

Abstract
5-Aminolevulinate synthase (ALAS) controls the rate-limiting step of heme biosynthesis in mammals by catalyzing the condensation of succinyl-coenzyme A and glycine to produce 5-aminolevulinate, coenzyme-A (CoA), and carbon dioxide. ALAS is a member of the α-oxoamine synthase family of pyridoxal 5′-phosphate (PLP)-dependent enzymes and shares high degree of structural similarity and reaction mechanism with the other members of the family. The X-ray crystal structure of ALAS from Rhodobacter capsulatus reveals that the alkanoate component of succinyl-CoA is coordinated by a conserved arginine and a threonine. The functions of the corresponding acyl-CoA-binding residues in murine erthyroid ALAS (R85 and T430) in relation to acyl-CoA binding and substrate discrimination were examined using site-directed mutagenesis and a series of CoA-derivatives. The catalytic efficiency of the R85L variant with octanoyl-CoA was 66-fold higher than that of the wild-type protein, supporting the proposal of this residue as key in discriminating substrate binding. Substitution of the acyl-CoA-binding residues with hydrophobic amino acids caused a ligand-induced negative dichroic band at 420 nm in the CD spectra, suggesting that these residues affect substrate-mediated changes to the PLP microenvironment. Transient kinetic analyses of the R85K variant-catalyzed reactions confirm that this substitution decreases microscopic rates associated with formation and decay of a key reaction intermediate and show that the nature of the acyl-CoA tail seriously affect product binding. These results show that the bifurcate interaction of the carboxylate moiety of succinyl-CoA with R85 and T430 is an important determinant in ALAS function and may play a role in substrate specificity.
Keywords: 5-aminolevulinate, 5-aminolevulinate synthase, heme, pyridoxal, pyridoxal 5′-phosphate, α-oxoamine synthase family, phenolic oxygen, quinonoid intermediate
Introduction
5-Aminolevulinate synthase (ALAS; EC 2.3.1.37) is a pyridoxal 5′-phosphate (PLP)-dependent enzyme consisting of two identical subunits, each containing one molecule of covalently bound PLP. ALAS catalyzes the Claisen-like condensation of glycine and succinyl-CoA to yield carbon dioxide (CO2), CoA, and 5-amino-4-oxopentanoate (5-aminolevulinate; ALA), and represents the first step of porphyrin biosynthesis in animals, fungi, and some bacteria. The structural and mechanistic properties of ALAS are markedly similar to those of 8-amino-7-oxononanoate synthase (AONS), serine palmitoyl transferase (SPT), and 2-amino-3-ketobutyrate-CoA ligase (KBL).1–3
The X-ray crystal structure of the holo form of Rhodobacter capsulatus ALAS was solved at 2.1 Å resolution and also as enzyme-substrate complexes with either glycine (2.7 Å) or succinyl-CoA (2.8 Å).4 ALAS is classified as a member of the α-oxoamine synthase subfamily of fold type I PLP-dependent enzymes. AONS, SPT, and KBL are the other members and represent the closest structural relatives, with the enzymes of the subfamily sharing a Cα root mean square deviation of ∼1.5 Å.5,6 The reaction chemistries are also highly similar, all involving small amino acids, CoA esters, and 1,3-aminoketones. AONS catalyzes the committed step in biotin biosynthesis,7 SPT catalyzes the first step of sphingolipid biosynthesis,8 and KBL catalyzes the degradation of threonine.9
Despite the remarkable structural and mechanistic similarities in this important group of enzymes, the molecular mechanisms underlying substrate specificity remain largely unexplored. SPTs utilize palmitoyl-CoA as the preferred physiological substrate,10 however, Han et al. have shown that the SPT of a coccolithovirus is more active when utilizing myristoyl-CoA, a substrate similar to palmitoyl-CoA, but shorter by two carbons.11 Before the elucidation of the X-ray crystal structure of R. capsulatus ALAS, the bacterial enzyme-catalyzed reaction was examined with nonphysiological acyl-CoA derivatives as substrates.12 Results of this investigation indicate that some naturally occurring three, four, and five carbon CoA thioesters can act as substrates and that both acyl chain length and hydrophilicity of the acyl-CoA substrate are important factors in determining specificity. The CoA substrate specificity of ALAS is of interest due to the localization of the eukaryotic enzyme in the inner mitochondrial matrix. Specifically, 90% of cellular acetyl-CoA and between 92 and 97% of short and long chain acyl-CoAs are located within this organelle,13 providing an abundant supply of possible alternative substrates for murine erythroid ALAS (meALAS). As such, promiscuous reactions with alternative CoA substrates would produce highly reactive 1-3-aminoketones, of presently unknown biological significance, that could potentially dimerize and form toxic dihydropyrazines.14,15
Previous investigations regarding the binding of the amino acid substrate of ALAS and substrate specificity led to the conclusion that the ALAS active site only accommodates the smallest naturally occurring amino acid, namely glycine.16 Variants of R. sphaeroides ALAS in which the glycine-binding threonine (T83) is replaced with the subtly smaller amino acid serine show a dramatic improvement in acceptance of nonphysiological amino acid substrates.16 This finding along with the crystal structures suggest that steric factors within the glycine-binding region of the active site are the major determinants of amino acid substrate specificity.
The ALAS active site is within a cleft at the subunit interface and is delimited by a β-strand bent around the PLP cofactor, in which the pyridinium ring of the cofactor lies at the bottom of the cavity.4 Connection between the surface of the enzyme and the active site is by an amphipathic channel, which is occupied by succinyl-CoA in the substrate-bound structure (see Fig. 1). Two distinct moieties of succinyl-CoA interact with the enzyme: the solvent accessible adenosyl component and the buried succinate. The alkanoic acid moiety of succinyl-CoA is bound to the active site via a strong hydrogen bond network that stabilizes a closed enzyme conformation (Lendrihas et al., submitted).4,17 At the end of a hydrophobic tunnel, the guanidino group of the highly conserved R21 (R85 in meALAS) donates a hydrogen bond to the carboxylate constituent of succinyl-CoA (see Fig. 1). Simultaneously, the hydroxyl group of the conserved T365 (T430 in meALAS), which is positioned at the apex of a conformationally dynamic active site loop, bridges both the carboxylic acid moiety of succinyl-CoA and the side chain of R21 to complete a hydrogen bonding triad (see Fig. 1).4 Accordingly, the chemical characteristics of the acyl-tail of the CoA substrate may be a determining factor for the enzyme in discriminating substrate entry into the active site.
