from the central atom absorber as well as the backscattering element j. The phase shift occurs due to the presence of atomic potentials that the photoelectron experiences as it traverses the potential of the absorber atom, the potential of the backscattering atom, and then back through the potential of the absorber atom.
In real systems, there is an inherent static disorder due to a distribution of distances R
aj, and dynamic disorder due to thermal vibrations of the absorbing and scattering atoms. Equation 4 is modified to include this disorder term or the Debye–Waller factor 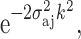 where
where 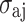 is the root-mean-square deviation to give the following equation:
is the root-mean-square deviation to give the following equation:
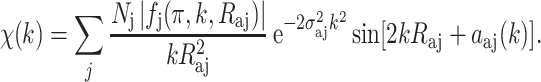 |
5 |
The loss of photoelectrons to inelastic scattering processes can be accounted for by including a term, 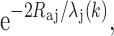 which reflects losses due to inelastic scattering, where λ
j
(k) is the electron mean free path. Thus, the EXAFS contribution from each backscattering atom j is a damped sine wave in k-space, with an amplitude, and a phase, which are both dependent on k. Additionally, S
20 is introduced as an amplitude reduction factor due to shake-up/shake-off processes at the central atom(s). This factor can be set for fits, on the basis of fits to model compounds. Thus, the following EXAFS equation is used to fit the experimental Fourier isolates using N, R, and σ
2 as variable parameters,
which reflects losses due to inelastic scattering, where λ
j
(k) is the electron mean free path. Thus, the EXAFS contribution from each backscattering atom j is a damped sine wave in k-space, with an amplitude, and a phase, which are both dependent on k. Additionally, S
20 is introduced as an amplitude reduction factor due to shake-up/shake-off processes at the central atom(s). This factor can be set for fits, on the basis of fits to model compounds. Thus, the following EXAFS equation is used to fit the experimental Fourier isolates using N, R, and σ
2 as variable parameters,
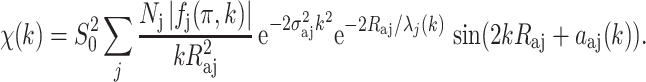 |
6 |
From the phase of each sine wave [2kR aj + α aj(k)], the absorber–backscatterer distance R aj can be determined if the phase shift α aj(k) is known. The phase shift is obtained either from theoretical calculations or empirically from compounds characterized by crystallography with the specific absorber–backscatterer pair of atoms. The phase shift α aj(k) depends on both the absorber and the scatterer atoms. As one knows the absorbing atom in an EXAFS experiment, an estimation of the phase shift can be used in identifying the scattering atom.
The amplitude function contains the Debye–Waller factor and N j, the number of backscatterers at R aj. These two parameters are highly correlated, which makes the determination of N j difficult. The backscattering amplitude function f j(π, k) depends on the atomic number of the scattering atom, and scattering intensity increases with the electron density (i.e., atomic number) of the scattering atom. In principle, this can be used to identify the scattering atoms. In practice, however, the phase shift and backscattering amplitude function, both of which are dependent on the identity of the backscattering atom, can be used only to identify scattering atoms that are well separated by atomic number (Rehr and Albers 2000).
The EXAFS fit-quality is evaluated using two different parameters Φ and ε 2.
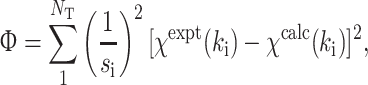 |
7 |
where N
T is the total number of data points collected, 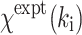 is the experimental EXAFS amplitude at k
i, and
is the experimental EXAFS amplitude at k
i, and 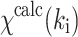 is the theoretical EXAFS amplitude at k
i. The normalization factor s
i is given by
is the theoretical EXAFS amplitude at k
i. The normalization factor s
i is given by
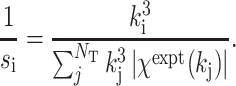 |
8 |
The ε 2 error takes into account the number of variable parameters p in the fit and the number of independent data points N ind is given by
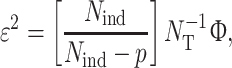 |
9 |
which is estimated from the Nyquist sampling theorem,
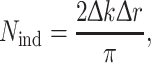 |
10 |
where ∆k is the k-range of the data and ∆r is the width of the Fourier-filtered peak in Å. ε 2 provides a gauge of whether the addition of another shell to the fit is justified. A detailed description of error analysis is presented by Lytle et al. (1989).
The importance of the EXAFS technique to the biochemist or structural biologist depends directly on the fact that the EXAFS modulations contain information about the distance between the absorbing and backscattering atoms within a distance of about 5 Å, as well as the identity and number of the backscattering atoms. Essentially, EXAFS analysis is used to determine the radial distribution of atoms around a particular absorbing atom, thus providing a probe for the local structure in the vicinity of the absorbing atom; for example, the metal in the active site of an enzyme. These vector lengths (distances) can be determined to a precision of 0.02 Å and much more precisely than by conventional X-ray crystallography.
Advantages and limitations of XAS
We summarized the advantages and the limitations (Eisenberger and Brown 1979) of the XAS method as follows.
