

Abstract
The molecular chaperone, Hsp90, is an essential eukaryotic protein that assists in the maturation and activation of client proteins. Hsp90 function depends upon the binding and hydrolysis of ATP, which causes large conformational rearrangements in the chaperone. Hsp90 is highly conserved from bacteria to eukaryotes, and similar nucleotide-dependent conformations have been demonstrated for the bacterial, yeast, and human proteins. There are, however, important species-specific differences in the ability of nucleotide to shift the conformation from one state to another. Although the role of nucleotide in conformation has been well studied for the cytosolic yeast and human proteins, the conformations found in the absence of nucleotide are less well understood. In contrast to cytosolic Hsp90, crystal structures of the endoplasmic reticulum homolog, Grp94, show the same conformation in the presence of both ADP and AMPPNP. This conformation differs from the yeast AMPPNP-bound crystal state, suggesting that Grp94 may have a different conformational cycle. In this study, we use small angle X-ray scattering and rigid body modeling to study the nucleotide free states of cytosolic yeast and human Hsp90s, as well as mouse Grp94. We show that all three proteins adopt an extended, chair-like conformation distinct from the extended conformation observed for the bacterial Hsp90. For Grp94, we also show that nucleotide causes a small shift toward the crystal state, although the extended state persists as the major population. These results provide the first evidence that Grp94 shares a conformational state with other Hsp90 homologs.
Keywords: Hsp90, human, yeast, Grp94, SAXS, apo conformation, solution states
Introduction
The essential eukaryotic protein, Hsp90, is a member of the large class of proteins known as molecular chaperones. Molecular chaperones are required for maintaining the correctly folded state of proteins within the cell. For many chaperones, this function is accomplished by the recognition of hydrophobic surfaces on the substrate protein and subsequent rounds of ATP hydrolysis-dependent binding and release.1 Hsp60 (GroEL) and Hsp70 (DnaK), two well-studied members of this class, function early in the protein folding pathway by interacting with nascent polypeptide chains and promoting their folding.2,3 In contrast, Hsp90 appears to act largely at later stages of the folding pathway interacting with substrates (clients) that are largely folded.4 The interaction with Hsp90 facilitates the binding of ligands or partner proteins to the client proteins.5,6 Despite this understanding of Hsp90's function, the nature of the interaction with client proteins remains elusive.
Hsp90 is highly conserved from bacteria to eukaryotes. While client proteins are just beginning to be identified for the bacterial Hsp90 (HtpG), a large and growing set have been discovered for the eukaryotic Hsp90s including steroid hormone receptors, protein kinases, nitric oxide synthase, and telomerase.7,8 Many of these substrates are oncogenic, and small molecule inhibitors of Hsp90 that disrupt its interaction with these oncoproteins are showing promise as novel chemotherapeutics.9–11
Hsp90 is an obligate dimer consisting of three domains per monomer. The C-terminal domain (CTD) is the site of dimerization, the middle domain (MD) has been implicated in client binding, and ATP binds to the N-terminal domain (NTD).12 Crystal structures of Hsp90 from different organisms have demonstrated the large conformational range of Hsp90 with an open, nucleotide-free (apo) form (bacteria), a closed NTD dimerized state in the presence of the nonhydrolyzable ATP analog, AMPPNP (yeast), and an even more compact state when ADP is bound (bacteria).13,14 These structures suggest a nucleotide-driven conformational cycle important for Hsp90 function. Negative-stain EM analysis has confirmed the presence of all three conformational states for the bacterial (HtpG), the yeast (Hsc82), and the cytosolic human (Hsp90α) Hsp90s, indicating a common pathway for Hsp90 function in different organisms.15
Structural and biochemical studies have shown that the conformational cycle is more complex and less deterministic than previously thought. In solution, these different states exist in equilibrium with one another and nucleotide acts to shift the equilibrium rather than specifically determining the conformation.15–18 Although the conformational states appear to be a conserved aspect of Hsp90's function, the positioning of the conformational equilibria between the states and the response to nucleotide appear to be extremely homolog specific. Based on a recent EM study,15 Hsp90α has an equilibrium that is heavily biased toward the open state even in the presence of AMPPNP, whereas the equilibrium for Hsc82 is shifted predominantly toward the closed state with the addition of AMPPNP. HtpG is positioned to maintain maximum levels of both conformations in the presence of AMPPNP.15 Because of the differences in the conformational equilibrium seen in the presence of nucleotide, it is reasonable to assume that differences also exist for the apo conformational states. The differing effects of nucleotide may arise from differences in the apo conformations, but this possibility has yet to be examined.
Less is known about the conformational dynamics of the endoplasmic reticulum (ER) homolog, Grp94. Grp94's distinct set of client proteins are found mainly as part of the secretory pathway or as transmembrane proteins,19,20 and many of these client proteins contain disulfide bonds. Grp94 also has a unique insertion of five residues in the active site lid,21 a flexible region adjacent to the nucleotide binding pocket, suggesting that its response to nucleotide may be different from other Hsp90s. The crystal structure of Grp94 bound to either ADP or AMPPPNP shows a distinct conformation that is intermediate to the open apo and closed AMPPNP-bound states observed in other homologs.22 This structure further supports the hypothesis that Grp94 has a unique conformational cycle when compared with other Hsp90s. Kinetic studies have demonstrated that Grp94 favors an open state in the presence of nucleotide much like human Hsp90α,23 but it is unclear how this “open” state relates to the apo states of other homologs.
Although the nucleotide bound states have been extensively characterized for several Hsp90 homologs and show strong conservation across species, there is less data available for the structures of the apo forms of Hsp90. Our previous small angle X-ray scattering (SAXS) study of apo HtpG16 revealed a more extended conformation in solution than in the crystal structure [Fig. 1(A)]. A recent SAXS and cryo-EM study of pig Hsp90 also showed a solution structure different from the known apo HtpG crystal structure.24 The pig Hsp90 study also demonstrated the presence of two apo conformations in solution and provided molecular envelopes for both conformations. Both conformations are distinct from the HtpG solution structure indicating that the apo structures are not conserved between bacteria and higher eukaryotes. Although the SAXS/cryo-EM study provides an important look at the apo structure of a eukaryotic Hsp90, the resultant surface representations yield only limited information about the orientation of individual domains, and the relative amounts of the two populations were not quantified.
Figure 1.