Figure 1.
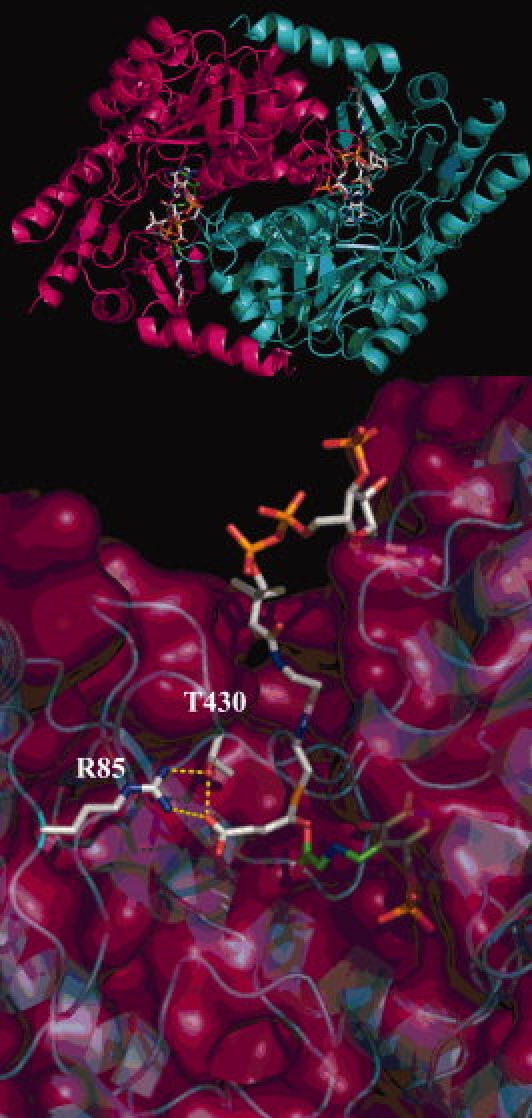
The acyl-CoA binding cleft in R. capsulatus ALAS. The ALAS dimer appears above the hydrogen bond network maintained between the alkanoic acid component of succinyl-CoA and the side chains of the conserved residues (R21 and T365) is indicated by dashed yellow lines. The PLP cofactor, succinyl-CoA substrate and the corresponding R and T residues (R85 and T430) are shown in stick format.
In this study, we investigate the role of the conserved R85 and T430 residues of meALAS in recognition and binding of the acyl-CoA substrate in relation to catalysis. Substitutions of the conserved residues with more hydrophobic amino acids (i.e., R85L and T430V) were introduced to examine the effect of hydrophobicity and steric hindrance on specificity toward the CoA-derived substrate. Such a difference would alter the aliphaticity of the substrate-binding cleft, which in turn, could affect the acyl chain-binding properties of this channel. The results presented here for the R85 and T430 variants of meALAS show that these residues are involved in both the orientation and binding of the succinyl-CoA substrate in the active site and may also, following the substrate binding, assist in enzyme closure.
Results
Kinetic characterization of the R85 and R85/T430 variants
The steady-state kinetic parameters of the ALAS variants were determined and the results are summarized in Table I. Wild-type ALAS was active with all of the CoA-derivatives tested. The Km for octanoyl-CoA was the lowest with a value of 0.51 μM compared with 2.9 μM for succinyl-CoA. This decreased value contributed to a catalytic efficiency value that was two-fold higher than the reaction completed with the physiological substrate succinyl-CoA. Glutaryl-CoA and β-hydroxybutyryl-CoA were the least catalytically efficient, due to six-fold increases in the Km for both substrates. Changing R85 to leucine (R85L) imparted dramatic changes with respect to the physiological substrate succinyl-CoA. The kcat associated with the R85L-catalyzed reaction using succinyl-CoA as the substrate decreased greater than 11-fold, whereas the 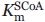 increased seven-fold. The 66-fold increase in catalytic efficiency toward octanoyl-CoA as well as the four-fold increase found with butyryl-CoA for the R85L variant highlight a shift toward acceptance of more hydrophobic CoA-derivatives within the acyl-CoA-binding cleft of this enzyme. The replacement of R85 with lysine (R85K) yielded a turnover number that was similar to that of the wild-type enzyme. When the different acyl-CoA substrates were tested, this pattern continued with the exception of the reaction with glutaryl-CoA. This CoA-derivative reacted slower than the physiological substrate succinyl-CoA as evidenced by a 21-fold decrease in kcat. Among the catalytic efficiencies calculated for R85K, octanoyl-CoA yielded the highest with a value 13-fold greater than that of the same reaction containing succinyl-CoA. The steady-state data obtained from the double variant (R85L/T430V) suggest that steric hindrance as well as hydrophobicity of the active site are important determinants for preference of CoA-derivative. Affinity for octanoyl-CoA in the double variant is diminished, resulting in a two-fold higher Km. Conversely, the reaction of the double variant with butyryl-CoA gave a Km value of 0.54 μM, a value indistinguishable from octanoyl-CoA in the R85L variant, and 31-fold lower than octanoyl-CoA in the double variant. The double variant-catalyzed reaction with glutaryl-CoA, a derivative similar to succinyl-CoA but longer by one methylene carbon, showed undetectable activity as measured under the assay conditions tested. These data are shown graphically as normalized specificity constants in Figure 2, in which the ratio of the catalytic efficiency of each variant for a particular substrate is compared with the catalytic efficiency of the variant with succinyl-CoA.
