

Abstract
In the last two decades, Magic Angle Spinning (MAS) NMR has created its own niche in studies involving photosynthetic membrane protein complexes, owing to its ability to provide structural and functional information at atomic resolution of membrane proteins when in the membrane, in the natural environment. The light-harvesting two (LH2) transmembrane complex from Rhodopseudomonas acidophila is used to illustrate the procedure of the technique applicable in photosynthesis research. One- and two-dimensional solid-state NMR experiments involving 13C- and 15N-labeled LH2 complexes allow to make a sequence-specific assignment of NMR signals, which forms the basis for resolving structural details and the assessment of charge transfer, electronic delocalization effects, and functional strain in the ground state.
Keywords: Magic angle spinning NMR, LH2 complex, Electronic structure, Membrane protein, Isotope labeling
Introduction
Solid-state magic angle spinning (MAS) NMR provides a versatile method for the determination of structure for ordered systems without translation symmetry, such as proteins, macromolecular complexes, aggregates, or membrane systems. With the continued difficulty in crystallizing membrane proteins, solid-state NMR spectroscopy is becoming an important method in the analysis of this important class of proteins. For MAS NMR, protein crystallization is not necessary. The homogeneous environment in the protein sample and a local order are sufficient. In addition, use of stable isotopes in combination with MAS NMR offers prospects for the study of larger and more complex biomolecules, such as large membrane-bound photosynthetic complexes, in their undisturbed native form. In photosynthetic research, a variety of structural details have been obtained using MAS NMR (de Groot 2008). For instance, structural and functional details from light-harvesting pigments (Boender et al. 1995; van Rossum et al. 2002), and pigment–protein complexes (Egorova-Zachernyuk et al. 2001; Alia et al. 2001, 2004; van Gammeren et al. 2004, 2005a, b; Ganapathy et al. 2007) and reaction center complexes (Prakash et al. 2005; Diller et al. 2007; Daviso et al. 2008; Alia et al. 2009) have been resolved with atomic resolution using MAS NMR in conjunction with stable-isotope labeling. Very recently, the structure of a member of the chlorosome class of light-harvesting antennae was determined and compared with the wild type (WT) to resolve how the biological light-harvesting function of the chlorosome is established, an important step on the way to artificial photosynthesis (Ganapathy et al. 2009). This article is devoted to summarize the research into the direction of comprehensive protein assessment using the LH2 antenna system as a model protein using MAS NMR. First, a brief theoretical background of the MAS NMR technique is presented. Subsequently, a variety of model experiments performed by MAS NMR for LH2 complex will be discussed to illustrate the versatility of MAS NMR as a biophysical technique in photosynthesis.
Theoretical background
MAS NMR is a technique for obtaining high resolution NMR data from solids. For an extensive introduction to the technique, the reader is referred to the existing literature (Duer 2004). This section serves to guide the interested student to this background literature. Contrary to solution NMR, where anisotropic interactions are averaged by the rapid tumbling of molecules, in solid-state NMR, interactions such as the chemical shift and dipolar coupling dominate. As a consequence, the spectral line width of nuclei in solids is rather broad. In order to overcome this problem in the solid state, MAS NMR is applied. In MAS NMR, a sample is rotated rapidly around an axis at the magic angle θ m = 54.74° with the static field (Andrew et al. 1958; Lowe 1959) to effectively suppress chemical shift broadening. In order to describe the MAS NMR experiment, the Hamiltonian
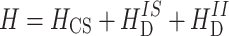 |
1 |
is used.  is the chemical shielding term,
is the chemical shielding term, 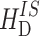 represents the heteronuclear dipolar couplings, and
represents the heteronuclear dipolar couplings, and 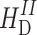 describes the homonuclear dipolar couplings. The chemical shielding affects the NMR frequency, which is determined by the Zeeman interaction
describes the homonuclear dipolar couplings. The chemical shielding affects the NMR frequency, which is determined by the Zeeman interaction
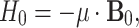 |
2 |
between a nuclear magnetic moment μ and the external static magnetic field 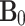 . The μ can be expressed in terms of the nuclear spin operator I as
. The μ can be expressed in terms of the nuclear spin operator I as 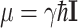 , and Eq. 2 can be rewritten as
, and Eq. 2 can be rewritten as
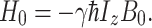 |
3 |
Although the Zeeman interaction is the most dominant interaction and generally determines the quantization (z-)axis in the theoretical description, it contains little structural information in itself and is removed from the description by a transformation to a frame that is rotating at the NMR frequency along the z-axis (Abragam 1961). In the solid-state, the nuclear spin interactions are anisotropic and can be described by second-rank tensors. This makes solid-state NMR a very rich field to explore, for the study of molecular structure and for functional spectroscopy investigations.
The chemical shielding Hamiltonian is written as
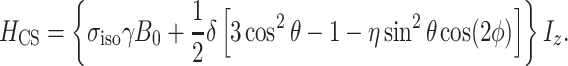 |
4 |
The chemical shielding and its anisotropy are represented by a tensor σ that is most conveniently represented in the coordinate system in which it is diagonal. This is in the principal axis system (PAS), which is an axis frame defined in such a way that the symmetric part of the shielding tensor is diagonal, and the principal values of the shielding tensor can be given as
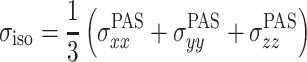 |
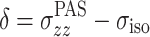 |
5 |
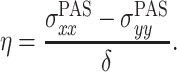 |
Here, 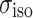 is the isotropic value, δ is the anisotropy, and η is the asymmetry parameter (Duer 2004; Schmidt-Rohr and Spiess 1994).
is the isotropic value, δ is the anisotropy, and η is the asymmetry parameter (Duer 2004; Schmidt-Rohr and Spiess 1994).