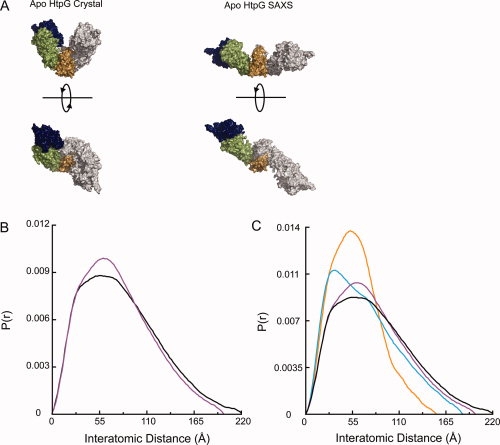
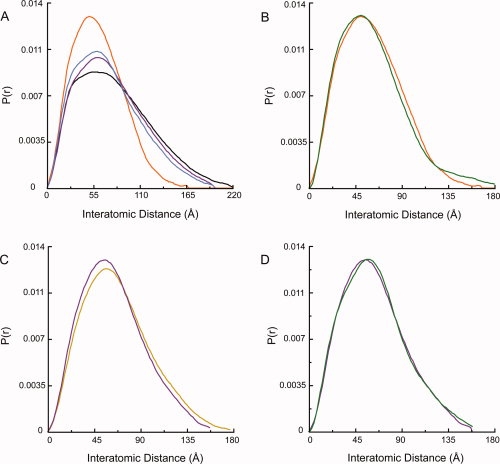
Human Hsp90α and yeast Hsc82 in solution have an extended conformation distinct from the bacterial HtpG structure. (A) Two previously described conformations of apo HtpG.14,16 (B) Comparison of the P(r) functions for apo Hsc82 (black) and apo Hsp90α (purple). (C) Hsc82 (black) and Hsp90α (purple) have a larger Dmax than either known apo bacterial Hsp90 solution state [high pH,16 blue; low pH (Krukenberg et al., in press), orange]. The human and yeast proteins also display a distinctive shape to their P(r) curves.
As a result of the already elucidated differences between Hsp90 homologs, especially differences in conformational equilibria in the presence of nucleotide, it is essential to have a better understanding of the structures of the different cytosolic and organelle Hsp90 apo forms and the conformational equilibria that may exist in the absence of nucleotide. To address this, we have used SAXS and rigid body modeling to investigate the domain orientations of the apo states of yeast Hsc82, human cytosolic Hsp90α, and the mouse ER Grp94. Our method also allows for the deconvolution of multiple populations found in solution. We show that the apo states of the eukaryotic homologs are all primarily populated by a similar extended state distinct from the bacterial extended state. We also examine the nucleotide bound state of Grp94. This study provides the first evidence that Grp94, despite its differences, populates a conformation similar to cytosolic Hsp90 and indicates that nucleotide only subtly shifts the equilibrium toward more closed states.
Results
In solution, apo Hsc82 and Hsp90α are in a conformation distinct from HtpG
Previous SAXS and EM studies have shown an extended conformation for apo Hsp90 in solution.15,16,24 The SAXS model for HtpG shows a conformation where a rigid body rotation about the MD-CTD domain interface opens the NTD-MD domain angle by about 30° when compared with the crystal structure creating a larger cleft between the two monomers. The NTD is also rotated away from the MD to create a more open and extended conformation [Fig. 1(A)].16 Negative-stain EM images of yeast and human Hsp90 also show an open conformation in the absence of nucleotide.15 Recent cryo-EM and SAXS reconstructions of pig Hsp9024 show an extended conformation distinct from HtpG. The differences between the pig and bacterial structures suggest a broader array of open conformations across species than expected. Given its different kinetics and equilibria, the apo Hsc82 (yeast) conformation may also be significantly different from either HtpG or pig Hsp90, but its structure remains largely uncharacterized.
To investigate the differences between cytosolic human and yeast Hsp90, we collected SAXS data for both yeast Hsc82 and human Hsp90α in the absence of nucleotide and used rigid body modeling to develop atomic models of the apo states. Crystallographic studies have revealed that the underlying domain structure is highly conserved; the variability seen between Hsp90 homologs and between different nucleotide states is largely in the quaternary arrangement of the domains. By using rigid body refinement, we take advantage of these known structures to better position the domains with respect to one another. This method provides more information about the resulting conformations than possible with the standard ab initio modeling methods or the existing EM reconstructions. Moreover, the precision of the SAXS data also allows for the deconvolution of resultant distance distribution functions into a linear combination of coexisting conformations. Although the cryo-EM images of pig Hsp90 revealed two distinct conformations, the relative amounts of the two populations were not determined. The SAXS methodology we have developed allows for the quantification of separate populations in solution, and with the variation in relative populations seen in the presence of nucleotide, this analysis is especially important for a complete understanding of Hsp90.
The SAXS scattering data (I(Q)) was collected for Q = 0.008–0.3 (Q = 4πsin θ/λ). A Guinier plot of the data indicates that the samples are well behaved with no observable aggregation (Supporting Information Figure S1). The data were converted to the distance distribution function (P(r)) by Fourier transform. The P(r) function provides a histogram of the interatomic distances found within the molecule giving information on shape as well as maximum end-to-end distance, and it is particularly sensitive to changes in the solution state of the protein. As seen in Figure 1(B), Hsc82 and Hsp90α have quite similar P(r) functions. The Hsp90α P(r) also compares well with the previously shown P(r) for pig Hsp90.24 Hsc82 does populate a small number of larger distances as seen by a maximum distance of 220 Å when compared with 200 Å for Hsp90α. Hsc82 also has a less pronounced shoulder at 35 Å. Overall, the shapes of the curves indicate a very similar solution structure or set of structures. Apo HtpG exists in a pH-dependent conformational equilibrium between an open-extended state and a more closed state (Krukenberg et al., in press). As shown in Figure 1(C), the P(r) curves make it very clear that neither HtpG apo nor Hsp90α conformation matches the solution conformations seen for Hsc82. In general, the eukaryotic Hsp90s contain many larger interatomic distances suggesting a larger opening angle for eukaryotic Hsp90s. The shape of the peaks for Hsc82 and Hsp90α are also distinct from HtpG implying that other conformational differences exist between the homologs.