increased seven-fold. The 66-fold increase in catalytic efficiency toward octanoyl-CoA as well as the four-fold increase found with butyryl-CoA for the R85L variant highlight a shift toward acceptance of more hydrophobic CoA-derivatives within the acyl-CoA-binding cleft of this enzyme. The replacement of R85 with lysine (R85K) yielded a turnover number that was similar to that of the wild-type enzyme. When the different acyl-CoA substrates were tested, this pattern continued with the exception of the reaction with glutaryl-CoA. This CoA-derivative reacted slower than the physiological substrate succinyl-CoA as evidenced by a 21-fold decrease in kcat. Among the catalytic efficiencies calculated for R85K, octanoyl-CoA yielded the highest with a value 13-fold greater than that of the same reaction containing succinyl-CoA. The steady-state data obtained from the double variant (R85L/T430V) suggest that steric hindrance as well as hydrophobicity of the active site are important determinants for preference of CoA-derivative. Affinity for octanoyl-CoA in the double variant is diminished, resulting in a two-fold higher Km. Conversely, the reaction of the double variant with butyryl-CoA gave a Km value of 0.54 μM, a value indistinguishable from octanoyl-CoA in the R85L variant, and 31-fold lower than octanoyl-CoA in the double variant. The double variant-catalyzed reaction with glutaryl-CoA, a derivative similar to succinyl-CoA but longer by one methylene carbon, showed undetectable activity as measured under the assay conditions tested. These data are shown graphically as normalized specificity constants in Figure 2, in which the ratio of the catalytic efficiency of each variant for a particular substrate is compared with the catalytic efficiency of the variant with succinyl-CoA.
Table I.
Comparison of Steady-State Kinetic Constants for Wild-type ALAS, R85K, R85L, and R85L/T430V with CoA Derivatives as Substrates
| Parameter | Wild-type ALAS | R85K | R85L | R85L/T430V |
|---|---|---|---|---|
| Succinyl-CoA as a substrate | ||||
| kcat, min−1 | 10.0 ± 0.2 | 6.4 ± 0.2 | 0.94 ± 0.10 | 0.11 ± 0.03 |
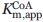 , μM , μM
|
2.9 ± 0.1 | 12.4 ± 0.6 | 20.3 ± 1.0 | 9.6 ± 0.8 |
|
kcat/, min−1 μM−1
|
3.6 ± 0.2 | 0.53 ± 0.04 | 0.050 ± 0.003 | 0.010 ± 0.002 |
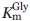 , mM , mM
|
24.2 ± 0.4 | 20.0 ± 0.4 | 63.1 ± 2.2 | 98.4 ± 3.5 |
|
kcat/, min−1 mM−1
|
0.43 ± 0.04 | 0.32 ± 0.06 | 0.010 ± 0.002 | 0.001 ± 0.002 |
| Octanoyl-CoA as a substrate | ||||
| kcat, min−1 | 3.4 ± 0.2 | 10.3 ± 0.4 | 1.8 ± 0.3 | (1.0 ± 0.1) × 10−3 |
|
, μM
|
0.51 ± 0.04 | 1.5 ± 0.04 | 0.55 ± 0.03 | 17.2 ± 0.6 |
|
kcat/, min−1 μM−1
|
6.8 ± 0.8 | 7.0 ± 0.2 | 3.3 ± 0.1 | (5.6 ± 0.2) × 10−5 |
|
, mM
|
17.1 ± 0.9 | 25.2 ± 1.1 | 55.2 ± 5.0 | 74.2 ± 3.3 |
|
kcat/, min−1 mM−1
|
0.14 ± 0.01 | 0.52 ± 0.02 | 0.030 ± 0.005 | (1.0 ± 0.08) × 10−5 |
| Butyryl-CoA as a Substrate | ||||
| kcat, min−1 | 6.3 ± 0.3 | 10.2 ± 0.3 | 2.01 ± 0.08 | 0.060 ± 0.002 |
|
, μM
|
6.1 ± 0.09 | 2.7 ± 0.1 | 9.3 ± 1.0 | 0.54 ± 0.03 |
|
kcat/, min−1 μM−1
|
1.0 ± 0.1 | 3.7 ± 0.4 | 0.21 ± 0.04 | 0.03 ± 0.002 |
|
, mM
|
29.3 ± 4.6 | 17.2 ± 0.7 | 70.3 ± 6.6 | 88.2 ± 3.9 |
|
kcat/, min−1 mM−1
|
0.26 ± 0.03 | 0.50 ± 0.01 | 0.030 ± 0.005 | (6.1 ± 0.8) × 10−4 |
| β-Hydroxybutyryl-CoA as a Substrate | ||||
| kcat, min−1 | 4.0 ± 0.8 | 2.8 ± 0.1 | 0.65 ± 0.03 | (1.0 ± 0.2) × 10−4 |
|
, μM
|
9.8 ± 1.0 | 5.5 ± 0.2 | 6.1 ± 0.8 | 74.2 ± 0.6 |
|
kcat/, min−1 μM−1
|
0.41 ± 0.04 | 0.51 ± 0.04 | 0.11 ± 0.02 | (1.3 ± 0.4) × 10−6 |
|
, mM
|
22.1 ± 0.8 | 18.2 ± 3.2 | 59.1 ± 4.7 | 92.2 ± 6.0 |
|
kcat/, min−1 mM−1
|
0.17 ± 0.06 | 0.14 ± 0.03 | 0.011 ± 0.003 | (1.0 ± 0.3) × 10−6 |
| Glutaryl-CoA as a Substrate | ||||
| kcat, min−1 | 7.0 ± 0.4 | 0.30 ± 0.03 | 2.2 ± 0.1 | n/d |
|
, μM
|
17.0 ± 1.6 | 7.5 ± 0.6 | 30.1 ± 1.1 | n/d |
|
kcat/, min−1 μM−1
|
0.41 ± 0.05 | 0.04 ± 0.003 | 0.07 ± 0.006 | n/d |
|
, mM
|
28.4 ± 0.8 | 21.2 ± 0.2 | 70.6 ± 4.2 | n/d |
|
kcat/, min−1 mM−1
|
0.29 ± 0.02 | 0.010 ± 0.002 | (8.0 ± 0.7) × 10−4 | n/d |
n/d, not determined.