The dipolar interaction between two spins arises by virtue of the small magnetic field each spin creates around itself. The truncated heteronuclear dipolar Hamiltonian is given by
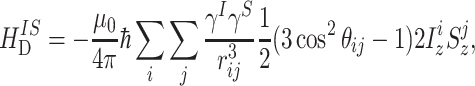 |
6 |
while the truncated homonuclear dipolar Hamiltonian is described by
 |
7 |
where r ij is the magnitude of the distance vector r ij between the nuclei i and j, and θ ij is the angle between r ij and the z-axis. In NMR, the general convention is to denote the abundant spins as the I spins and the rare spins as the S spins (Schmidt-Rohr and Spiess 1994).
The dependence on the molecular orientation in Eqs. 4, 6, and 7 is of the form (3cos2 θ − 1), where θ is the angle that describes the orientation of the spin interaction tensor, which could be the chemical shielding tensor in case of the chemical shielding interaction, or the dipolar coupling tensor in the case of the dipolar coupling interaction.
MAS is an elegant technique that averages all anisotropic interactions described by second-rank tenors, if the rotation frequency exceeds the largest coupling of the spin species considered. The experimental setup is indicated schematically in Fig. 1. A solid sample is placed in a rotor and mechanically rotated at a high frequency about an axis oriented at the magic angle of 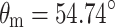 with respect to the static magnetic field B
0 (Andrew et al. 1958; Lowe 1959) yielding (3cos2
θ − 1 = 0). When the sample is spun at the magic angle, the anisotropic part produces NMR sidebands and with fast rotation, the sidebands are shifted away, and the spectrum consists of narrow lines at the isotropic shifts. Only the term σisoγB
0 in Eq. 4 remains, and high resolution spectra are obtained in solid state. In practice, the dipolar interactions
with respect to the static magnetic field B
0 (Andrew et al. 1958; Lowe 1959) yielding (3cos2
θ − 1 = 0). When the sample is spun at the magic angle, the anisotropic part produces NMR sidebands and with fast rotation, the sidebands are shifted away, and the spectrum consists of narrow lines at the isotropic shifts. Only the term σisoγB
0 in Eq. 4 remains, and high resolution spectra are obtained in solid state. In practice, the dipolar interactions 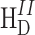 are not averaged for an abundant proton system where the chemical shift dispersion is small as compared to the dipolar interactions.
are not averaged for an abundant proton system where the chemical shift dispersion is small as compared to the dipolar interactions.
Fig. 1.

Schematic representation of the MAS technique. The spinning axis of the sample is at an angle of 54.74º (magic angle) with respect to the static magnetic field B0
Cross polarization
The elemental composition of organic and biomolecules is primarily hydrogen, carbon, nitrogen, and oxygen, of which the first three elements are spin 1/2. Proton spins having a large natural abundance also have a high gyromagnetic ratio γ, which are the two main factors that determine the sensitivity of an NMR experiment. Hence, protons have the highest sensitivity of all the naturally occurring spins. However, the homonuclear dipolar couplings between 1H spins are considerable. In addition, the topology of protons in molecules is such that they form a dense network of strongly coupled spins, with effective overall couplings of ~50 kHz. These dipolar interactions induce severe line broadening in solids. Even with MAS, high resolution 1H NMR spectroscopy is still difficult in solids.
Low abundance, e.g., for 13C and 15N, on the other hand, inevitably results in less-sensitive NMR spectra, and less signal-to-noise (S/N) ratio. In addition, the relaxation times of dilute nuclei are rather long, due to the absence of homonuclear dipolar interactions that induce relaxation transitions. In solid-state NMR, isotope labeling is often used when enhanced sensitivity is required. It is possible to further enhance the peak resolution and signal intensity in the MAS experiment by the transfer of the 1H transverse magnetization to a dilute spin species via CP in combination with high power proton decoupling (Bennett et al. 1995; Hartmann and Hahn 1962; Pines et al. 1973; Schaefer and Stejskal 1976).
The separation between the spin up and spin down energy levels for 1H exceeds the splitting for 13C, for example, given by  . The 1H polarization in the magnetic field B
0 is, therefore, larger than the 13C polarization. In the magnetic field B
0, it is not possible to transfer longitudinal magnetization from 1H to 13C (Fig. 2a). If an rf field B
1 is applied (Fig. 2b), however, with a phase x, and the polarization is also along x, then the energy difference between the states
. The 1H polarization in the magnetic field B
0 is, therefore, larger than the 13C polarization. In the magnetic field B
0, it is not possible to transfer longitudinal magnetization from 1H to 13C (Fig. 2a). If an rf field B
1 is applied (Fig. 2b), however, with a phase x, and the polarization is also along x, then the energy difference between the states 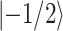 and
and 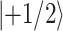 in the rotating frame can be varied independently for different nuclear species, which makes the transfer of transverse magnetization possible. Resonance occurs between 1H and 13C, if
in the rotating frame can be varied independently for different nuclear species, which makes the transfer of transverse magnetization possible. Resonance occurs between 1H and 13C, if
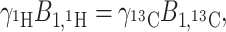 |
8 |
which is known as the Hartman–Hahn condition (Hartmann and Hahn 1962).
Fig. 2.