Advantages
X-ray absorption spectroscopy (XAS) is element specific, so one can focus on one element without interference from other elements present in the sample. In a protein, which has more than one metal like cytochrome oxidase (Cu and Fe), or nitrogenase (Fe and Mo), it is possible to study the structural environment of each metal atom selectively. The element specificity and the fact that it is always possible to obtain an X-ray spectrum of an element also means that one ‘sees’ all of the metal of interest, which is present in the sample. This makes it imperative that one is sure of the biochemical homogeneity of the sample and, if there is more than one site for the same metal, to resolve the structural parameters of the different sites.
Another important advantage of XAS is that the metal of interest is never ‘silent’ with respect to X-ray absorption spectra. The system could be ‘silent’ with respect to EPR, optical, or other spectroscopic methods, but one can always probe the metal site structure by XAS.
-
X-ray absorption spectroscopy (XAS) is not limited by the state of the sample, because it is sensitive only to the local metal site structure. The sample can be prepared as a powder, a solution or, as is done most often, as a frozen solution for biological samples. It is not necessary to obtain single crystals of the material to examine the local structure of the metal. However, having oriented crystals such as membranes and single crystals significantly increases the structural information obtained from the XAS method. This will be discussed in a latter section.
The more important aspect is that one can either trap intermediates in the enzymatic cycle or modify the site by the addition of inhibitors or substrate or generate other chemical modifications. Such samples can be made as frozen solutions, avoiding the problems of trying to obtain single crystals. The study by this technique of trapped intermediates and treated samples has yielded insights into the mechanism of the reaction involved, in several biological systems.
Damage to biological samples by X-rays is cause for serious concern for X-ray crystallography and XAS experiments. However, with the right precautions one can successfully perform these experiments leaving the materials largely intact. The most serious damage is produced by the reaction with free radicals and hydrated electrons that are produced in biological samples by X-rays. The diffusion of the free radicals and hydrated electrons can be minimized by the use of low temperatures. The use of a liquid He flow cryostat or liquid He cryostream, where the samples are at atmospheric pressure in a He gas atmosphere, has greatly reduced the risk of sample damage by X-rays. XAS experiments require a lower X-ray dose than X-ray crystallography, and radiation damage can be precisely monitored and controlled, thus allowing for data collection from an intact metal cluster (Yano et al. 2005b; Corbett et al. 2007).
Limitations
It is also important to realize the intrinsic limitations of EXAFS, beyond those of a purely experimental nature. A frequent problem is the inability to distinguish between scattering atoms with little difference in atomic number (C, N, O or S, Cl, or Mn, Fe). Care must also be exercised when deciding between atoms that are apart in Z, as frequently, it is possible to obtain equally good fits using backscattering atoms which are very different in Z (e.g., Mn or Cl), but which are at different distances from the absorbing atom. This is more acute when dealing with Fourier peaks at greater distances. In bridged multinuclear centers, it is not always possible to unequivocally assign the Fourier peaks at >3 Å (Scott and Eidsness 1988).
Distances are usually the most reliably determined structural parameters from EXAFS. But the range of data that can be collected, often-times due to practical reasons like the presence of the K-edge of another metal, limits the resolution of distance determinations to between 0.1 and 0.2 Å. Also it is difficult to determine whether a Fourier peak should be fit to one distance with a relatively large disorder parameter or to two distances, each having a small disorder parameter. Careful statistical analysis, taking into consideration the degrees of freedom in the fits, should precede any such analysis. The resolution in the distance Δr can be estimated from the relation that ΔrΔk ~ 1 (see “Range-extended XAS”).
Determination of coordination numbers or number of backscatterers is fraught with difficulties. The Debye–Waller factor is strongly correlated with the coordination number and one must have recourse to other information to narrow the range that is possible from curve-fitting analysis alone. It is useful to compare the spectra from the unknown complex to some known model complexes (assuming that there is evidence that the structure resembles that of the model complex) and then use Debye–Waller parameters obtained from the model complexes in the fits. This method works reasonably well, when the structure of the system being studied is well-modeled by inorganic complexes.
X-ray absorption spectroscopy studies of photosystem II
One of the advantages of XAS is that one can potentially study the chemical events from each element which is involved in the reaction. In the OEC, Mn, Ca, and possibly Cl are the key elements we can focus on, in order to obtain the mechanistic information during the catalytic cycle. The XAS results, with emphasis on results from our laboratory, will be used to highlight the utility of the technique for the study of the Mn4Ca cluster in PS II.
Mn XAS
The geometric and electronic structural changes of the OEC have been studied intensively using Mn XAS. Figure 3 shows the Mn K-edge spectrum of each S-state of spinach PS II after deconvolution of the spectra obtained from consecutive flash illumination into pure S-state spectra, and their second derivative spectra (Messinger et al. 2001). Traditionally, the inflection point of the rising Mn K main edge (electron 1s to 4p transition) has been used as an indicator of the oxidation states in the field of XAS. The edge positions for each of the S-states have been quantitated by measuring the inflection point energy (IPE), given by the zero-crossing of the second derivative. Extensive model compound studies have shown that, when Mn is oxidized by one electron in a set of Mn model compounds with similar ligands, the IPE shifts 1–2 eV to higher energy (Visser et al. 2001). Clear differences in absorption edge energy attributed to Mn oxidation were seen in the S0 → S1 and S1 → S2 transitions in the OEC, but the absorption edges for S2 and S3 did not show a significant difference. These results were taken to indicate the absence of Mn oxidation during the S2 → S3 transition, although different interpretation exists. However, one has to be aware that the edge position cannot be simply an indicator of only the oxidation state and it is problematic to conclude oxidation state changes based only on the XANES inflection point. Due to the size of the metal 4p orbital, this orbital overlaps with p orbitals of the ligands, either through σ- or π-bonding. Consequently, XANES is sensitive not only to the oxidation state but also to the ligand environment of the metal. Additionally, no definite theory is available for calculating main K-edge spectra for transition-metal complexes, owing to several factors that affect the metal p-density. Information about metal–atom oxidation state changes that is less influenced by the nature of coordinating ligands is obtained from the XES (see the next article by Bergmann and Glatzel, this issue).