Figure 2.
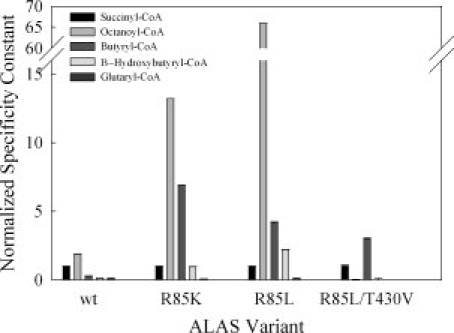
Comparison of normalized specificity constants for murine eALAS variants with different CoA substrates. The specificity constant 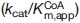 with succinyl-CoA as substrate was arbitrarily defined as equal to 1.00 for each ALAS variant.
with succinyl-CoA as substrate was arbitrarily defined as equal to 1.00 for each ALAS variant.
Circular dichroism spectroscopy
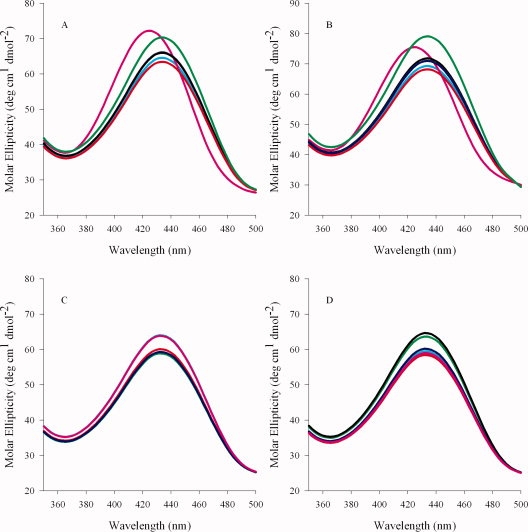
To verify whether the R85K, R85L, or R85L/T430V amino acid substitutions introduced substantial changes in secondary structure, CD spectra in the far-UV region (200–270 nm) were recorded for the wild-type and variant enzymes (data not shown). All enzymes displayed similar CD spectra indicating that the introduced residue exchanges did not result in gross differences in the overall conformation of the ALAS protein. The enzyme-bound cofactor gives rise to a defined CD spectrum in the visible region (300–500 nm) because of anisotropic interactions between the amino acid side chains and the chromophore in the active site (see Fig. 3). The CD spectrum thus contains information about the asymmetric orientation of the bound PLP cofactor in the active site, including the aldimine linkage between the PLP cofactor and the active site lysine (Scheme 1).18,19 The visible CD spectra of wild-type ALAS [Fig. 3(A)] show that in the presence of either succinyl- or octanoyl-CoA there is a positive cotton effect. However, close inspection of the results indicates that there is a distinct difference in the CD maximum in relation to that of wild-type ALAS; the wavelength maximum for succinyl-CoA blue shifts 15 nm to ∼420 nm, whereas the octanoyl-CoA dichroic band at ∼435 nm increases. The spectra observed for the R85K variant are different [Fig. 3(B)]. The changes caused by succinyl-CoA binding to the R85K variant mimic those observed upon octanoyl-CoA interacting with wild-type ALAS, in that the spectral differences between the two are unremarkable. Substitutions of polar residues with nonpolar amino acids yielded proteins (R85L and R85L/T430V) in which ligand binding induces changes in the PLP environment and, consequently, display CD spectra (visible region) that are markedly different from those of wild-type ALAS and the R85K variant. Within the R85L enzyme, succinyl-CoA binding does not cause a shift in the microenvironment of the chromophore [Fig. 3(C)]. However, all of the remaining CoA-derivatives cause a negative cotton effect, decreasing the amplitude of the dichroic band ∼5θ. Steric hindrance and enhanced hydrophobicity of the acyl-CoA-binding cleft are hypothesized to be the most exaggerated in the double variant. This theory is supported by the CD data which suggest that R85L/T430V is affected most by the binding of octanoyl- and butyryl-CoA [Fig. 3(D)]. The positive cotton effect observed in the spectra for the enzyme in the presence of these two hydrophobic ligands could suggest that the exclusion of water and degree of active site aliphaticity are crucial components for determining substrate specificity. Overall, a comparison of the CD spectra of the wild-type and R85K variant enzymes with those of the hydrophobic variants (R85L and R85L/T430V) show that the distinct change in the microenvironment of the PLP pocket correlates well with the degree of hydrophobicity inherent to the bound ligand.
Figure 3.
Visible circular dichroism spectra of wild-type ALAS and the R85 and R85/T430 variants. (A) wild-type ALAS, (B) R85K, (C) R85L, and (D) R85L/T430V. Spectra of the holoenzymes are in cyan. Spectra of the holoenzymes (100 μM) in the presence of 100 μM CoA derivative are indicated according to the following color scheme: pink, succinyl-CoA; green, octanoyl-CoA; blue, β-hydroxybutyryl-CoA; black, butyryl-CoA; and red, glutaryl-CoA.
Scheme 1.
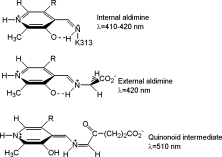
Absorbance maxima associated with chemical species in the ALAS-catalyzed reaction.  .
.