Cytosolic yeast and human Hsp90 are predominantly in an extended chair-shaped conformation
Even though the crystal structures of Hsp90 have vastly different overall shapes, the structures within each domain are maintained between the different structures. This suggests that the dynamic motions of Hsp90 can be accurately modeled using rigid body motions of the domains. The known structures therefore provide a large number of possibilities for domain–domain configurations, greatly facilitating the modeling of the yeast and human data. Also, in these structures, the NM interface (between the NTD and MD) is generally more tightly packed than the MC interface (between the MD and CTD), suggesting that most of the flexibility originates at the MC interface and the NM interface may be characterized by discreet conformations. To understand the structural origins of the yeast and human apo Hsp90 P(r) curves, we systematically varied rotations about either the NM interface or the MC interface, comparing calculated P(r) curves with those experimentally observed. Because variations at the MC interface have the largest effect, we began our search by treating the NM domains as rigid bodies that pivot about the MC interface. This is also in keeping with EM studies which indicate that the largest differences in conformation occur at the MC interface. Given that numerous NM configurations have already been solved, we initially used the previously described NM domain configurations from the yeast AMPPNP-bound crystal state,13 the HtpG solution state,16 the apo HtpG crystal state, and the extended ADP-bound HtpG crystal state.14 These conformations provided a good sampling of the conformational space accessible to the NM domain. Using the library of NM configurations, residue 530 (Hsc82 numbering) on the flexible linker connecting the MD to the CTD was chosen as the hinge point, and conformational space was systematically and exhaustively searched at regular angles about a general rotation axis while maintaining twofold dimer symmetry.16 At the end of the grid search, the rotations and translations about the pivot point were further refined by several cycles of least-squares minimization. Regardless of the starting MC angle, the extended NM domain, seen in the tetramer form of the full-length HtpG crystal bound to ADP, consistently gave a better fit to the scattering data for both Hsc82 and Hsp90α than the other NM domains (Supporting Information Figure S2 and data not shown). The fact that fully extended NM domain is correct for both Hsc82 and Hsp90α suggests that any differences between the two homologs occur largely at the MC interface.
Although the fits were tolerable, the remaining disagreements indicate that the model does not fully explain the experimental data. Using the extended NM domain, the MC interface was further refined. Additional refinement of the NM angles did little to improve the fits. Throughout the rounds of refinement, we observed a tendency of the NM domains to translate away from the CTD disrupting the MC interface. All attempts to restrain the translations resulted in worse fits (e.g., R = 12% without translation and ∼8% if translations are allowed, Supporting Information Figure S3C). Examination of the models showed that a rotation of the two CTDs as one unit by ∼45° about the symmetry axis could restore contacts between the MD and CTD. The dimer interface is unchanged by the rotation. In the crystal structure, the MC interface is quite small and linked by a long, highly mobile tether suggesting that significant CTD rotation is possible. Although the exact rotation is not known, this new CTD orientation is much more consistent with ab initio models [Fig. 3(A)], supporting the key structural interpretations of the scattering data. The ab initio model shown in Figure 3 is an average of 20 independent Gasbor25 reconstructions. An average model from 20 independent DAMMIN26 reconstructions is nearly equivalent (data not shown), indicating the robustness of the solution. The CTD rotation is also consistent with EM studies of apo pig, human, and yeast Hsp90.15,24
Figure 3.
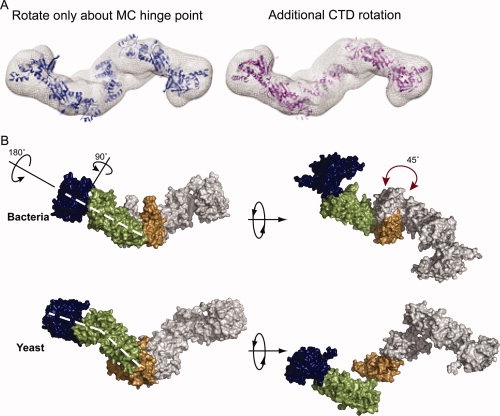
The apo yeast structure is distinct from the extended bacterial solution state. (A) When conformational space is searched only by rotations and translations about the hinge point, the resulting model loses the MC interace (blue). Rotation of the CTDs around the twofold axis by ∼45° (pink) restores the MD/CTD connection and better fits the Gasbor density (mesh). (B) In the yeast structure, the NTD is maximally extended by a ∼90° rotation away from the MD. The NM domains are also rotated away from one another by ∼180° as shown. These rotations cause a bend in the arms of the yeast protein not seen in HtpG (denoted by the dashed white lines). A rotation of the CTD (shown by the red arrow) causes a change in the point of contact between the MD and CTD leading to an angular chair-like structure.
Even with the CTD reconfiguration, the best fits for Hsc82 and Hsp90α still show significant deviation from the experimental data (Supporting Information Figure S3; Hsc82, R = 8.1%; Hsp90α, R = 8.9%). Noting that the previous EM study of pig Hsp90 showed two conformations,24 we tested for the presence of an additional conformation by fitting our data with a linear combination of two conformations. The open-extended state was further refined about residue 530, whereas the second conformation, represented by a known structure, was held fixed. At each refinement step, the best linear combination of the two states was determined. For the fixed structure, all known crystal structures were tested, as well as the bacterial apo SAXS structure and the two crystallographic tetramers observed for HtpG (see Materials and Methods section). The best open-extended state resulting from the single population searches was also held fixed, whereas a second population of the extended state was further refined. The best fits were obtained with a linear combination of an extended chair-like conformation (having a slightly smaller opening angle than found in the single population searches) and a small population of the crystallographic apo tetramer as seen in the HtpG crystal structure (Fig. 2 and Supporting Information Figure S3D; Hsc82, R = 5.9%; Hsp90α, R = 5.7%). The independently derived extended structure for Hsp90α is remarkably similar to the conformation found for Hsc82. The only difference between the two conformations is a small rotation (15°) of the Hsp90α NM domains toward the CTD. The extended conformation also correlates well with the previously described “flying seagull” SAXS volume for pig Hsp90.24 Attempts to model the data with additional extended conformations did not result in better fits to the data. Also, changes in monomer orientations in the extended model lead to worse fits to the scattering data, suggesting that the extended conformation is well defined in solution. Secondary structure rearrangements within the domains probably occurs, especially at the domain interfaces, but the resolution of the SAXS data limits our analysis to the rigid body motions of the domains.
Figure 2.
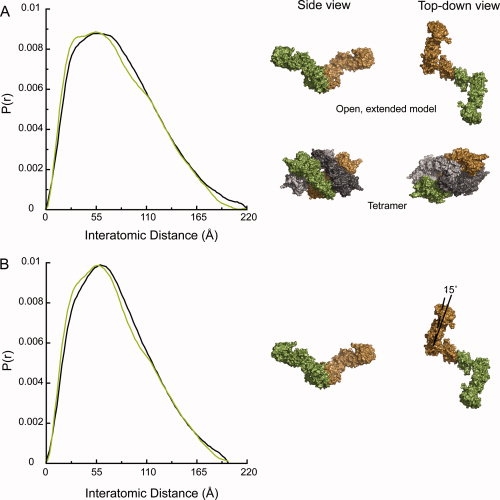
Hsc82 and Hsp90α are best modeled with similar extended conformations and a small population of tetramer. (A) The Hsc82 solution data (black) are fit with a mixture of two populations (green). The mixture is 93% of the shown open, extended model and 7% of the shown tetramer (R = 5.9%). The tetramer is the crystallographic tetramer found in the apo bacterial structure.14 (B) Hsp90α solution data (black) is fit with a mixture of 90% of the shown open, extended model and 10% of the tetramer shown in Panel A (R = 5.7%). The extended model for Hsp90α has a 15° rotation of the NM domains toward each other when compared with the Hsc82 model. One monomer is depicted in green and the other in brown. In the tetramer, the second biological dimer is shown in gray.