Fig. 3.

a The Mn K-edge spectra of spinach PS II (BBY), from the S0 through S3 states (top) and their second derivative spectra (bottom). The magnitude of the inflection point energy shift for the S0 to S1 (2.1 eV) and S1 and S2 (1.1 eV) is much larger than the shift for the S2 to S3 transition (0.3 eV). The inset shows the pre-edge (1s to 3d transition) from the S-states is enlarged and shown above the Mn K-edge spectra. b The Fourier transform (FT) from a PS II sample in the S1 state. The three FT Peak I corresponds to Mn-bridging and terminal ligand (N/O) distances at 1.8–2.0 Å, Peak II is from Mn–Mn distances (2 at ~2.7 and 1 at ~2.8 Å), and FT Peak III is from Mn–Mn distance at ~3.3 Å and Mn–Ca distances at ~3.4 Å
The EXAFS is interpretable as shells at 1.8 and 2.0 Å (Peak I) attributable to N or O atoms and a shell at ~2.7–2.8 Å (Peak II) from Mn to Mn interactions. An additional shell from Mn was seen at 3.3 Å (Peak III; Fig. 3). The Mn EXAFS spectra changes upon the S-state transitions, particularly from the S2 to S3 state transition, suggesting that the OEC goes through structural changes triggered by the oxidation state changes and protonation/deprotonation events.
Co-factor XAS
The S-state catalytic cycle can be studied also by co-factor XAS studies (Cinco et al. 2002). One Ca is known to be a part of the OEC, and this has been proven by Ca XAS studies and from X-ray crystallography using the anomalous diffraction technique. Regarding Cl, there is no spectroscopic evidence at least in the S1 state that the Cl is a direct ligand to the OEC, although several biochemical studies suggest a critical role for one tightly bound Cl in maintaining oxygen-evolving activity.
In general, the requirements of X-ray spectroscopy place some restrictions with respect to sample preparation and experimental conditions. Ca and Cl in some sense fall into this category. The investigation of light elements can present difficulties due to the presence of an aqueous medium and the pervasive occurrence of C, N, and O in biological materials. In X-ray energy regions, where atmospheric gases absorb, samples must be placed in an atmosphere of helium or in vacuum. For elements like Ca and Cl, which can occur in a wide variety of environments in biological materials, it is particularly challenging to remove sources of background signals that greatly complicate interpreting the results.
Another strategy to study the role of such light element co-factor(s) is to replace it with heavier element(s). Ca can be replaced chemically or biosynthetically with Sr without losing its enzymatic activity. Similarly, Cl can be substituted with Br. XAS measurements at the Sr K-edge (16,200 eV; Cinco et al. 1998; Pushkar et al. 2008) or Br K-edge (13,600 eV; Haumann et al. 2006) compared to those at lower-energy Ca K-edge (~4,050 eV) or Cl K-edge (~2,850 eV) are less prone to X-ray damage, if we compare the X-ray damage by the total number of X-ray photons per area at each energy. It is also preferred for experimental reasons as X-rays at higher energies are attenuated less by the air path, the buffer solution in which the sample is made, and the cryostat windows.
Range-extended XAS
In general, EXAFS spectra of systems which contain adjacent elements in the periodic table have a limited EXAFS range due to the presence of the rising edge of the next element, thus limiting the EXAFS distance resolution. For the Mn K-edge EXAFS studies of PS II, the absorption edge of Fe in PS II limits the EXAFS energy range (Fig. 4). Traditional EXAFS spectra of PS II samples are collected as an excitation spectrum by electronically windowing the Kα fluorescence (2p to 1s, at 5,899 eV) from the Mn atom. The solid-state detectors that have been used over the past decade have a resolution of about 150–200 eV (FWHM) at the Mn K-edge, making it impossible to discriminate Mn fluorescence from that of Fe Kα fluorescence (at 6,404 eV). The presence of the obligatory 2–3Fe/PS II (Fe edge at 7,120 eV) limits, the data to a k-range of ~11.5 Å−1 (k = 0.51 ΔE1/2, the Mn edge is at 6,540 eV and ΔE = 580 eV). The Mn–Mn and Mn–ligand distances that can be resolved in a typical EXAFS experiment are given by
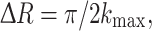 |
11 |
where k max is the maximum energy of the photoelectron of Mn.
Fig. 4.