ALA binding kinetics monitored by the transient intrinsic protein fluorescence
To determine whether a change in the degree of hydrophobicity of the acyl-CoA-binding pocket could influence the product binding and concomitant quenching of the intrinsic protein fluorescence, wild-type ALAS and the enzyme variants were rapidly mixed with excess ALA and the changes in intrinsic protein fluorescence were monitored. The observed pseudo first-order decay rates of these kinetic traces were dependent on ALA concentration, and differed among the enzymes tested (see Fig. 4). In all cases the change in observed rate as a function of ALA concentration was hyperbolic, indicating two binding steps. Presumably, these two steps are indicative of rapid formation of an initial collision complex followed by a slower shift to the closed conformation observed in the crystal structures of the R. capsulatus enzyme. Although the resolved ALA “off” rates (i.e., k−1) coincide with the kcat values determined through steady-state kinetics, suggesting that a conformational change associated with ALA release defines kcat for each enzyme, the collision complex dissociation constants of the variants are increased. The resolved on and off rates for the reaction between ALA and wild-type ALAS were k1 = 0.120 ± 0.015 s−1 and k−1 = 0.140 ± 0.005 s−1, respectively, with a KD of 500 ± 16 μM. The rates for the reaction of the R85K variant with ALA were approximately two-fold lower with k1 = 0.090 ± 0.003 s−1 and k−1 0.079 ± 0.003 s−1; however, the dissociation constant was three-fold higher at 1470 ± 30 μM, indicating decreased affinity for ALA. Among the more hydrophobic variants, the rates of protein quenching with ALA were the most rapid for R85L with on and off rates of 0.039 ± 0.009 s−1 and 0.024 ± 0.003 s−1, respectively, values three-fold lower that those of the wild-type enzyme. A KD value of 1130 ± 27 μM corresponding to a three-fold greater dissociation of ALA from the R85L variant than that of wild-type ALAS was calculated. The double variant bound ALA less tightly, with a KD 13-fold lower than that for wild-type ALAS and an estimated value of 6710 ± 33 μM. The on and off rates for R85L/T430V were also the lowest of the enzymes tested, with rates measuring k1 = 0.007 ± 0.0001 s−1, and k−1 0.005 ± 0.001 s−1, respectively.
Figure 4.
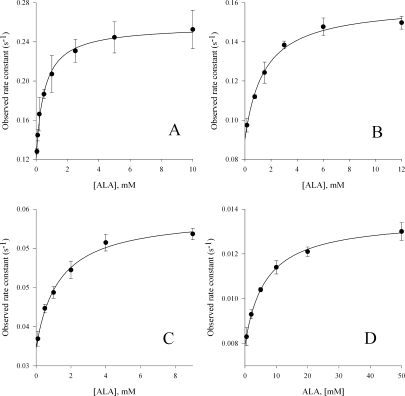
Reaction of wild-type ALAS, R85K, R85L, and R85L/T430V (5 μM) with ALA. The observed rate constants were calculated by fitting the decrease in intrinsic protein fluorescence over time to Eq. (2) for a single exponential process. (A) wild-type ALAS; (B) R85K; (C) R85L; and (D) R85L/T430V.
Presteady-state reaction of the variant enzyme-glycine complexes with acyl-CoA-derivatives
The R85L and R85L/T430V variant enzyme-glycine complexes did not yield a measurable absorbance change at 510 nm, previously assigned to a quinonoid reaction intermediate (Scheme 1), when rapidly mixed with any of the five acyl-CoA derivatives tested.17,20,21 Consequently, the investigation of the transient kinetics associated with the formation and decay of the quinonoid intermediate was limited to the reactions catalyzed by wild-type ALAS and the R85K variant, both of which display a quantifiable absorbance change at 510 nm upon the addition of the acyl-CoA substrates to the enzyme-glycine complexes. The rates associated with the lifetime of the quinonoid intermediate, measured during the enzyme catalyzed reactions, are indicated in Table II. The reaction time courses, monitored as absorbance changes at 510 nm, were fitted to a sequential, three-step mechanism outlined by Eq. (1). An initial burst of quinonoid intermediate formation, followed by a two-step decay was characteristic to each of the enzymes tested (see Fig. 5). Of all the acyl-CoA derivatives examined, only β-hydroxybutyryl-CoA, when rapidly mixed with the wild-type enzyme-glycine complex, failed to produce an absorbance change at 510 nm. Overall, faster rates of quinonoid intermediate formation were observed for the wild-type enzyme, when compared with the R85K variant (Table II). The rates of quinonoid formation when octanoyl-CoA was used as the substrate (2.3 and 1.9 s−1) were of the same order of magnitude as the values recorded for succinyl-CoA (6.0 and 4.2 s−1) for both the wild-type and R85K enzymes, respectively. These data support the increased catalytic efficiency observed from the experiments performed in the steady-state. Further comparison of the reaction catalyzed by wild-type ALAS with butyryl-CoA versus glutaryl-CoA showed that quinonoid intermediate formation was accelerated 90%. A similar enhancement was observed for the R85K-catalyzed reaction which showed a 70% increase in the rate of quinonoid intermediate formation with butyryl-CoA versus glutaryl-CoA. The preference for butyryl-CoA over glutartyl CoA suggests that in addition to the hydrogen bonding properties of R85 and T430, the amino acids that line the hydrophobic tunnel leading to the terminal guanidino group may play a role in substrate acceptance and orientation. Curiously, only the R85K-glycine complex when rapidly mixed with β-hydroxybutyryl-CoA gave a time-dependent absorbance change at 510 nm and rate associated with quinonoid intermediate formation (0.12 s−1). This is in stark contrast to the observations made of wild-type ALAS, where no quantifiable change with this substrate was detected. This slow rate may be explained by the mixed polarity of the substrate tail, an attribute which simultaneously imparts hydrogen bonding character, as well as aliphaticity to the acyl-CoA-binding cleft of the variant enzyme.
Table II.