Energy levels of the 1H and 13C spins: a In the laboratory frame the transfer of magnetization is not possible; b In the rotating frame, the transfer of magnetization is possible as the energy separation is determined by the rf field. The matching condition is then fulfilled
Homonuclear correlation spectroscopy
The CP MAS experiment with two-pulse phase modulation (TPPM) decoupling is the starting point for many advanced pulse sequences. In order to resolve signals and for de novo structure determination of solids, homonuclear correlation NMR spectroscopy of multi-spin labeled molecules is necessary. The polarization transfer between spins is governed by the high-field truncated Hamiltonian for the homonuclear dipolar coupling (Ernst et al. 1987)
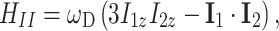 |
9 |
with
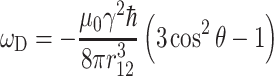 |
10 |
Here γ is the gyromagnetic ratio, r 12 the distance between the spins, and θ the angle between the internuclear distance vector and the external field. Dipolar couplings are averaged by MAS and can be reintroduced during a mixing interval to generate correlated spin states. The sequence of a 13C–13C radio frequency-driven recoupling (RFDR) MAS correlation experiment is shown in Fig. 3a (Bennett et al. 1992). Following CP, the 13C spins precess under heteronuclear decoupling during t 1 to give a high resolution. During τ m, however, the dipolar 13C–13C couplings have to be reintroduced to promote transfer of magnetization. The magnetization is first stored along z by a π/2 pulse. The actual recoupling is achieved by a series of π pulses, which are synchronized with the rotor period. The evolution of the spin state ρ is described by the commutator.
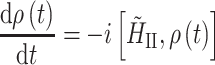 |
11 |
Fig. 3.

a RFDR Pulse sequence for 2D homonuclear correlation spectroscopy: Following CP, the 13C spins precess during t 1. During a mixing period, 13C–13C couplings are reintroduced by a rotor-synchronized train of π pulses. The NMR signal is collected during t 2. b 2D 1H–13C LG-CP hetcor experiment: Following 1H excitation, homo- nuclear decoupling (LG) is applied during the 1H precession period t 1. After CP transfer, the 13C signal is detected in t 2 with heteronuclear decoupling (DEC) to decouple the 1H spins
Here, 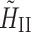 is a spin Hamiltonian describing the effective, reintroduced homonuclear dipolar interactions. This yields
is a spin Hamiltonian describing the effective, reintroduced homonuclear dipolar interactions. This yields
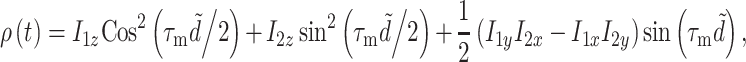 |
12 |
where 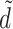 depends on the dipolar coupling strength (Bennett et al. 1992). The first two terms indicate transfer of longitudinal magnetization, while the third term represents double quantum states, which can be eliminated by phase cycling. If a short mixing time τ
m ~ 1 ms is used, only correlations between spins separated by one bond are promoted, which is the optimal condition for the assignment of the chemical shifts.
depends on the dipolar coupling strength (Bennett et al. 1992). The first two terms indicate transfer of longitudinal magnetization, while the third term represents double quantum states, which can be eliminated by phase cycling. If a short mixing time τ
m ~ 1 ms is used, only correlations between spins separated by one bond are promoted, which is the optimal condition for the assignment of the chemical shifts.
Intermolecular transfer between 13C spins with RFDR is difficult due to rapid relayed spin diffusion along the multispin 13C-labeled molecular network (Boender et al. 1995). An alternative is to generate 13C–13C correlations by 1H spin diffusion (Mulder et al. 1998). In a CP3 or CHHC proton-mediated spin diffusion experiment, the 13C magnetization is transferred back to 1H after the first precession interval. Next, 1H spin diffusion is allowed to take place during a mixing period. Finally, the signal is transferred again to 13C by a third CP step and detected. In this way, mixing by the strong 1H dipolar interactions is combined with the high resolution of a 13C MAS spectrum. An effective transfer range, d max, can be determined for short mixing times, and intermolecular distance constraints can be resolved with this sequence (de Boer et al. 2002). In practice, a limited number of such constraints can be very useful for elucidating the structure of solids.
Heteronuclear correlation spectroscopy
Another class of experiments that is widely used in biological solid-state NMR is 1H–13C heteronuclear correlation spectroscopy. A straightforward 1H–13C correlation experiment consists of the CP scheme, where t 1 is inserted after the first 1H π/2 pulse and the CP interval constitutes the mixing step. This is known as wideline separation, since broad 1H lines in the indirect dimension are separated by correlation with 13C shifts in the direct dimension (Schmidt-Rohr and Spiess 1994).
Modern FSLG MAS NMR methods also provide a direct correlation of proton signals of the protein with 13C responses (van Rossum et al. 1997). For heteronuclear transfer of magnetization, LG–CP methods are most convenient to improve the 1H resolution (Fig. 3b). The LG homonuclear decoupling effectively produces isolated heteronuclear spin pairs for efficient transfer and is very stable since the performance is determined by the frequency or phase switching, which can be done very accurately with a modern NMR spectrometer (van Rossum et al. 2000; Ladizhansky et al. 2003). For instance, the FSLG techniques employ off-resonance rf irradiation to generate an effective rf field inclined at the magic angle (Bielecki et al. 1989; Lee and Goldburg 1965). With the 2D LG/MAS experiment in Fig. 3b spectra can be obtained with a good resolution in both dimensions (van Rossum et al. 1997). Another version uses phase-modulated Lee–Goldburg (PMLG) decoupling, which is also easy to implement (Vinogradov et al. 1999). The effective
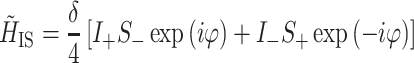 |
13 |
was introduced to describe a coupled 1H–13C spin pair during LG–CP (van Rossum et al. 2000). Here, I ± and S ± are spin operators in a tilted frame for the 1H and 13C spin, respectively. The dipolar coupling, δ, is given by
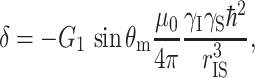 |
14 |
with G 1 a geometrical factor and r IS the distance between the spins. The coherent build-up of the 13C signal S(t) is then described by (van Rossum et al. 2000)
 |
15 |
From the build-up of S(t), the dipolar coupling can be determined. This technique yields accurate distances up to a few angstroms. Since the dipolar couplings scale with r −3, the effects of long-distance interactions are obscured by strong short-range interactions. For longer CP times, the magnetization transfer is incoherent due to the many spin interactions and due to relaxation. Although accurate intermolecular distances are difficult to determine in chlorophylls, incoherent long-range transfer proceeds over an effective maximum transfer range d max, which depends on the length of the mixing period (van Rossum et al. 2002).