a Left (top): X-ray fluorescence of Mn and Fe. The multi-crystal monochromator with 1 eV resolution is tuned to the Mn Kα1 peak (red spectrum). Left (below): fluorescence peaks of Mn and Fe as detected using Ge-detector. The fluorescence peaks are convoluted with the electronic window resolution of 150–200 eV of the Ge-detector (black and green spectra for Mn and Fe fluorescence). Note different energy scales for the schemes shown above and below. Iron is an obligatory element in functional PS II complexes. Right: Comparison of the traditional Mn K-edge EXAFS spectrum (blue) from the S1 state PS II sample obtained with a traditional 30-element energy-discriminating Ge-detector with a spectrum collected using the high-resolution crystal monochromator (note the absence of Fe contribution). The dashed line at k = 11.5 Å−1 denotes the spectral limit of a conventional EXAFS experiment owing to the iron edge. Use of the high-resolution detector eliminates the interference of Fe and removes the limit of the energy range for Mn EXAFS data collection. b The comparison of the k-space Mn EXAFS collected with a crystal monochromator and a Ge-detector. The range of data, as indicated by k max, is inversely proportional to the resolution of the data
The use of a high-resolution crystal monochromator (see the article by Bergmann and Glatzel, this issue) allows us to selectively separate the Mn K fluorescence from that of Fe (Fig. 4), resulting in the collection of data to higher photoelectron energies and leading to increased distance resolution of 0.1 Å. The new detection scheme produces distinct advantages: (1) improvement in the distance resolution, and (2) more precise determination in the numbers of metal–metal vectors.
These data resolve a distance heterogeneity in the short Mn–Mn distances of the S1 and S2 state and thereby provide firm evidence for three Mn–Mn distances between ~2.7 and ~2.8 Å (Yano et al. 2005a; Pushkar et al. 2007). This result gives clear criteria for selecting and refining possible structures from the repertoire of proposed models based on spectroscopic and diffraction data.
Polarized XAS
Polarized XAS studies on oriented membranes
Membrane proteins like PS II can be oriented on a substrate such that the lipid membrane planes are roughly parallel to the substrate surface. This imparts a one-dimensional order to these samples, while the z-axis for each membrane (collinear with the membrane normal) is roughly parallel to the substrate normal, the x and y axes remain disordered. Exploiting the plane-polarized nature of synchrotron radiation, spectra can be collected at different angles between the substrate normal and the X-ray E vector. The dichroism, which is the dependence of the intensity of the absorber–backscatterer pairs present in the oriented samples as a function of the polarization of the X-rays, is reflected in, and can be extracted from, the resulting X-ray absorption spectra (George et al. 1989, 1993). The EXAFS of the oriented PS II samples exhibits distinct dichroism, from which we have deduced the relative orientations of several interatomic vector directions relative to the membrane normal and derived a topological representation of the metal sites in the OEC (Mukerji et al. 1994; Dau et al. 1995; Cinco et al. 2004; Pushkar et al. 2007).
To a first order approximation, the angle dependence of the EXAFS is proportional to cos2(θ ER), with θ ER being the angle between the X-ray electric field vector (E) and the absorber–backscatter vector (R) (Fig. 5a). In turn, θ ER is composed of the detection angle θ and the angle ϕ between R and M, the membrane normal. Due to the rotational symmetry of the layered membranes, the angle ϕ defines a cone around the membrane normal M. When membranes are layered on a flat substrate, the preferential orientation of M is parallel to the underling substrate normal (S). For those imperfectly stacked sheets, the probability (P α) of finding an angle α between M and S is the product of sinα and the order function P ord(α), which is maximal at α = 0°. P ord(α) is approximated by a Gaussian distribution whose half-width is the mosaic spread (Ω) or the disorder angle. Here, the mosaic spread is assumed to account for the disorder between the membrane normal and substrate normal, while the spread of R relative to M is negligible. For an ensemble of A–B vectors (R), the magnitude of the EXAFS is related to the P α-weighted integration over all possible orientations of M (α- and β-integration) and along the cone of the possible directions of R (γ-integration).
Fig. 5.

a Scheme for the vectors and angles involved in X-ray absorption linear dichroism of one-dimensionally oriented (PS II) membrane fragments on a flat surface. For the polarized EXAFS experiment, spectra are measured for several values of θ (angle between the X-ray electric field vector E and the substrate normal S); θ ER is the angle between, E and the absorber–scatterer vector, R. θER is composed of the detection angle θ and the angle ϕ between R and M, the absorber–backscatterer vector and the membrane normal. Because of the rotational symmetry of the layered membranes, the angle ϕ defines a cone around the membrane normal, M. When membranes are layered on a flat substrate, the preferential orientation of M is parallel to the underlying substrate normal, S. For an ensemble of R vectors, the magnitude of the EXAFS is related to the P α-weighted integration over all possible orientations of M (α- and β-integration) and along the cone of possible directions of R (γ-integration). b Mn K-edge EXAFS spectra (k 3-weighted) from oriented PS II membrane samples in the S1 state obtained with a high-resolution spectrometer (range-extended EXAFS) at orientations of 15° (green solid line) and 75° (red dashed line) of the sample normal with respect to the X-ray E-vector. The orientation of the X-ray E-vector with respect to the membrane normal is shown as an inset. c The structural information from the dichroism of FT peak III is illustrated showing the orientation of the average Mn–Ca vector in relation to the Mn–Mn vector. The cones represent a range for the average Mn–Ca vector(s) along the membrane normal, and the Mn–Mn vector toward the membrane plane, respectively
The N app found from EXAFS curve-fitting on oriented samples at particular θ is related to the coordination number of an isotropic sample N iso by the following equation:
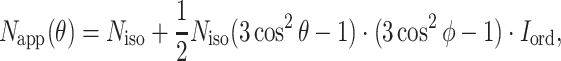 |
12 |
where I ord is the order integral:
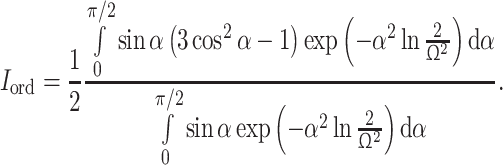 |
13 |
By fitting the θ-dependence of N app by nonlinear regression analysis, the average relative orientation ϕ and N app can be obtained.