Rates of Quinonoid Intermediate Formation and Decay Under Single-Turnover Conditions
| Parameter | Succinyl (s−1) | Octanoyl (s−1) | Butyryl (s−1) | β-Hydroxybutyryl (s−1) | Glutaryl (s−1) |
|---|---|---|---|---|---|
| Wild-type ALAS | |||||
| Qf | 6.0 ± 0.6 | 2.3 ± 0.4 | 0.41 ± 0.04 | n/d | 0.220 ± 0.03 |
| Qd1 | 2.00 ± 0.30 | 0.071 ± 0.004 | 0.070 ± 0.005 | n/d | 0.091 ± 0.005 |
| Qd2 | 0.072 ± 0.005 | 0.020 ± 0.003 | 0.011 ± 0.003 | n/d | 0.0010 ± 0.0004 |
| R85K | |||||
| Qf | 4.20 ± 0.1 | 1.91 ± 0.20 | 0.323 ± 0.020 | 0.121 ± 0.021 | 0.192 ± 0.042 |
| Qd1 | 1.10 ± 0.1 | 0.11 ± 0.02 | 0.002 ± 0.0001 | 0.022 ± 0.005 | 0.134 ± 0.071 |
| Qd2 | 0.050 ± 0.004 | 0.070 ± 0.003 | 0.041 ± 0.007 | 0.013 ± 0.001 | 0.0010 ± 0.0001 |
n/d, not determined; Qf, quinonoid intermediate formation; Qd1, first step of quinonoid intermediate decay; Qd2, second step of quinonoid intermediate decay.
Figure 5.
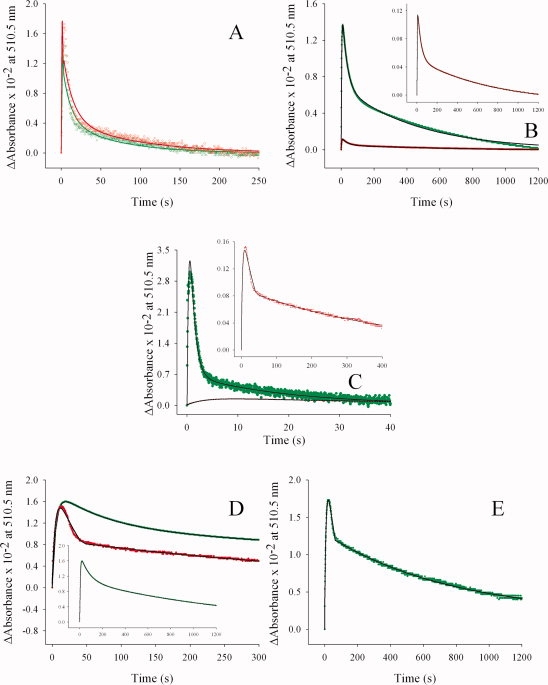
Reaction of wild-type ALAS- and R85K-glycine complexes with different CoA derivatives under single turnover conditions. The data (•• in green/•• in red) are overlaid with the line representing the best-fit curve ( ). The rate constants for the three step sequence corresponding to both enzymes are listed in Table II. Green and red data points correspond with wild-type ALAS and the R85K variant, respectively. (A) octanoyl-CoA, (B) butyryl-CoA, (C) succinyl-CoA, (D) glutaryl-CoA, and (E) β-hydroxybutyryl-CoA.
). The rate constants for the three step sequence corresponding to both enzymes are listed in Table II. Green and red data points correspond with wild-type ALAS and the R85K variant, respectively. (A) octanoyl-CoA, (B) butyryl-CoA, (C) succinyl-CoA, (D) glutaryl-CoA, and (E) β-hydroxybutyryl-CoA.
Discussion
The reactions catalyzed by the highly related members of the α-oxoamine synthase subfamily of PLP-dependent enzymes can be compared with respect to the specificity of the acyl-CoA substrate due to the elucidation of the three-dimensional structures of subfamily members together with mutagenesis, spectroscopic and kinetic methods.1,4–6,22 Both of the variant enzymes constructed for the arginine residue (R85L and R85K) as well as the doubly mutated enzyme (R85L/T430V) were expressed, overproduced, and then purified as holoenzymes, indicating that cofactor binding by the apoprotein was not affected by the introduction of the amino acid substitutions. However, replacement of the invariant threonine residue with valine (T430V) resulted in a poorly expressed, unstable, and proteolytically susceptible enzyme that could never be purified to homogeneity (data not shown). All of the purifiable variants were active with the physiological substrate succinyl-CoA. As the threonine to valine replacement at position 430 appears to dramatically affect protein stability, we suggest that T430 is essential not only for optimal molecular recognition of succinyl-CoA, but also for stable folding. R85 may be less crucial for proper enzyme function, a finding supported by the crystallographic data for SPT.6 Given that this enzyme lacks the arginine residue implicated in salt bridge formation with the carboxylate group of CoA substrates in the other three members of the α-oxoamine synthase subfamily and utilizes palmitoyl-CoA, an acyl-CoA derivative of increased aliphaticity, it is proposed that acyl-CoA binding in ALAS may be driven by noncovalent interactions between the two residues and the substrate. However, considering the structural and mechanistic data for ALAS and SPT, turnover likely remains orchestrated by amino acids that are proximal to the site of α-carbon bond scission.17,23
Comparison of the active sites of AONS and ALAS showed that the coordination of the acyl-CoA substrate is assisted by way of pantetheine association with the enzyme face and tail interactions with the buried hydrophobic tunnel.1,4,6 Both R85 and T430 coordinate the carboxylate tail of the acyl-CoA substrate in meALAS. The steady-state kinetic analysis of the variants (R85K, R85L, and R85L/T430V) with the family of CoA-derivatives showed that the apparent Michaelis parameters () are dramatically different when compared with those of wild-type ALAS. Acyl-CoA substrates of increased hydrophobicity (e.g., octanoyl- and butyryl-CoA) demonstrated greater affinity for the variants where the substituted amino acid was aliphatic in nature (R85L and R85L/T430V). The 36-fold decrease in the () for octanoyl-CoA in the R85L variant leads us to suggest that the exclusion of water from the acyl-CoA-binding tunnel is a determining feature of substrate binding. Further, in the double variant, an 18-fold reduction in the Michaelis constant for butyryl-CoA also supports this hypothesis. The introduction of valine at position 430 would reduce the diameter of the hydrophobic tunnel, making steric hindrance a more significant consideration for substrate binding. These differences identified between the variant enzymes and wild-type ALAS suggest that reaction specificity is driven by the chemical characteristics of the CoA-derived tail and the hydrogen-bonding potential of the invariant acyl-CoA-binding residues, a phenomenon recognized in the acyl-CoA thioesterases of the peroxisome.24,25