A comparison of the yeast open model with the extended state of bacterial Hsp90 shows the yeast protein in a much more extended conformation than the bacterial protein [Fig. 3(B)]. The NTD of the yeast structure, which is equivalent to the extended HtpG ADP NM crystal structure,14 is rotated away from the middle domain by ∼90° when compared with the bacterial structure. The NM domains of the yeast structure were then rotated away from one another by ∼180° while the opening angle remained nearly the same as in the bacterial structure. These two rotations result in a conformation where the arms of the dimer are bent when viewed from the side as opposed to the linear arrangement seen in bacteria [indicated by dashed white line in Fig. 3(B)]. Another striking difference is that the yeast dimerization domain (CTD) must be rotated ∼45° from the bacterial position [marked by the red arrows in Fig. 3(B)] causing the NM domain to contact the CTD at a different position. Because of the large domain rearrangements that occur between the bacterial and yeast structures, repacking of the MC interface in yeast probably occurs. At the low resolution of the SAXS data, these changes at the MC interface are undetectable. The net result is that the NM domains of the yeast protein are stretched much further apart forming a more angular structure. Overall, the bacterial protein is more linear in its conformation.
Although the amount of tetramer (Hsc82, 7%; Hsp90α, 10%), is small, this addition significantly improves the fit to the data (Fig. 2 and Supporting Information Figure S3). The data can be fit nearly equivalently within a range of 5–10% apo tetramer. Adding in either more or less apo tetramer does, however, significantly compromise the fit to the experimental data (Supporting Information Figure S4). The presence of a tetramer was also confirmed by size-exclusion chromatography and multiangle light scattering for Hsp90α. Small amounts of crosslinker (0.1% glutaraldehyde) were required to trap the tetramer under these conditions (data not shown). This level of crosslinker stabilizes transient complexes without significantly building up higher-order structures. The requirement for a crosslinker indicates that the tetramer is formed only by weak/transient interactions. Interactions with the column matrix could alter these weak interactions, making it difficult to quantify the amount of tetramer by gel filtration chromatography. This result further underscores the sensitivity of the SAXS data to subtle changes in solution state. The biological dimers that form the apo tetramer are similar to the second conformation observed by cryo-EM for pig Hsp90.24 The only difference between the two structures is that the NTD is more extended in the cryo-EM reconstruction than in the crystal structure. This conformation may be more prone to self-association given the MC angle and the high concentration conditions of SAXS. The lower concentrations required for EM would shift the equilibrium toward the dimer form as was reported.
Grp94 populates a similar extended conformation in the absence of nucleotide
The extent to which the conformational cycle of the ER homolog, Grp94, differs from other cytosolic homologs remains an open question. Recent crystal structures in the presence of ADP or the nonhydrolyzable AMPPNP revealed a unique V-shaped conformation that is more closed than the previously described apo states but that lacks NTD dimerization as seen in the yeast AMPPMP structure.22 In the Grp94 crystal, the NTD-MD conformation is virtually identical to that found in the apo HtpG structure, but the MD-CTD angle is markedly smaller for Grp94 and closely approximates that for the low pH state of apo HtpG found in solution (Krukenberg et al., in press). It is of significant interest to know how the solution structure of Grp94 is compared with the cytosolic yeast and human proteins and whether or not Grp94 has a different conformational cycle than other Hsp90s. To answer these questions, scattering data were collected for Grp94 in the absence of nucleotide.
To begin our analysis of the Grp94 SAXS data, we compared the experimental data to the P(r) predicted from the Grp94 crystal structure and our apo eukaryotic Hsp90 data (shown here as Hsc82). The three P(r) curves are significantly different from one another as seen in Figure 4(A). Although all have peaks around 55 Å, Grp94 in solution has a much greater number of larger interatomic distances than the Grp94 crystal structure and fewer large distances than the yeast apo state in solution. This difference is also reflected in very different values of Dmax: 180 Å for Grp94 in solution when compared with 110 Å for the Grp94 crystal state and 220 Å for Hsc82 in solution. Although the larger Grp94 distances are reminiscent of those for Hsc82 and Hsp90α, the data suggest that Grp94 on average populates a somewhat more compact conformation or set of conformations than its cytosolic homologs.
Figure 4.
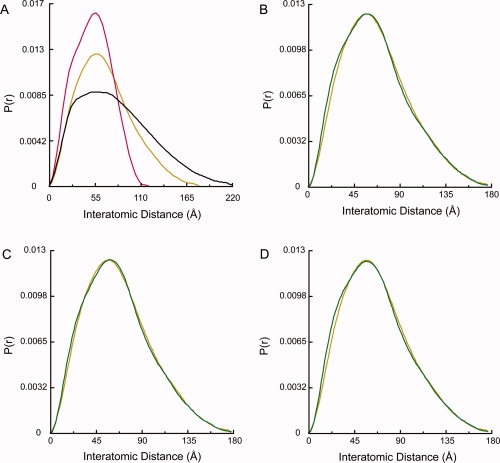
The apo Grp94 solution data is a mixture of primarily an extended state with small amounts of apo tetramer and a closed state. (A) The apo Grp94 solution data (gold) is different from both the Hsc82 data (black) and the Grp94 crystal state22 (red). (B) The Grp94 data (gold) is fit by a mixture of 77% extended, 10% apo tetramer, and 13% Grp94 crystal state (green, R = 3.9%). The extended model is shown in Figure 5. (C) The data (gold) are fit by varied amounts of each population with the two endpoints represented by the data in (B) and 77% extended and 23% apo tetramer (green, R = 2.8%). (D) The data (gold) can be equally well fit using the yeast AMPPNP-bound crystal state13 instead of the Grp94 crystal state. In green is the fit using 77% extended, 10% tetramer, and 13% yeast “ATP” state (R = 4.1%).