Figure 5b shows the orientation of the membranes with respect to the X-ray E-vector and an example of the polarized spectrum from PS II.
However, as the samples are ordered in only one dimension, the dichroism information is available only in the form of an angle with respect to the membrane normal. For EXAFS measurements, this means that the absorber–backscatterer vectors can lie anywhere on a cone defined by the angle the vector forms with the membrane normal as shown in Fig. 5c.
Polarized XAS studies using single crystals of PS II
Further refinement can be performed if samples with three-dimensional order, i.e., single crystals, are examined instead of oriented membranes. Single-crystal X-ray spectroscopy has been performed on model complexes (Pickering and George 1995) and metallo-proteins (Scott et al. 1982; Flank et al. 1986; George et al. 1999). These studies have been able to significantly expand the X-ray absorption spectroscopic information available for these systems over what is gleaned from studies of isotropic samples. An example of polarized XANES and EXAFS spectra from a Mn(V) complex is shown in Fig. 6a and b.
Fig. 6.

Polarized Mn XAS spectra of Mn(V)-oxo compound (inset). a Polarized XANES spectra. The pre-edge peak is most intense when the X-ray E-vector is parallel to the Mn-oxo bond. b Polarized EXAFS spectra in the two extreme orientations. The distinct dichroism in the XANES and EXAFS spectra show the utility of the polarized XAS methodology
This type of analysis can also be useful for systems, where a high-resolution X-ray crystal structure is not available, such as PS II. Examination of the orientation dependence of the EXAFS of single crystals will provide structural information about the Mn sites at resolution higher than will be practically obtainable from single-crystal X-ray diffraction. Performing single-crystal EXAFS experiments can help to refine the low-resolution structure of the OEC by revealing information such as the angle(s) between the di-μ-oxo-bridged Mn–Mn vectors (~2.7 Å), as well as the relative orientation between the mono-μ-oxo Mn–Mn vector (~3.3 Å) and the di-μ-oxo-bridged Mn–Mn vectors. The directions of the Mn–Mn vectors in conjunction with the electron density derived from X-ray crystallography promises to refine the structure of the Mn complex to a resolution that neither method has presently achieved.
Figure 7 shows the experimental setup for collecting single-crystal XAS data from PS II at SSRL BL 9-3. It consists of a kappa goniometer, a 30-element Ge-detector for collecting XAS data, and a CCD or a MAR 345 imaging plate detector placed behind the sample for in situ collection of diffraction data to determine of the crystal orientation. The crystals are cooled using a liquid He cryostream.
Fig. 7.

X-ray spectroscopy and diffraction set-up for PS II single crystals. The MAR345 is behind the sample, which is cooled by a liquid He cryostream to 10 K. The 30-element Ge-detector is perpendicular to the direction of the beam
We have shown that the polarized EXAFS data from the single crystals of PS II improve the resolution of the distances and the determination of the directions of the vectors of the Mn complex, thus leading to a more refined structure of the Mn cluster (Yano et al. 2006).
Mn K-edge pre-edge spectra and DFT calculations
In “X-ray absorption spectroscopy studies of photosystem II”, we discussed the oxidation states of the OEC obtained from Mn XANES. In order to obtain a more detailed view of the electronic structure at the metal site, it is preferable to probe the lowest unoccupied metal 3d orbitals. The pre-edge spectra arise from excitations of 1s electron into 3d orbitals that are mainly localized around the metal ion. It shows the immediate surrounding of the excited ion through the Coulomb interaction between the core hole and the valence electrons within a short range. This pre-edge feature is a quadrupole-allowed transition; it occurs at a lower energy than the main edge transitions with approximately 1% of the intensity of the dipole-allowed main-edge transition. The transition can gain intensity by the metal 4p mixing, when the metal–ligand environment is distorted from a centro-symmetric to a non-centro-symmetric coordination. The spectra reflect coordination number, ligand environment, and oxidation state of metals. In fact, the pre-edge spectra of PS II noticeably change during the S-state transitions (Messinger et al. 2001). In the single-crystal XANES of PS II S1 state, the pre-edge spectra show a characteristic dichroism (Yano et al. 2006). Additionally, the nature of the S4 state can be studied by the pre-edge feature if a high-valent Mn, such as Mn(V), is involved in the transition. In order to understand the pre-edge feature and obtain the electronic configuration, however, one needs to investigate various model compounds and combine experimental data with theoretical calculations based on the ligand field and/or Density-functional theories.