The chemical characteristics of the acyl-CoA tail are a determining factor in the substrate specificity of another family of enzymes that utilize related substrates in turnover, the crotonase family.26,27 Among those enzymes, octanoyl-CoA has been shown to bind in a characteristic bent conformation.26 This substrate conformation is accomplished by two structurally conserved hydrogen bond-donating groups to the carbonyl moiety of the substrate and through the entropically driven loss of water coordinated by the hydrophobic amino acids that line the binding cleft.26 The CD data for butyryl-CoA with the double variant in addition to the substrate configuration observed in enzymes that physiologically utilize octanoyl-CoA led us to suggest that octanoyl-CoA, which differs from butyryl-CoA by a four methylene bridge, most likely bends in the ALAS active site. This hypothesis is further supported by the data obtained for both wild-type ALAS and R85L with octanoyl-CoA. For wild-type ALAS, the catalytic efficiency for the nonphysiological substrate octanoyl-CoA is ∼100% greater than that of succinyl-CoA (Table I and Fig. 2). Congruently, the specificity constant for octanoyl-CoA compared with that of succinyl-CoA in the R85L variant is 66-fold higher, indicating a significant change in substrate specificity. Therefore, we hypothesize that octanoyl-CoA, devoid of a salt bridge to anchor with the guanidino group of R85, likely bends, excludes water from the active site, and assists in enzyme closure, a conformation postulated to be essential for turnover.17
Interestingly, the specificity constant for the wild-type enzyme with octanoyl-CoA is higher than the physiological substrate, succinyl-CoA, albeit at the cost of a three-fold reduction in the specificity constant for the other substrate, glycine. Nevertheless, this finding and the activity of the enzyme with the other CoA esters tested here lead us to raise the question as to what extent ALAS may catalyze formation of 1-3-aminoketones other than ALA in vivo. This is currently unknown, but may warrant further investigation. Conceivably, the potential toxicity, associated with the generation of other aminoketones rather than ALA, could be minimized through the action of a regulatory acyl-CoA-binding protein. In fact, studies have demonstrated that the acyl-CoA binding protein binds long-chain acyl-CoA esters with high specificity and affinity (with Kd values of 1–10 nM)28; hence by interacting with acyl-CoA utilizing enzymes, the acyl-CoA-binding protein may provide a mechanism for control of free acyl-CoA esters and regulation of the activity of acyl-CoA utilizing enzymes. Further, substrate specificity in vivo might be enhanced via a substrate channeling mechanism involving interaction between ALAS and succinyl-CoA synthetase.29 The requirement for such a mechanism is emphasized by the evidence presented here.
The chromophoric properties of the PLP cofactor provide a valuable probe to examine alterations in the cofactor-binding cleft of ALAS. As PLP is not a chiral molecule, the cotton effect of PLP bound in the active site must result from certain asymmetric distortion of the PLP molecule through interaction with the enzyme.30 Binding of acyl-CoA substrates to ALAS (or ALAS variants) most likely induced changes, even if subtle, in the PLP-protein interaction, as reflected by the different visible CD spectra (see Fig. 3). Curiously, for each of the enzymes tested, the most noticeable spectral deviations from the spectra obtained in the absence of substrate were produced by CoA-derivatives with tails that matched the chemical nature of the amino acid substitution introduced in ALAS. In the R85L/T430V double variant, binding of either octanoyl- or butyryl-CoA induced a marked positive cotton effect (see Fig. 3). A definite interpretation for the relationship between substrate binding and induced cotton effect of the PLP cofactor cannot be provided at this point. Perhaps the observed effects could be explained, in part, by a shift in the enzyme conformational equilibrium toward the closed conformation, which has been hypothesized for the wild-type enzyme to be energetically driven by succinyl-CoA binding (Lendrihas et al., unpublished data).17,20 However, a decrease in active site diameter, triggered by the entropic loss of water upon the binding of a more hydrophobic substrate, could also explain the change in the cofactor microenvironment as evidenced by the CD spectra. It is therefore premature to assign the observed CD spectral differences to a particular molecular event (e.g., a protein conformational change, a PLP reorientation, or a perturbation of the electronic system of the chromophore). Nevertheless, the CD spectra in the UV region of all the variants were indistinguishable from that of the wild-type enzyme (data not shown), indicating no gross alterations in secondary structure, and thus suggesting that the differences observed in the visible CD spectra, upon substrate binding, are confined to the PLP-binding cleft. Although specific contributions by single amino acid substitutions cannot be easily disentangled using CD, the CD spectral differences in the visible range among ALAS variants with the family of CoA-derivatives of different hydrophobicities lead us to propose that interactions between key residues (e.g., R85 and T430) that bind the tail of CoA play a role in determining substrate specificity.