As mentioned in the previous section, the large homonuclear dipolar couplings of protons make their direct detection difficult. It is possible to improve the proton resolution using the LG technique (Lee and Goldburg 1965). The basic principle of this technique is to irradiate the protons continuously with an off-resonance rf field, in such a way that the total effective field 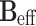 in the rotating frame is inclined at the magic angle
in the rotating frame is inclined at the magic angle 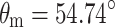 with respect to the static magnetic field B
0 along the z-axis. The LG condition is given by
with respect to the static magnetic field B
0 along the z-axis. The LG condition is given by
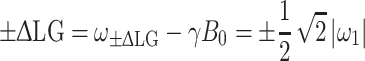 |
16 |
with 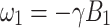 (Lee and Goldburg 1965).
(Lee and Goldburg 1965).
In the 2D MAS LG-CP sequence for heteronuclear 1H–13C detection the FSLG pulse protocol is used for homonuclear decoupling (Bielecki et al. 1989). In this experiment the frequency is switched between 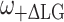 and
and 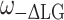 and the phase is switched between
and the phase is switched between 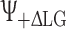 and
and 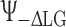 such that
such that  .
.
LH2 complex
The LH2 complex is a peripheral photosynthetic antenna complex. It serves to absorb light and to transfer the excited state energy to the LH1-reaction center complex. The structure of the LH2 complex has been resolved at high resolution by X-ray methods (Cogdell et al. 1999; McDermott et al. 1995; Papiz et al. 2003). LH2 from the purple bacterium Rhodopseudomonas acidophila strain 10050 is built from nine identical monomeric repeating units forming a ring with nine-fold symmetry (Fig. 4a). Each monomer consists of two helical polypeptide subunits, three molecules of BChl a, and two carotenoids (Fig. 4b). The polypeptide segments are called the α-subunit and β-subunit and consist of 53 and 41 amino acid residues, respectively. The BChl a cofactors are denoted by their prominent absorption maxima as B800, αB850, and βB850. The B800 pigments are axially coordinated at their central Mg ion by the carboxyl-αM1 at the N-terminus of the α-subunit, forming a weakly coupled nine-membered ring where the separation between the B800 molecules is approximately 21 Å. Their spectral properties are consistent with their being individual molecules. The pigments which absorb at 850 nm are arranged quite differently. αB850 and βB850 are arranged as a closely coupled dimer, are sandwiched between each α- and β-subunit pair, and are axially coordinated at their central Mg ion by βH30 and αH31 respectively (Fig. 4c). In LH2 antennae these dimers form a continuous overlapping ring of 18 pigments that is subject to moderate structural heterogeneity on the scale of optical spectroscopy, while appearing nearly crystalline in the NMR (Novoderezhkin et al. 2003, 2006; van Gammeren et al. 2005b). The LH2 complex serves as a model for studying membrane proteins by using MAS NMR spectroscopy (van Gammeren et al. 2004, 2005a, b). In the following section we will give some examples of how MAS NMR can be used to probe the structure and obtain functional information from membrane bound LH2 complexes.
Fig. 4.

Arrangement of histidines in LH2 of Rps. acidophila. The helices are represented by ribbons. a Top view; b Side view of one of the protomers of LH2; c A portion of the ring showing distances between the δ and ε carbons of β-His 30 and α-His 31 and the central Mg atoms of coordinated B850 molecules. The aliphatic chains of BChl have been omitted for clarity; d The nomenclature of the histidine
MAS NMR in combination with pattern labeling for the sequence specific assignment of NMR signals
The sequence-specific assignment of chemical shifts is an essential step for comprehensive studies of proteins by NMR. Correlation pulse sequences for inter-residue transfer across the peptide bond (NCOCα), and for intra-residue transfer into the sidechains (NCαCX) were applied for editing of spectra and resolving correlations between adjacent amino acids in the sequence (Fig. 5). Pattern labeling reduces the number of correlation signals and decreases the linewidth of these signals compared to the uniformly labeled samples, which enables to resolve the narrowly distributed correlation signals of the backbone carbons and nitrogens involved in the long α-helical transmembrane segments. [1,2,3,4–13C], [1,4–13C] and [2,3–13C] succinic acid were chemically labeled and used for the biosynthetic preparation of site-directed isotopically 13C enriched LH2 complexes from the Rhodopseudomonas acidophila strain 10050. 2D PDSD correlation spectroscopy was used to show that carbonyl carbons in the protein backbone were labeled by [1,4–13C]-succinic acid, while the Cα and Cβ carbons of the residues were labeled by [2,3–13C]-succinic acid in the growth medium (van Gammeren et al. 2004). In addition, leucine and isoleucine residues can be labeled using a uniformly labeled amino acid mixture in the medium (van Gammeren et al. 2004).
Fig. 5.