Figure 8a shows the solution pre-edge spectrum of a five-coordinated Mn(V)-oxo model complex (Yano et al. 2007; the polarized XANES of the same complex is shown in Fig. 6a). Due to the strong axial distortion of the Mn site symmetry from the octahedral environment, a formally forbidden pre-edge (1s to 3d) transition gains intensity through a 3d z–4p z mixing mechanism and a strong pre-edge peak is observed. However, the pre-edge intensity is sensitive to the ligand environment as demonstrated in Fig. 8b by time-dependent DFT calculation of the theoretical models, in which the addition of the sixth ligand is investigated. The addition of a weak sixth ligand like water weakens the pre-edge intensity by a factor of ~2, while the addition of a stronger ligand, such as hydroxide or carboxylate, weaken the peak intensity by a factor of ~5 relative to the five-coordinated Mn(V) compounds.
Fig. 8.

Comparison of the TD-DFT calculated Mn K-edge spectra of the Mn(V)-oxo(DCB) complex (top) as compared to Mn(V)-oxo(H2O)4, Mn(V)-oxo(H2O)5, Mn(V)-oxo(H2O)4(OH), and Mn(V)-oxo(H2O)4(CH3COO) (bottom). The effect of the addition of a sixth ligand and/or lengthening of the Mn–O ligand distance on the intensity of the pre-edge feature is significant
New directions
Several unresolved problems are being addressed through new directions in XAS that are being pursued in different laboratories.
Time-dependent XAS with sub-μs time resolution opens the possibility of identifying and characterizing intermediates in the individual S-state transitions that have not yet been documented (Haumann et al. 2005). Of particular interest is the series of events on the ms time scale that accompany the formation of dioxygen during the S4 to S0 transition.
The combination of XAS with X-ray microscopy has shown great promise in studying very small localized domains of larger biological systems, and the possibility for combining imaging with spectroscopy. Another powerful approach has been the combined in situ use of XAS along with other methods, such as X-ray diffraction, electrochemistry, UV/Vis or FTIR/Raman spectroscopy. This methodology has allowed for monitoring of changes in the system and also the integrity of the sample. These methodologies are being applied to substrate binding studies and for following the course of catalytic reactions.
Acknowledgments
The research presented here was supported by the NIH Grant GM 55302, and by the Director, Office of Science, Office of Basic Energy Sciences (OBES), Division of Chemical Sciences, Geosciences, and Biosciences of the Department of Energy (DOE) under Contract DE-AC02-05CH11231. Synchrotron facilities were provided by the Stanford Synchrotron Radiation Laboratory (SSRL), the Advanced Light Source (ALS), and the Advanced Photon Source (APS) operated by DOE OBES. The SSRL Biomedical Technology program is supported by NIH, the National Center for Research Resources (NCRR), and the DOE Office of Biological and Environmental Research.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- PS II
Photosystem II
- XAS
X-ray absorption spectroscopy
- EXAFS
X-ray absorption fine structure
- EPR
Electron paramagnetic resonance
- XANES
X-ray absorption near-edge structure
Contributor Information
Junko Yano, Phone: +1-510-4864366, FAX: +1-510-4867668, Email: jyano@lbl.gov.
Vittal K. Yachandra, Phone: +1-510-4864963, FAX: +1-510-4867668, Email: vkyachandra@lbl.gov
References
- Cinco RM, Robblee JH, Rompel A, Fernandez C, Yachandra VK, Sauer K, Klein MP. Strontium EXAFS reveals the proximity of calcium to the manganese cluster of oxygen-evolving photosystem II. J Phys Chem B. 1998;102:8248–8256. doi: 10.1021/jp981658q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinco RM, Rompel A, Visser H, Aromi G, Christou G, Sauer K, Klein MP, Yachandra VK. Comparison of the manganese cluster in oxygen-evolving photosystem II with distorted cubane manganese compounds through X-ray absorption spectroscopy. Inorg Chem. 1999;38:5988–5998. doi: 10.1021/ic991003j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinco RM, Holman KLM, Robblee JH, Yano J, Pizarro SA, Bellacchio E, Sauer K, Yachandra VK. Calcium EXAFS establishes the Mn-Ca cluster in the oxygen-evolving complex of photosystem II. Biochemistry. 2002;41:12928–12933. doi: 10.1021/bi026569p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinco RM, Robblee JH, Messinger J, Fernandez C, Holman KLM, Sauer K, Yachandra VK. Orientation of calcium in the Mn4Ca cluster of the oxygen-evolving complex determined using polarized strontium EXAFS of photosystem II membranes. Biochemistry. 2004;43:13271–13282. doi: 10.1021/bi036308v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett MC, Latimer MJ, Poulos TL, Sevrioukova IF, Hodgson KO, Hedman B. Photoreduction of the active site of the metalloprotein putidaredoxin by synchrotron radiation. Acta Crystallogr Sect D. 2007;63:951–960. doi: 10.1107/S0907444907035160. [DOI] [PubMed] [Google Scholar]