To test this hypothesis, rigid body modeling was conducted in the same way as described earlier. Based on preliminary negative-stain EM studies with Grp94, we also included a third population represented by the Grp94 crystal structure when modeling the Grp94 data. As seen for Hsc82 and Hsp90α, the Grp94 data are well fit with an open state with the same extended NM domain [Fig. 4(B), R = 3.9%]. The open state observed for Grp94 is unexpectedly similar to the open states observed for both Hsc82 and Hsp90α (Fig. 5). Although the MC open angles of the three structures are the same, the arms of the Grp94 state are not as extended as either Hsp90α or Hsc82. When compared with Hsc82, the arms of the Grp94-extended state are rotated toward each other by 20°. The Grp94 solution state also includes 10% apo tetramer and 13% Grp94 crystal structure [Fig. 4(B)]. As with the Hsc82 and Hsp90α, the data is also well fit with varied ratios of the minor conformations. The best fits to the data are found within the range of 10–23% apo tetramer with the fraction of Grp94 crystal structure varying from 13 to 0%, respectively. Representative fits of the two end points on the continuum are shown in Figure 4(B,C). The larger population of the more compact states (tetramer or crystal structure) is responsible for the intermediate P(r) profile observed in Figure 4(A).
Figure 5.
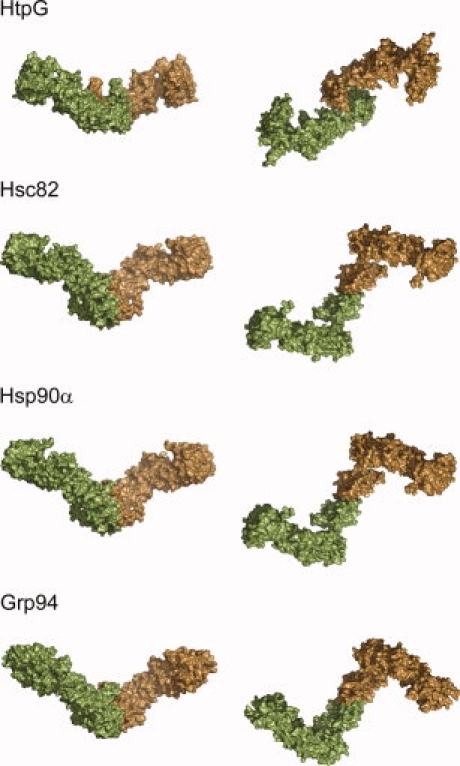
Apo yeast Hsc82, human Hsp90α, and mouse Grp94 are distinct from bacterial HtpG in solution. The side view shown in the left column demonstrates a similar open angle for all homologs, whereas the eukaryotic homologs have a more distinctive bend in the NM domains. The second view is a 90° rotation of the first, and this top–down view shows that the Hsc82, Hsp90α, and Grp94 structures share the same CTD rotation and extended NM domain when compared with HtpG. These differences give the eukaryotic structures a chair-like shape instead of the linear structure seen for HtpG.
The negative-stain EM confirmed that Grp94 is predominantly in an extended conformation in solution (Fig. 6) but also show a few particles similar to a closed state (Fig. 6, arrows). Because the P(r) profiles of the Grp94 and yeast AMPPNP crystal structures are highly similar (Supporting Information Figure S5C), we tested the yeast AMPPNP structure as the third population instead of the Grp94 crystal structure. Both crystal structures gave nearly identical fits to the experimental data as seen in Figure 4(D) (R = 4.1% with the yeast AMPPNP structure), suggesting that the two structures are indistinguishable in solution at these low levels. Other structures, including the high pH bacterial solution state,16 the apo crystal structure,14 and the ADP-compact state14 were also tested in combination with the extended state and the tetramer. Only the inclusion of the Grp94 crystal state and the AMPPNP-bound crystal state as the third population resulted in good fits to the scattering data.
Figure 6.
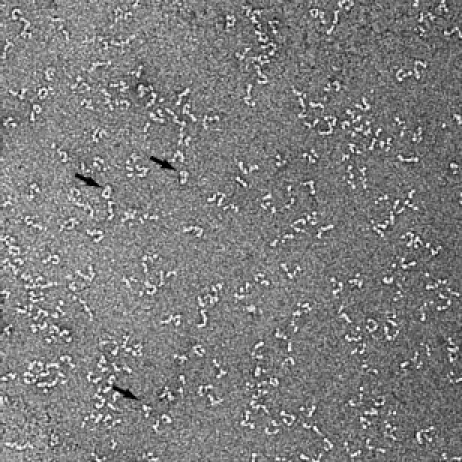
Negative-stain EM also shows that apo Grp94 is largely populated by an open extended state. A smaller population of a closed conformation is also present and indicated by the arrows.
Although the very best SAXS fits were obtained with only the extended state and the tetramer, the presence of closed structures in the negative-stain EM images and the nearly equivalent fits support the presence of a third conformation. Based on the EM, the closed state is more likely represented by the Grp94 crystal structure than the AMPPNP state, but given the small number of closed particles this assignment remains ambiguous. Another possibility is that Grp94 samples both the Grp94 crystal state and the yeast AMPPNP crystal state simultaneously.
Because the ER functions as a Ca2+ storage organelle and Grp94 has been reported as a Ca2+ binding protein,27,28 we also tested the effect of Ca2+ on Grp94 conformation. SAXS data revealed that, under our conditions, Ca2+ has a negligible effect on the conformational equilibrium (data not shown).
Nucleotide has a minimal role on the solution conformation of Grp94
Like cytosolic yeast and human Hsp90s, Grp94 requires ATP for function. To compare the effect of nucleotide on each homolog, we collected SAXS data for Hsc82, Hsp90α, and Grp94 in the presence of 10 mM AMPPNP. It has been previously demonstrated that ATP shifts the conformation of yeast Hsc82 to the closed state, and human Hsp90 remains largely in the open state in the presence of nucleotide,15 and our solution data confirm this observation [Fig. 7(A)]. Further analysis of the yeast Hsc82 AMPPNP data [Fig. 7(B)] shows that under the SAXS conditions, the data is fit with 39% extended state, 56% yeast AMPPNP-bound crystal state, and 5% tetramer (R = 5.4%). As with the apo data, a continuum of 5–10% tetramer is equally compatible with the observed scattering data (data not shown).
Figure 7.
AMPPNP has the largest effect on the conformation of Hsc82. (A) AMPPNP-bound Hsc82 (orange) is significantly more compact than apo Hsc82 (black). AMPPNP-bound Hsp90α (blue) is highly similar to apo Hsp90α (purple) and is best fit with the same mixture as apo Hsp90α. (B) AMPPNP-bound Hsc82 (orange) is best fit by a mixture (green) of 39% extended state, 56% yeast “ATP” crystal state, and 5% apo tetramer (R = 5.4%). (C) A comparison of apo Grp94 (gold) and AMPPNP-bound Grp94 (purple). (D) AMPPNP-bound Grp94 (purple) is fit with a mixture (green) of 58% extended state, 24% Grp94 crystal state, and 18% apo tetramer (R = 3.3%).