In the upper panels two regions from homonuclear 13C–13C PDSD correlation spectra collected from 2,3-LH2 (red) and AA-LH2 (black) are shown. The upper left panel contains cross peaks between aliphatic and carbonyl carbons, while the upper right panel shows correlations between sidechain aliphatic carbons. In the upper right panel the aliphatic responses are shown. In the middle panel, the aliphatic region of the NCACX spectra of 2,3-LH2 (red) and AA-LH2 (black) are shown. Finally, in the lower panel the NCACX spectrum of a 1,2,3,4-LH2 sample is shown
The pattern labeling allows for the residual assignment of the LH2 α-helical transmembrane protein complex. Correlations between nearby residues and between residues and the labeled BChl a cofactors, provided by the 13C–13C correlation experiments using a 500 ms spin diffusion period, were utilized to arrive at sequence specific chemical shift assignments for 76 residues of the 94 residues of the monomeric unit of the LH2 complex. An example of the sequence specific assignment of LH2 is shown in Fig. 5. Here the LH2 were labeled with either [2,3-13C]-succinic acid (2,3-LH2), [1,2,3,4-13C] succinic acid (1,2,3,4-LH2) or with uniformly 13C-labeled amino acids (AA-LH2). In the upper left part of Fig. 5, a few responses are observed for 2,3-LH2, belonging to H, Q and E residues. The responses from AA-LH2 in the carbonyl area are from I, L, A, G and V. The blue spectrum in the carbonyl region comprises carbonyl responses from 1,2,3,4-LH2. The dashed lines in the upper right panel indicate correlations involving the αT38 and four P residues for the 2,3-LH2, and correlations involving βI16 for the AA-LH2. Here we follow the notation in (van Gammeren et al. 2005b). The residues that are labeled via both nutrient sources are also indicated in this panel. The data in all panels are aligned and correlations involving αT38, βI16 and the 4P residues are indicated with dashed lines for the two different samples. The responses of the G residues are indicated with a rectangular box. Assignments were obtained from 2D PDSD 13C–13C correlation datasets with mixing times of 20 and 500 ms and band selective 13C–15N correlation spectroscopy by alignment of the NCA signals with the carbonyl area of the PDSD spectrum (van Gammeren et al. 2005b).
Following the sequence specific assignment, it is possible to get access to four classes of distance constraints, (i) along the helix for assignment of signals, (ii) between helix side chains and cofactors, (iii) between amino acids of two subunits that form the monomer, and (iv) between amino acids of different monomers (Ganapathy et al. 2007). Since [2,3-13C]-succinic acid is a precursor for the biosynthesis of BChls in photosynthetic bacteria, most of the ring functionalities of the BChls in the 2,3-LH2 sample that interact with the protein matrix are labeled and αC121/βV28/βA29/βH30 and βC121/αA27/αV30/αH31 intermolecular correlations were resolved with a PDSD spectrum with a mixing time of 500 ms (van Gammeren et al. 2005a). The red arrow in Fig. 6 indicates an inter-helical inter-monomeric correlation between the α1V10 and α2A13 residues, the green arrow shows inter-helical intra-monomeric correlations between the βT2 and αP12 residues, the orange arrows indicate cofactor-residue contacts between the αB850 cofactor and the βH30 residue as well as the B800 cofactor and βG18 residue and the remaining blue arrows point to inter-residue correlations along the helix (Ganapathy et al. 2007).
Fig. 6.

Distance restraints obtained by MAS NMR for the LH2 antenna complex, projected on the 1NKZ PDB structure. The βB850 cofactor is omitted to provide a better view on the restraints
Finally, the resonance assignments for the helices in the LH2 complex can be compared with random coil values in the liquid state. The resulting chemical shift differences are called secondary chemical shifts and generally correlate with the backbone torsion angles ψ. However, the LH2 membrane protein forms a complex topology with primary, secondary, tertiary, and quaternary structure, and several of the secondary shifts are outside the range of values commonly encountered across proteins. Recent analyses of MAS NMR secondary shifts have shown that in the strongly condensed and rigid LH2 system, the higher order stabilization of the tertiary and quaternary structure, possibly in synergy with the dielectric properties, leads to localized points of physical frustration that are involved in tuning the light-harvesting function (van Gammeren et al. 2005a; Wawrzyniak et al. 2008). In this way, the analysis of the secondary shifts provide access to guiding principles of how a 3D nanostructured arrangement can tune its functional properties by self-organization.
Electronic structure and protonation state of axial histidines in LH2 complex
In the LH2 complex, the two conserved histidines βH30 and αH31 stabilize the B850 ring assembly (Scholes et al. 1999) (Fig. 4b, c), and understanding of the electronic structure of the His/B850 complexes is important for understanding the mechanism of exciton transfer over the BChl ring and the transfer rate from the B800 to the B850. Quantum electronic delocalization couples to distortions of the protein-cofactor “smart” matrix to enhance the transfer rate from the B800 to the B850 in a robust process (Jang et al. 2007). On excitation with blue light, the B800 band is populated, and the transfer to the B850 takes place on a time scale of 0.7–3 ps (Grondelle and Novoderezhkin 2006). While the intraband B800 and interband B800–B850 electronic coherences decay rapidly, the B850 intraband coherence lasts several picaseconds in a wavepacket that is delocalized over several B850 BChls. In order to probe the electronic and protonic states of axial histidines, MAS NMR has been applied in conjunction with site-specific isotope labeling of histidine residues in LH2 complex (Alia et al. 2001, 2004). By means of 1D 15N MAS NMR, our group has shown that the τ nitrogen of β-His30 and α-His31 ligate to the Mg2+ of the B850 BChl a molecules. The hydrogen bonding status of the π nitrogen was reflected by the resonance shift in the 1D 15N spectra. In addition, a 2D homonuclear (13C–13C) MAS NMR experiment, using a phase-sensitive RFDR pulse sequence and a double CP/MAS experiment performed on U–15N and 13C labeled LH2, revealed that axial histidines in LH2 complex carry partial positive charge in an overall neutral Histidine/B850 complex (Alia et al. 2001) (Fig. 7). With DFT calculations these effects were analyzed in detail, and it was established that the histidines are subject to protein-induced strain that forces the histidine imidazole side chain in the positive charge-type electronic configuration as a result of the higher order self-assembly process (Wawrzyniak et al. 2008).