- Dau H, Andrews JC, Roelofs TA, Latimer MJ, Liang W, Yachandra VK, Sauer K, Klein MP. Structural consequences of ammonia binding to the manganese cluster of the photosynthetic oxygen-evolving complex: an X-ray absorption study of isotropic and oriented photosystem II particles. Biochemistry. 1995;34:5274–5287. doi: 10.1021/bi00015a043. [DOI] [PubMed] [Google Scholar]
- Eisenberger P, Brown GS. Study of disordered systems by EXAFS: limitations. Solid State Commun. 1979;29:481–484. doi: 10.1016/0038-1098(79)90790-7. [DOI] [Google Scholar]
- Eisenberger P, Kincaid BM. EXAFS: new horizons in structure determinations. Science. 1978;200:1441–1447. doi: 10.1126/science.663627. [DOI] [PubMed] [Google Scholar]
- Flank AM, Weininger M, Mortenson LE, Cramer SP. Single-crystal EXAFS of nitrogenase. J Am Chem Soc. 1986;108:1049. doi: 10.1021/ja00265a034. [DOI] [Google Scholar]
- George GN, Prince RC, Cramer SP. The manganese site of the photosynthetic water-splitting enzyme. Science. 1989;243:789–791. doi: 10.1126/science.2916124. [DOI] [PubMed] [Google Scholar]
- George GN, Cramer SP, Frey TG, Prince RC. X-ray absorption spectroscopy of oriented cytochrome oxidase. Biochim Biophys Acta. 1993;1142:240–252. doi: 10.1016/0005-2728(93)90152-6. [DOI] [PubMed] [Google Scholar]
- George GN, Pickering IJ, Kisker C. X-ray absorption spectroscopy of chicken sulfite oxidase crystals. Inorg Chem. 1999;38:2539. doi: 10.1021/ic981451b. [DOI] [Google Scholar]
- Haumann M, Liebisch P, Muller C, Barra M, Grabolle M, Dau H. Photosynthetic O2 formation tracked by time-resolved X-ray experiments. Science. 2005;310:1019–1021. doi: 10.1126/science.1117551. [DOI] [PubMed] [Google Scholar]
- Haumann M, Barra M, Loja P, Loscher S, Krivanek R, Grundmeier A, Andreasson LE, Dau H. Bromide does not bind to the Mn4Ca complex in its S1 state in Cl−-depleted and Br−-reconstituted oxygen-evolving photosystem II: evidence from X-ray absorption spectroscopy at the Br K-edge. Biochemistry. 2006;45:13101–13107. doi: 10.1021/bi061308r. [DOI] [PubMed] [Google Scholar]
- Koningsberger DC, Prins R, editors. X-ray absorption: principles, applications, techniques of EXAFS, SEXAFS and XANES. New York: Wiley; 1988. [Google Scholar]
- Latimer MJ, DeRose VJ, Mukerji I, Yachandra VK, Sauer K, Klein MP. Evidence for the proximity of calcium to the manganese cluster of photosystem II: determination by X-ray absorption spectroscopy. Biochemistry. 1995;34:10898–10909. doi: 10.1021/bi00034a024. [DOI] [PubMed] [Google Scholar]
- Lytle FW, Sayers DE, Stern EA. Report of the international workshop on standards and criteria in X-ray absorption-spectroscopy (1988), Brookhaven National Laboratory. Physica B. 1989;158:701–722. doi: 10.1016/0921-4526(89)90351-7. [DOI] [Google Scholar]
- Messinger J, Robblee JH, Bergmann U, Fernandez C, Glatzel P, Visser H, Cinco RM, McFarlane KL, Bellacchio E, Pizarro SA, Cramer SP, Sauer K, Klein MP, Yachandra VK. Absence of Mn-centered oxidation in the S2 → S3 transition: implications for the mechanism of photosynthetic water oxidation. J Am Chem Soc. 2001;123:7804–7820. doi: 10.1021/ja004307+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukerji I, Andrews JC, Derose VJ, Latimer MJ, Yachandra VK, Sauer K, Klein MP. Orientation of the oxygen-evolving manganese complex in a photosystem-II membrane preparation: an X-ray-absorption spectroscopy study. Biochemistry. 1994;33:9712–9721. doi: 10.1021/bi00198a042. [DOI] [PubMed] [Google Scholar]
- Penner-Hahn JE. Structural characterization of the Mn site in the photosynthetic oxygen-evolving complex. Struct Bond. 1998;90:1–36. [Google Scholar]
- Pickering IJ, George GN. Polarized X-ray-absorption spectroscopy of cupric chloride dihydrate. Inorg Chem. 1995;34:3142–3152. doi: 10.1021/ic00116a004. [DOI] [Google Scholar]