For Grp94, the presence of 10 mM AMPPNP causes a small shift in the P(r) curve [Fig. 7(C)]. This change in P(r) correlates with a small shift in the conformational equilibrium toward the closed state. Scattering data for Grp94 in the presence of AMPPNP is fit by a combination of 58% extended, 18% apo tetramer, and 24% of the Grp94 crystal structure [Fig. 7(D), R = 3.3%]. AMPPNP-bound Grp94 data is well fit with 10–18% tetramer and 27–24% Grp94 crystal structure, respectively. As with the apo Grp94 data, the fits are recapitulated while using the yeast AMPPNP structure instead of the Grp94 crystal structure (Supporting Information Figure S5) and are marginally better with the yeast structure (R = 2.0%). Although the amounts of the different populations are variable, it is clear that slightly more of either the Grp94 or yeast AMPPNP crystal state is present when nucleotide is bound. Data collected in the presence of ADP are essentially indistinguishable from the AMPPNP data (data not shown).
Discussion
In this study, we have used SAXS to characterize the apo conformations of yeast Hsc82 and ER Grp94 and compare them to human Hsp90α. Grp94 has long been thought to function in a unique manner when compared with the cytosolic homologs, but the differing responses of the cytosolic Hsp90s to nucleotide suggest that the apo conformations may be varied even among closely related homologs. Surprisingly, we have shown that all three homologs exist in a highly similar extended conformation in the absence of nucleotide, and that this conformation is quite distinct from the extended apo bacterial conformation (Fig. 5). All of the proteins from bacteria to humans have a similar open angle forming the cleft of the dimer, but a twist of the dimerization domain about the twofold axis and a rotation of the NM domains away from the plane of symmetry result in the arms of the eukaryotic proteins being stretched much further apart from one another in a chair-like conformation. For the eukaryotic Hsp90s, an additional rotation of the NTD away from the MD results in an even larger arm extension. This extended conformation differs from the more linear configuration of the bacterial protein and could potentially allow for a larger interaction platform for client proteins. The eukaryotic structures obtained here are in good agreement with the flying seagull form observed for pig apo Hsp90.24
Although Hsp90α, Hsc82, and Grp94 all have comparable extended conformations, there are subtle differences between the structures. All three structures share the same NM domain configuration, whereas the angle between the MD and CTD differs slightly for each protein. Hsc82 has the most extended conformation with the largest angle between the MD and CTD. Hsp90α is somewhat less extended with the NM domains rotated toward the CTD by 15°, and Grp94 represents the least extended conformation with the NM domain rotated 20° toward the CTD when compared with the Hsc82 structure. The differences between the conformations indicate that the structures of the different homologs may have been optimized for interaction with their specific client proteins.
The SAXS data presented here also indicate that apo Hsc82, Hsp90α, and Grp94 all exist in equilibrium between the extended state and a small population of an alternate conformation best represented by the crystallographic apo HtpG tetramer. Given their exposed hydrophobic interfaces, it is not surprising that tetramers can form at high concentration. All attempts to model the data using another dimer conformation failed to produce the correct shape observed around the P(r) peak. While we also tried the ADP tetramer observed in the HtpG crystallographic asymmetric unit, the fits were much better using a small amount of the crystallographic packing conformation seen in apo HtpG. The biological dimer within this tetramer is in good agreement with the second less populated dimer population seen in the cryo-EM images of pig Hsp90.24 The presence of a second apo population is also in good agreement with recent FRET studies demonstrating the presence of at least two open conformations for yeast Hsp90.18,29 The higher concentrations of the SAXS samples may promote the tetramer formation, whereas the apo crystal structure dimer is observed under the lower concentration EM conditions. Accordingly, the tetramer appears transient at lower concentrations but can be trapped with low levels of glutaraldehyde and observed by size-exclusion chromatography. These data suggest that apo conformational equilibrium includes a very extended conformation having a rotated CTD and a minor population of a second less extended conformation having a CTD orientation similar to the crystal structure. The addition of nucleotide shifts the equilibrium toward more closed states having a CTD orientation like that observed in the crystal structures.
Unlike cytosolic yeast and human Hsp90, apo Grp94 also populates at low level a third, more closed, conformation represented by either the known Grp94 ADP/AMPPNP crystal structures or the yeast AMPPNP crystal structure. Our EM data support this conclusion. Interestingly, an early EM study of Grp94 using rotary shadowing also described both an extended and compact conformation for Grp94.30 Because the P(r) curves calculated from these crystal structures are so similar to one another, SAXS data cannot be used to distinguish between them at the low levels observed. Analysis of the EM images does, however, suggest that the Grp94 crystal structure is more relevant although the presence of both closed states cannot be ruled out as a possibility. All of these states appear to be largely isoenergetic, highlighting the stochastic nature of the Hsp90 chaperone cycle. Hsp90 exists in equilibrium between multiple conformations and the binding of nucleotide functions to shift the preexisting equilibrium as opposed to determining a specific conformation. The binding of cochaperones and client proteins may further regulate the cycle by affecting the conformational equilibrium of Hsp90. To answer this question, further studies of the interaction of Hsp90 with other proteins will be required.
In previous studies, it has been shown that Ca2+ influences the binding of peptides to Grp94.31 Therefore, we tested the ability of Ca2+ to affect the confirmation of Grp94. Under the conditions reported here, Ca2+ does not affect the conformation of Grp94. Unlike Ca2+, AMPPNP and ADP do have a slight affect on the conformational equilibria, favoring a slightly larger population of the more closed yeast AMPPNP or Grp94 crystal structures. Although AMPPNP stabilizes the compact state to some extent, the equilibrium remains solidly in favor of the extended state. Although unexpected, this observation is in strong agreement with a previous kinetic analysis of Grp94 showing Grp94 to be 97% in an open state in the presence of nucleotide.23
As previously observed,15,23 AMPPNP-bound Hsc82 populates the closed AMPPNP-bound crystal state to a larger extent than Grp94 or Hsp90α. This result was confirmed with our SAXS data indicating that Hsc82 is 60% in the closed conformation in the presence of nucleotide. This number is lower than the 80% previously reported from kinetic and EM studies, and the difference may be a result of different experimental conditions. All of the SAXS data were collected at 25°C, whereas the kinetic analysis was done at 37°C. The EM samples were also incubated at 37°C before data collection. It has been established that the ATPase rate of yeast Hsp90 increases with temperature,23 and this may explain the different levels of the closed state seen using SAXS or other methods.