Fig. 7.

a 2-D homonuclear (13C–13C) and b heteronuclear (1H–13C) dipolar correlation spectrum of [13C6,15N3]-histidine labeled LH2 complex collected in a field of 17.6 T. The spectrum was recorded with a spinning frequency ω r/2π = 12 kHz at a temperature of 230 K. The 1H homonuclear interactions in b were suppressed with PMLG irradiation during proton evolution, applying a RF power corresponding with a nutation frequency of 74.4 kHz. Cross peaks from the cationic histidines (Type 2A) are indicated by (′) and cross peaks from the histidines bound with B850 (Type 2B) are indicated by (*)
In addition to charge transfer, 2D heteronuclear (1H–13C) MAS experiments can assess the electronic delocalization and overlap in a chlorophyll ring. A 2D heteronuclear (1H–13C) MAS NMR experiment was performed using a 2D PMLG decoupled heteronuclear sequence (Alia et al. 2004). Due to significant ring current shift effects experienced by protons of axial histidine present in close proximity to the conjugated porphyrin ring system of B850, their resonances were clearly separated from the resonances of non-axial histidines. The magnitude of these ring current shifts, 2–4 ppm, provides convincing evidence that in the electronic ground state the supermolecular π–π interactions in the assembly of 18 B850 ring in LH2 are very moderate, since they do not quench the ring currents for the individual BChl a/Histidine complexes (Alia et al. 2004).
Histidine residues are main ligands to B(Chl) in all known reaction centers. It appears that histidine has the strongest effect in changing the midpoint potential in the ground state of chlorophylls involved in charge separation (Ivancich et al. 1998). The characterization of histidine signals from LH2 antenna systems and models provides the basis for a detailed structural analysis of the histidines interacting with chlorophyll donor molecules that are involved in charge separation in reaction centers (Alia et al. 2009).
In conclusion, MAS NMR is an area of technological growth, for resolving structure and for structure–function studies. The technology provides access to photosynthetic assemblies in the natural membrane environment, when they are inaccessible to X-ray and other diffraction methods. Going beyond X-ray, with MAS NMR it is possible to resolve molecular mechanisms in the ground state, which are behind the function of these important systems in Nature.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- BChl
Bacteriochlorophyll
- CP
Cross polarization
- FSLG
Frequency switched Lee–Goldburg
- HETCOR
Heteronuclear correlation
- LG
Lee–Goldburg
- LH2
Light-harvesting complex 2
- MAS
Magic angle spinning
- NMR
Nuclear magnetic resonance
- PDSD
Proton-driven spin diffusion
- PMLG
Phase modulated Lee Goldburg
- RFDR
Radio frequency-driven recoupling
- TPPM
Two-pulse phase modulation
References
- Abragam A. Principles of nuclear magnetism. Oxford: Oxford University Press; 1961. [Google Scholar]
- Alia, Matysik J, Soede-Huijbregts C, Baldus M, Raap J, Lugtenburg J, Gast P, van Gorkom HJ, Hoff AJ, de Groot HJM. Ultrahigh field MAS NMR dipolar correlation spectroscopy of the histidine residues in light-harvesting complex II from photosynthetic bacteria reveals partial internal charge transfer in the B850/His complex. J Am Chem Soc. 2001;123:4803–4809. doi: 10.1021/ja002591z. [DOI] [PubMed] [Google Scholar]
- Alia, Matysik J, de Boer I, Gast P, van Gorkom HJ, de Groot HJM. Heteronuclear 2D (1H–13C) MAS NMR resolves the electronic structure of coordinated histidines in light-harvesting complex II: assessment of charge transfer and electronic delocalization effect. J Biomol NMR. 2004;28:157–164. doi: 10.1023/B:JNMR.0000013842.72291.48. [DOI] [PubMed] [Google Scholar]
- Alia A, Wawrzyniak PK, Janssen GJ, Buda F, Matysik J, de Groot HJM. Differential charge polarization of axial histidines in bacterial reaction centers balances the asymmetry of the special pair. J Am Chem Soc. 2009;131:9626–9627. doi: 10.1021/ja9028507. [DOI] [PubMed] [Google Scholar]
- Andrew ER, Bradbury A, Eades RG. Nuclear magnetic resonance spectra from a crystal rotated at high speed. Nature. 1958;182:1659. doi: 10.1038/1821659a0. [DOI] [Google Scholar]
- Bennett AE, Ok JH, Griffin RG, Vega S. Chemical-shift correlation spectroscopy in rotating solids—radio frequency-driven dipolar recoupling and longitudinal exchange. J Chem Phys. 1992;96:8624–8627. doi: 10.1063/1.462267. [DOI] [Google Scholar]
- Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear decoupling in rotating solids. J Chem Phys. 1995;103:6951–6958. doi: 10.1063/1.470372. [DOI] [Google Scholar]