- Pizarro SA, Glatzel P, Visser H, Robblee JH, Christou G, Bergmann U, Yachandra VK. Mn oxidation states in tri- and tetra-nuclear Mn compounds structurally relevant to photosystem II: Mn K-edge X-ray absorption and Kβ X-ray emission spectroscopy studies. Phys Chem Chem Phys. 2004;6:4864–4870. doi: 10.1039/B407513G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushkar Y, Yano J, Glatzel P, Messinger J, Lewis A, Sauer K, Bergmann U, Yachandra V. Structure and orientation of the Mn4Ca cluster in plant photosystem II membranes studied by polarized range-extended X-ray absorption spectroscopy. J Biol Chem. 2007;282:7198–7208. doi: 10.1074/jbc.M610505200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushkar Y, Yano J, Sauer K, Boussac A, Yachandra VK. Structural changes in the Mn4Ca cluster and the mechanism of photosynthetic water splitting. Proc Natl Acad Sci USA. 2008;105:1879–1884. doi: 10.1073/pnas.0707092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehr JJ, Albers RC. Theoretical approaches to X-ray absorption fine structure. Rev Mod Phys. 2000;72:621–654. doi: 10.1103/RevModPhys.72.621. [DOI] [Google Scholar]
- Sauer K, Yano J, Yachandra VK. X-ray spectroscopy of the photosynthetic oxygen-evolving complex. Coord Chem Rev. 2008;252:318–335. doi: 10.1016/j.ccr.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers DE, Stern EA, Lytle F. New technique for investigating noncrystalline structures: Fourier analysis of the extended X-ray-absorption fine structure. Phys Rev Lett. 1971;27:1204–1207. doi: 10.1103/PhysRevLett.27.1204. [DOI] [Google Scholar]
- Scott RA, Eidsness MK. The use of X-ray absorption spectroscopy for detection of metal-metal interactions. Application to copper-containing enzymes. Comments Inorg Chem. 1988;7:235–267. doi: 10.1080/02603598808072311. [DOI] [Google Scholar]
- Scott RA, Hahn JE, Doniach S, Freeman HC, Hodgson KO. Polarized X-ray absorption spectra of oriented plastocyanin single crystals. Investigation of methionine-copper coordination. J Am Chem Soc. 1982;104:5364–5369. doi: 10.1021/ja00384a020. [DOI] [Google Scholar]
- Shulman RG, Yafet Y, Eisenberger P, Blumberg WE. Observation and interpretation of X-ray absorption edges in iron compounds and proteins. Proc Natl Acad Sci USA. 1976;73:1384–1388. doi: 10.1073/pnas.73.5.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo BK. EXAFS: basic principles and data analysis. Berlin: Springer; 1986. [Google Scholar]
- Visser H, Anxolabehere-Mallart E, Bergmann U, Glatzel P, Robblee JH, Cramer SP, Girerd JJ, Sauer K, Klein MP, Yachandra VK. Mn K-edge XANES and Kβ XES studies of two Mn-oxo binuclear complexes: investigation of three different oxidation states relevant to the oxygen-evolving complex of photosystem II. J Am Chem Soc. 2001;123:7031–7039. doi: 10.1021/ja004306h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachandra VK. The catalytic manganese-cluster: organization of the metal ions. In: Wydrzynski T, Satoh S, editors. Photosystem II: the light-driven water: plastoquinone oxidoreductase. Dordrecht: Springer; 2005. pp. 235–260. [Google Scholar]
- Yachandra VK, Sauer K, Klein MP. Manganese cluster in photosynthesis: where plants oxidize water to dioxygen. Chem Rev. 1996;96:2927–2950. doi: 10.1021/cr950052k. [DOI] [PubMed] [Google Scholar]
- Yano J, Yachandra VK. Oxidation state changes of the Mn4Ca cluster in photosystem II. Photosynth Res. 2007;92:289–303. doi: 10.1007/s11120-007-9153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J, Yachandra VK. Where water is oxidized to dioxygen: Structure of the photosynthetic Mn4Ca cluster from X-ray spectroscopy. Inorg Chem. 2008;47:1711–1726. doi: 10.1021/ic7016837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J, Pushkar Y, Glatzel P, Lewis A, Sauer K, Messinger J, Bergmann U, Yachandra VK. High-resolution Mn EXAFS of the oxygen-evolving complex in photosystem II: structural implications for the Mn4Ca cluster. J Am Chem Soc. 2005;127:14974–14975. doi: 10.1021/ja054873a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J, Kern J, Irrgang K-D, Latimer MJ, Bergmann U, Glatzel P, Pushkar Y, Biesiadka J, Loll B, Sauer K, Messinger J, Zouni A, Yachandra VK. X-ray damage to the Mn4Ca complex in photosystem II crystals: a case study for metallo-protein X-ray crystallography. Proc Natl Acad Sci USA. 2005;102:12047–12052. doi: 10.1073/pnas.0505207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J, Kern J, Sauer K, Latimer M, Pushkar Y, Biesiadka J, Loll B, Saenger W, Messinger J, Zouni A, Yachandra VK. Where water is oxidized to dioxygen: structure of the photosynthetic Mn4Ca cluster. Science. 2006;314:821–825. doi: 10.1126/science.1128186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J, Robblee J, Pushkar Y, Marcus MA, Bendix J, Workman JM, Collins TJ, Solomon EI, George SD, Yachandra VK. Polarized X-ray absorption spectroscopy of single-crystal Mn(V) complexes relevant to the oxygen-evolving complex of photosystem II. J Am Chem Soc. 2007;129:12989–13000. doi: 10.1021/ja071286b. [DOI] [PMC free article] [PubMed] [Google Scholar]