It is especially interesting that Grp94 is able to populate an extended conformation when compared with the cytosolic Hsp90s. Only a few substrates of Grp94 have been demonstrated. Most of these substrates are cell-surface proteins or secreted proteins, and given the conditions of the ER many of these substrates contain disulfide bonds. This sets Grp94 client proteins apart from the Hsp90 client proteins found in the cytosol. Also, both Hsc82 and Hsp90α require cochaperones for function. These helper proteins are thought to assist in the loading of client proteins and in the regulation of the chaperone cycle, but unlike its cytosolic homologs, Grp94 has no known cochaperones. This data in combination with the alternative conformation seen in the crystal structure of Grp94 suggest a unique conformational and functional cycle for Grp94. The data presented here suggest that Grp94 may utilize some conformations similar to the cytosolic Hsp90s, but the regulation of these conformations may be very different. This idea is further supported by recent SAXS data demonstrating the ability of HtpG to populate a Grp94-like conformation (Krukenberg et al., in press) at low pH. Overall, the data supports a broader set of similarities between the eukaryotic proteins with important differences, such as the extent to which ATP shifts the equilibrium to a closed state, still existing between homologs. For the future it will be quite important to elucidate the structural basis for the observed equilibrium differences and to correlate these with the functional properties of the different homologs of Hsp90.
Materials and Methods
Protein expression and purification
Full-length Hsc82 and Hsp90α were purified as described previously.15,32 Truncated Grp94 (Δ1-55) was PCR amplified from cDNA of mouse Grp94 (ATCC) and subsequently cloned into the pET151/D-TOPO (Invitrogen), which includes a TEV-cleavable N-terminal His tag. Grp94 was purified from induced E. coli BL21(DE3) cells (Invitrogen) using Ni2+-NTA resin (Qiagen). His tags were removed by cleavage with TEV protease. Uncleaved protein was removed via a second Ni-NTA affinity column. The protein was further purified by anion exchange (MonoQ) and size exclusion chromatography on a Superdex S200 column (GE Healthcare). Protein was concentrated in 50 mM Tris, pH 8, 50 mM KCl using Ultrafree Biomax concentrators (Millipore) to a final concentration of 10 mg/mL based on UV280 absorption. Protein was flash-frozen in liquid nitrogen and stored at −80°C. Analytical size exclusion chromatography was performed to confirm the stability of Grp94 after storage at −80°C. The measured ATPase rates for all homologs were comparable with the published rates.22,33,34
SAXS data collection
SAXS data were collected at SSRL beamline 4-2 and at the SIBYLS beamline (12.3.1) at the advanced light source (ALS) as previously described.16 Data were collected for protein in 50 mM Tris pH 8, 50 mM KCl, 10 mM MgCl2, 1 mM DTT, and ±10 mM AMPPNP. For Grp94, data were also collected for samples with 10 mM ADP or 10 mM CaCl2. Other Grp94 samples contained CaCl2 in the absence of MgCl2 or no divalent cations. The full-length Grp94 was tested but no differences were seen between the full-length protein and the construct without the ER signal sequence.
SAXS data analysis
The buffer subtracted scattering data, I(Q), were scaled and merged using the program PRIMUS.35 The interatomic distance distribution functions (P(r)) were then calculated using the program GNOM.36Dmax was determined by constraining rmax to zero and then varying rmax. Dmax was chosen as the rmax, where the P(r) smoothly approached zero based upon perceptual criteria. Initial ab initio models were also calculated using the software Gasbor and DAMMIN.25,26 Using each program, 20 reconstructions were calculated and then averaged using the DAMAVER software package.37 Molecular modeling of the scattering data was then done using the in-house software PRFIT as previously described.16 Initial rounds of refinement resulted in models where the MD no longer contacted the CTD. This was corrected by rotating the two CTDs as a single body about the twofold axis until contact was restored with the MD. To test for the presence of a second population in solution, the modeled structure was refined in the presence of a second fixed structure. After each round of refinement, the optimal linear combination of the two structures was determined. For the fixed population, we tested the yeast “ATP” crystal structure (PDB code, 2CG9),13 the compact HtpG ADP crystal structure (2IOP),14 the low (unpublished data) and high pH HtpG solution states,16 the Grp94 crystal structure (2IOV),22 the crystallographic apo tetramer (2IOQ), and the crystallographic ADP teteramer (2IOP).14 The quality of all fits were measured with an R-factor metric similar to the one used in X-ray crystallography (R = Σ‖‖Pobs(r)| − |Pcalc(r)‖‖/Σ |Pobs(r)|). Coordinates for the resulting SAXS models can be found at http://www.msg.ucsf.edu:16080/agard/pdb/htpg.html.
Electron microscopy
Grp94 protein was negatively stained with uranyl formate on thin carbon-layered (40–50 Å thick) 400 mesh copper grids (Pelco) as previously described.15,38 Grp94 samples (200 nM) in 50 mM Tris pH 8.0, 50 mM KCl, 10 mM MgCl2, and 1 mM DTT were imaged using a Tecnai G2 Spirit TEM (FEI) operated at 120 keV. Micrograph images were recorded using a 4k × 4k CCD camera (Gatan) at 50,000× magnification with 2.2 Å pixel size.
Acknowledgments
The authors thank the staff at the Advanced Light Source SIBYLS beamline 12.3.1 (especially G. Hura) and at the Stanford Synchrotron Radiation Laboratory beamline 4-2 (especially H. Tsuruta). They also thank C. Cunningham and T. Street for their helpful discussions.
Glossary
Abbreviations:
- EM
electron microscopy
- ER
endoplasmic reticulum
- Grp94
endoplasmic reticulum Hsp90 homolog
- Hsc82
S. cerevisiea Hsp90 homolog
- Hsp90α
human heat shock protein 90
- HtpG
E. coli Hsp90 homolog
- SAXS
small angle X-ray scattering.