- Bielecki A, Kolbert AC, Levitt MH. Frequency-switched pulse sequences—homonuclear decoupling and dilute spin NMR in solids. Chem Phys Lett. 1989;155(4–5):341–346. doi: 10.1016/0009-2614(89)87166-0. [DOI] [Google Scholar]
- Boender GJ, Raap J, Prytulla S, Oschkinat H, de Groot HJM. MAS NMR structure refinement of uniformly 13C enriched chlorophyll-a/water aggregates with 2D dipolar correlation spectroscopy. Chem Phys Lett. 1995;237:502–508. doi: 10.1016/0009-2614(95)00357-A. [DOI] [Google Scholar]
- Cogdell RJ, Isaacs NW, Howard TD, McLuskey K, Fraser NJ, Prince SM. How photosynthetic bacteria harvest solar energy. J Bacteriol. 1999;181(13):3869–3879. doi: 10.1128/jb.181.13.3869-3879.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daviso E, Jeschke G, Matysik J. Photochemically induced dynamic nuclear polarization (Photo-CIDNP) Magic-Angle Spinning NMR. In: Aartsma TJ, Matysik J, editors. Biophysical techniques in photosynthesis II. Dordrecht: Springer Academic Publishers; 2008. pp. 385–399. [Google Scholar]
- De Boer I, Bosman L, Raap J, Oschkinat H, de Groot HJM. 2D 13C–13C MAS NMR correlation spectroscopy with mixing by true 1H spin diffusion reveals long-range intermolecular distance restraints in ultra high magnetic field. J Magn Reson. 2002;157:286–291. doi: 10.1006/jmre.2002.2588. [DOI] [PubMed] [Google Scholar]
- De Groot HJM. Magic Angle Spinning (MAS) NMR for structure determination in photosynthesis. In: Aartsma TJ, Matysik J, editors. Biophysical techniques in photosynthesis II. Dordrecht: Springer Academic Publishers; 2008. pp. 361–383. [Google Scholar]
- Diller A, Roy E, Gast P, et al. 15 N-photo-CIDNPN-photo-CIDNP MAS NMR analysis of the electron donor of photosystem II. Proc Natl Acad Sci USA. 2007;104:12843–12848. doi: 10.1073/pnas.0701763104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duer MJ. Introduction to solid-state NMR spectroscopy. Oxford: Blackwell Publishing Ltd.; 2004. [Google Scholar]
- Egorova-Zachernyuk TA, Hollander J, Fraser N, Gast P, Hoff AJ, Cogdell R, de Groot HJM, Baldus M. Heteronuclear 2D-correlations in a uniformly [C-13, N-15] labeled membrane-protein complex at ultra-high magnetic fields. J Biomol NMR. 2001;19:243–253. doi: 10.1023/A:1011235417465. [DOI] [PubMed] [Google Scholar]
- Ernst RR, Bodenhausen G, Wokaun A. Principles of nuclear magnetic resonance in one and two dimensions. Oxford: Clarendon Press; 1987. [Google Scholar]
- Ganapathy S, van Gammeren AJ, Hulsbergen FB, de Groot HJM. Probing secondary, tertiary, and quaternary structure along with protein-cofactor interactions for a helical transmembrane protein complex through 1H spin diffusion with MAS NMR spectroscopy. J Am Chem Soc. 2007;129:1504–1505. doi: 10.1021/ja0664436. [DOI] [PubMed] [Google Scholar]
- Ganapathy S, Oostergetel GT, Wawrzyniak PK, Reus M, Gomez Maqueo Chew A, Buda F, Boekema EJ, Bryant DA, Holzwarth AR, de Groot HJ. Alternating syn-anti bacteriochlorophylls form concentric helical nanotubes in chlorosomes. Proc Natl Acad Sci USA. 2009;106:8525–8530. doi: 10.1073/pnas.0903534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grondelle R, Novoderezhkin VI. Energy transfer in photosynthesis: experimental insights and quantitative models. Phys Chem Chem Phys. 2006;8:793–807. doi: 10.1039/b514032c. [DOI] [PubMed] [Google Scholar]
- Hartmann SR, Hahn EL. Nuclear double resonance in rotating frame. Physical Rev. 1962;128:2042–2053. doi: 10.1103/PhysRev.128.2042. [DOI] [Google Scholar]
- Ivancich A, Artz K, Williams JC, Allen JP, Mattioli TA. Effects of hydrogen bonds on the redox potential and electronic structure of the bacterial primary electron donor. Biochemistry. 1998;37:11812–11820. doi: 10.1021/bi9806908. [DOI] [PubMed] [Google Scholar]
- Jang S, Newton MD, Silbey RJ. Multichromophoric Förster resonance energy transfer from b800 to b850 in the light harvesting complex 2: evidence for subtle energetic optimization by purple bacteria. J Phys Chem B. 2007;111:6807–6814. doi: 10.1021/jp070111l. [DOI] [PubMed] [Google Scholar]
- Ladizhansky V, Vinogradov E, van Rossum BJ, de Groot HJM, Vega S. Multiple-spin effects in fast magic angle spinning Lee–Goldburg cross-polarization experiments in uniformly labeled compounds. J Chem Phys. 2003;118:5547–5557. doi: 10.1063/1.1517299. [DOI] [Google Scholar]
- Lee M, Goldburg WI. Nuclear-magnetic-resonance line narrowing by a rotating rf field. Phys Rev. 1965;140:A1261–A1271. doi: 10.1103/PhysRev.140.A1261. [DOI] [Google Scholar]
- Lowe IJ. Free induction decays of rotating solids. Phys Rev Let. 1959;2:285–287. doi: 10.1103/PhysRevLett.2.285. [DOI] [Google Scholar]