References
- 1.Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- 2.Weissman JS, Hohl CM, Kovalenko O, Kashi Y, Chen S, Braig K, Saibil HR, Fenton WA, Horwich AL. Mechanism of GroEL action: productive release of polypeptide from a sequestered position under GroES. Cell. 1995;83:577–587. doi: 10.1016/0092-8674(95)90098-5. [DOI] [PubMed] [Google Scholar]
- 3.Kurt N, Rajagopalan S, Cavagnero S. Effect of hsp70 chaperone on the folding and misfolding of polypeptides modeling an elongating protein chain. J Mol Biol. 2006;355:809–820. doi: 10.1016/j.jmb.2005.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLaughlin SH, Smith HW, Jackson SE. Stimulation of the weak ATPase activity of human hsp90 by a client protein. J Mol Biol. 2002;315:787–798. doi: 10.1006/jmbi.2001.5245. [DOI] [PubMed] [Google Scholar]
- 5.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 6.Brown MA, Zhu L, Schmidt C, Tucker PW. Hsp90—from signal transduction to cell transformation. Biochem Biophys Res Commun. 2007;363:241–246. doi: 10.1016/j.bbrc.2007.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richter K, Buchner J. Hsp90: chaperoning signal transduction. J Cell Physiol. 2001;188:281–290. doi: 10.1002/jcp.1131. [DOI] [PubMed] [Google Scholar]
- 8.Zhao R, Davey M, Hsu YC, Kaplanek P, Tong A, Parsons AB, Krogan N, Cagney G, Mai D, Greenblatt J, Boone C, Emili A, Houry WA. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell. 2005;120:715–727. doi: 10.1016/j.cell.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 9.Li Y, Zhang T, Schwartz SJ, Sun D. New developments in Hsp90 inhibitors as anti-cancer therapeutics: mechanisms, clinical perspective and more potential. Drug Resist Updat. 2009;12:17–27. doi: 10.1016/j.drup.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiosis G, Lucas B, Huezo H, Solit D, Basso A, Rosen N. Development of purine-scaffold small molecule inhibitors of Hsp90. Curr Cancer Drug Targets. 2003;3:371–376. doi: 10.2174/1568009033481778. [DOI] [PubMed] [Google Scholar]
- 11.Solit DB, Osman I, Polsky D, Panageas KS, Daud A, Goydos JS, Teitcher J, Wolchok JD, Germino FJ, Krown SE, Coit D, Rosen N, Chapman PB. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res. 2008;14:8302–8307. doi: 10.1158/1078-0432.CCR-08-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 13.Ali MM, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shiau AK, Harris SF, Southworth DR, Agard DA. Structural analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell. 2006;127:329–340. doi: 10.1016/j.cell.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 15.Southworth DR, Agard DA. Species-dependent ensembles of conserved conformational states define the Hsp90 chaperone ATPase cycle. Mol Cell. 2008;32:631–640. doi: 10.1016/j.molcel.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krukenberg KA, Forster F, Rice LM, Sali A, Agard DA. Multiple conformations of E. coli Hsp90 in solution: insights into the conformational dynamics of Hsp90. Structure. 2008;16:755–765. doi: 10.1016/j.str.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graf C, Stankiewicz M, Kramer G, Mayer MP. Spatially and kinetically resolved changes in the conformational dynamics of the Hsp90 chaperone machine. EMBO J. 2009;28:602–613. doi: 10.1038/emboj.2008.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mickler M, Hessling M, Ratzke C, Buchner J, Hugel T. The large conformational changes of Hsp90 are only weakly coupled to ATP hydrolysis. Nat Struct Mol Biol. 2009;16:281–286. doi: 10.1038/nsmb.1557. [DOI] [PubMed] [Google Scholar]
- 19.Randow F, Seed B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat Cell Biol. 2001;3:891–896. doi: 10.1038/ncb1001-891. [DOI] [PubMed] [Google Scholar]
- 20.Melnick J, Dul JL, Argon Y. Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature. 1994;370:373–375. doi: 10.1038/370373a0. [DOI] [PubMed] [Google Scholar]
- 21.Immormino RM, Dollins DE, Shaffer PL, Soldano KL, Walker MA, Gewirth DT. Ligand-induced conformational shift in the N-terminal domain of GRP94, an Hsp90 chaperone. J Biol Chem. 2004;279:46162–46171. doi: 10.1074/jbc.M405253200. [DOI] [PubMed] [Google Scholar]
- 22.Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol Cell. 2007;28:41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frey S, Leskovar A, Reinstein J, Buchner J. The ATPase cycle of the endoplasmic chaperone Grp94. J Biol Chem. 2007;282:35612–35620. doi: 10.1074/jbc.M704647200. [DOI] [PubMed] [Google Scholar]
- 24.Bron P, Giudice E, Rolland JP, Buey RM, Barbier P, Diaz JF, Peyrot V, Thomas D, Garnier C. Apo-Hsp90 coexists in two open conformational states in solution. Biol Cell. 2008;100:413–425. doi: 10.1042/BC20070149. [DOI] [PubMed] [Google Scholar]
- 25.Svergun DI, Petoukhov MV, Koch MH. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van PN, Peter F, Soling HD. Four intracisternal calcium-binding glycoproteins from rat liver microsomes with high affinity for calcium. No indication for calsequestrin-like proteins in inositol 1,4,5-trisphosphate-sensitive calcium sequestering rat liver vesicles. J Biol Chem. 1989;264:17494–17501. [PubMed] [Google Scholar]
- 28.Macer DR, Koch GL. Identification of a set of calcium-binding proteins in reticuloplasm, the luminal content of the endoplasmic reticulum. J Cell Sci. 1988;91:61–70. doi: 10.1242/jcs.91.1.61. [DOI] [PubMed] [Google Scholar]
- 29.Hessling M, Richter K, Buchner J. Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nat Struct Mol Biol. 2009;16:287–293. doi: 10.1038/nsmb.1565. [DOI] [PubMed] [Google Scholar]
- 30.Wearsch PA, Nicchitta CV. Endoplasmic reticulum chaperone GRP94 subunit assembly is regulated through a defined oligomerization domain. Biochemistry. 1996;35:16760–16769. doi: 10.1021/bi962068q. [DOI] [PubMed] [Google Scholar]
- 31.Biswas C, Ostrovsky O, Makarewich CA, Wanderling S, Gidalevitz T, Argon Y. The peptide-binding activity of GRP94 is regulated by calcium. Biochem J. 2007;405:233–241. doi: 10.1042/BJ20061867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cunningham CN, Krukenberg KA, Agard DA. Intra- and intermonomer interactions are required to synergistically facilitate ATP hydrolysis in Hsp90. J Biol Chem. 2008;283:21170–21178. doi: 10.1074/jbc.M800046200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLaughlin SH, Ventouras LA, Lobbezoo B, Jackson SE. Independent ATPase activity of hsp90 subunits creates a flexible assembly platform. J Mol Biol. 2004;344:813–826. doi: 10.1016/j.jmb.2004.09.055. [DOI] [PubMed] [Google Scholar]
- 34.Prodromou C, Panaretou B, Chohan S, Siligardi G, O'Brien R, Ladbury JE, Roe SM, Piper PW, Pearl LH. The ATPase cycle of Hsp90 drives a molecular ‘clamp’ via transient dimerization of the N-terminal domains. EMBO J. 2000;19:4383–4392. doi: 10.1093/emboj/19.16.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Konarev PV, Volkov VV, Sokolova AV, Koch MH, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Cryst. 2003;36:1277–1282. [Google Scholar]
- 36.Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr. 1992;25:495–503. [Google Scholar]
- 37.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohi M, Li Y, Cheng Y, Walz T. Negative staining and image classification—powerful tools in modern electron microscopy. Biol Proced Online. 2004;6:23–34. doi: 10.1251/bpo70. [DOI] [PMC free article] [PubMed] [Google Scholar]