- McDermott G, Prince SM, Freer AA, Hawthornthwaitelawless AM, Papiz MZ, Cogdell RJ, Isaacs NW. Crystal-structure of an integral membrane light-harvesting complex from photosynthetic bacteria. Nature. 1995;374:517–521. doi: 10.1038/374517a0. [DOI] [Google Scholar]
- Mulder FM, Heinen W, van Duin M, Lugtenburg J, de Groot HJM. Spin diffusion with 13C selection and detection for the characterization of morphology in labeled polymer blends with MAS NMR. J Am Chem Soc. 1998;120(49):12891–12894. doi: 10.1021/ja9809511. [DOI] [Google Scholar]
- Novoderezhkin V, Wendling M, van Grondelle R. Intra- and interband transfers in the B800–B850 antenna of Rhodospirillum molischianum: Redfield theory modeling of polarized pump-probe kinetics. J Phys Chem B. 2003;107:11534–11548. doi: 10.1021/jp035432l. [DOI] [Google Scholar]
- Novoderezhkin VI, Rutkauskas D, van Grondelle R. Dynamics of the emission spectrum of a single LH2 complex: Interplay of slow and fast nuclear motions. Biophys J. 2006;90:2890–2902. doi: 10.1529/biophysj.105.072652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papiz MZ, Prince SM, Howard T, Cogdell RJ, Isaacs NW. The structure and thermal motion of the B800–850 LH2 complex from Rps. acidophila at 2.0 (A)over-circle resolution and 100 K: new structural features and functionally relevant motions. J Mol Biol. 2003;326:1523–1538. doi: 10.1016/S0022-2836(03)00024-X. [DOI] [PubMed] [Google Scholar]
- Pines A, Gibby MG, Waugh JS. Proton-enhanced NMR of dilute spins in solids. J Chem Phys. 1973;59:569–590. doi: 10.1063/1.1680061. [DOI] [Google Scholar]
- Prakash S, Alia A, Gast P, et al. Magnetic field dependence of photo-CIDNP MAS NMR on photosynthetic reaction centers of Rhodobacter sphaeroides WT. J Am Chem Soc. 2005;127:14290–14298. doi: 10.1021/ja054015e. [DOI] [PubMed] [Google Scholar]
- Schaefer J, Stejskal EO. C-13 Nuclear magnetic-resonance of polymers spinning at magic angle. J Am Chem Soc. 1976;98:1031–1032. doi: 10.1021/ja00420a036. [DOI] [Google Scholar]
- Schmidt-Rohr K, Spiess W. Multidimensional solid-state NMR and polymers. London: Academic Press Ltd.; 1994. [Google Scholar]
- Scholes GD, Gould IR, Cogdell RJ, Fleming GR. J Phys Chem B. 1999;103:2543–2553. doi: 10.1021/jp9839753. [DOI] [Google Scholar]
- van Gammeren AJ, Hulsbergen FB, Hollander JG, de Groot HJM. Biosynthetic site-specific C-13 labeling of the light-harvesting 2 protein complex: a model for solid state NMR structure determination of transmembrane proteins. J Biomol NMR. 2004;30:267–274. doi: 10.1007/s10858-004-3736-7. [DOI] [PubMed] [Google Scholar]
- van Gammeren AJ, Hulsbergen FB, Hollander JG, de Groot HJM. Residual backbone and side-chain C-13 and N-15 resonance assignments of the intrinsic transmembrane light-harvesting 2 protein complex by solid-state Magic Angle Spinning NMR spectroscopy. J Biomol NMR. 2005;31:279–293. doi: 10.1007/s10858-005-1604-8. [DOI] [PubMed] [Google Scholar]
- van Gammeren AJ, Buda F, Hulsbergen FB, Kiihne S, Hollander JG, Egorova-Zachernyuk TA, Fraser NJ, Cogdell RJ, de Groot HJM. Selective chemical shift assignment of B800 and B850 bacteriochlorophylls in uniformly [C-13, N-15]-labeled light-harvesting complexes by solid-state NMR spectroscopy at ultra-high magnetic field. J Am Chem Soc. 2005;127:3213–3219. doi: 10.1021/ja044215a. [DOI] [PubMed] [Google Scholar]
- van Rossum BJ, Förster H, de Groot HJM. High-field and high-speed CP-MAS 13C NMR heteronuclear dipolar-correlation spectroscopy of solids with frequency-switched Lee–Goldburg homonuclear decoupling. J Magn Reson. 1997;124:516–519. doi: 10.1006/jmre.1996.1089. [DOI] [Google Scholar]
- van Rossum BJ, de Groot C, de Groot HJM, Ladizhansky V, Vega S. A method for measuring hetronuclear (1H–13C) distances in high speed MAS NMR. J Am Chem Soc. 2000;122:3465–3472. doi: 10.1021/ja992714j. [DOI] [Google Scholar]
- van Rossum BJ, Schulten EAM, Raap J, Oschkinat H, de Groot HJM. A 3-D structural model of solid self-assembled Chlorophyll a/H2O from multispin labeling and MAS NMR 2-D dipolar correlation spectroscopy in high magnetic field. J Magn Res. 2002;155:1–14. doi: 10.1006/jmre.2002.2502. [DOI] [PubMed] [Google Scholar]
- Vinogradov E, Madhu PK, Vega S. High-resolution proton solid-state NMR spectroscopy by phase-modulated Lee–Goldburg experiment. Chem Phys Lett. 1999;314:443–450. doi: 10.1016/S0009-2614(99)01174-4. [DOI] [Google Scholar]
- Wawrzyniak PK, Alia A, Schaap RG, Heemskerk MM, de Groot HJM, Buda F. Protein-induced geometric constraints and charge transfer in bacteriochlorophyll-histidine complexes in LH2. Phys Chem Chem Phys. 2008;10:6971–6978. doi: 10.1039/b810457c. [DOI] [PubMed] [Google Scholar]