The proposed kinetic mechanism of the ALAS-catalyzed reaction is limited by product release, or opening of the active site loop coincident with product release.17 Utilization of protein fluorescence to study the reverse catalytic reaction, that is, the reaction of the enzyme with the product, resolves the ALA “off” rate, and confirms it to be indistinguishable from kcat.17 With this in mind, we investigated whether mutations to the acyl-CoA-binding residues affect the rates of product release and/or perturb the enzyme-product equilibrium (see Fig. 4). Quenching of the ALAS intrinsic fluorescence with ALA obeyed first-order decay kinetics. For all the variant catalyzed-reactions, rates of product release and capture are diminished. The most dramatic decrease is seen in the double variant; this enzyme exhibited a 13-fold reduction in the “on” and “off” rates (k−1 and k1) for the reaction with ALA. As CoA-derivatives of increased hydrophobicity bind with greater affinity to the nonpolar variants (R85L and R85L/T430V), it is possible that the decreased affinity for the ALA product, a δ-amino acid, may be reversed for aminoketones of decreased polarity. In this scenario and in agreement with a mechanism in which a conformational step follows ligand binding, the affinity of the aminoketones toward ALAS would not be reduced. The Kd values would either increase or remain unchanged; this hypothesis, however, awaits further experimentation with aminoketones of differing hydrophobicity.
The three-dimensional structures of ALAS and AONS revealed a hydrogen bonding network between the invariant arginine and threonine residues and the carboxylate moieties of acyl-CoA substrates.1,4 Accordingly, how the use of chemically different acyl-CoA-derivatives would affect the transient kinetic parameters of the enzymes was evaluated. Single turnover reactions with a family of CoA-derivatives were used to determine the rates of quinonoid intermediate formation and decay. The reaction catalyzed by wild-type ALAS with glutaryl-CoA as the substrate exhibited a 30-fold lower rate corresponding to quinonoid intermediate formation versus the rate calculated with succinyl-CoA. One possible explanation for the retarded rate is that the binding of glutaryl-CoA affects the hydrophobic acyl-CoA-binding tunnel in such a way that cofactor-mediated electron transfer from the site of bond scission to the resonance-stabilized pyridinium ring of PLP is perturbed. This notion is further supported by the dramatic 70-fold reduction in the second step of quinonoid intermediate decay, with the absorbance change at 510 nm apparently stabilizing at a level that approaches steady-state [Fig. 5(D)]. The R85K variant demonstrated a change in the first step of quinonoid intermediate decay, with a 10-fold lower rate for both octanoyl-CoA and glutaryl-CoA when compared with the physiological substrate succinyl-CoA. When R85 is substituted with a lysine, the enzyme is chemically similar to wild-type ALAS in many respects, presumably because this conservative replacement retains the positive charge and hydrogen bonding properties. In addition, in silico protonation of the lysine amino group to create the ɛ-ammonium charge center would contribute charge and polar interactions characteristic of the guanidinium group.31,32 However, the molecular volume of the amino acid side chain is different, as is the electrostatic charge distribution. With respect to their n-alkyl moieties, the n-propylguanidine side chain of arginine is longer than the n-butylamine side chain of lysine by 1.6 Å.33 The R85K substitution could therefore accommodate the additional sp3 hybridized carbon atom present in glutaryl-CoA, allowing for a reduction in steric strain and/or unfavorable van der Waals interactions. Further, the increased hydrophobic tunnel length could also assist the bending of octanoyl-CoA within the cleft, a circumstance rationalized earlier.
In summary, the spectroscopic and kinetic studies detailed here demonstrate not only the role played by R85 and T430 in determining acyl-CoA substrate specificity, but also provide insight into the structure-function relationships in ALAS and the α-oxoamine synthase subfamily as a whole. Although R85 and T430 recognize a part of the substrate that is distal from the bulk of the ligand and the active site, the ability of all the enzymes to turnover nonphysiological substrates remains intact. Changing these residues to leucine and valine abates succinyl-CoA binding and catalytic efficiency, as well as increases the affinity of the enzyme for CoA-derivatives of greater aliphaticity. These observations indicate that a mutation-mediated decrease in substrate binding energy could be accountable for the enhanced affinity measured for the hydrophobic CoA-derivatives, instead of a direct mechanistic linkage between these residues and the site of bond cleavage. Certainly, the conserved amino acid duo (R85 and T430) are at some distance from the PLP-binding site, and therefore the coupling of substrate binding to di-α-carbon cleavage in the active site presumably involves coordinated movement of the enzyme upon acyl-CoA binding. However, further experiments to prove this await the development of tractable fluorescent probes and the elucidation of three-dimensional crystal structures with CoA-derivatives bound.
Materials
The following reagents were purchased from Sigma-Aldrich Chemical Company (St. Louis, MO): ampicillin, DEAE-Sephacel, Ultrogel AcA-44, β-mercaptoethanol, PLP, bovine serum albumin, succinyl-CoA, ALA-hydrochloride, α-ketoglutaric acid, α-ketoglutarate dehydrogenase, HEPES-free acid, MOPS, tricine, thiamine pyrophosphate, NAD+, and the bicinchoninic acid protein determination kit. Glucose, glycerol, glycine, disodium ethylenediamine tetraacetic acid dihydrate, ammonium sulfate, magnesium chloride hexahydrate, and potassium hydroxide were acquired from Fisher Scientific (Pittsburgh, PA). Sodium dodecyl sulfate polyacrylamide gel electrophoresis reagents were acquired from Bio-Rad. Sal I, Blp I, Xho I, Bam HI restriction enzymes, Vent DNA polymerase, and T4 DNA ligase were from New England Biolabs (Ipswich, MA). Oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA).
Methods
Mutagenesis
The pGF23 expression plasmid encodes the full-length sequence for the murine, mature eALAS.34 The R85L variant was generated using a previously described method.35 The mutagenic primers used for the R85L mutation were 5′-GAC CAC ACC TAC CTT GTG TTC AAG ACT GT-3′ and 5′-ACA GTC TTG AAC ACA AGG TAG GTG TGG TC-3′, with the introduced codon substitution italicized. The PCR-generated fragment containing the R85L mutation was used as a megaprimer in an independent round of PCR. The PCR-generated fragment was sequenced to verify the presence of the desired mutations. Subsequently, the PCR product was then digested with Sal I and Bam HI and subcloned into pGF23 digested similarly. The site-directed mutagenesis for the T430V variant was performed on the whole plasmid pGF23 using a previously described method.36 The mutagenic oligonucleotide for T430V was: 5′-ATC AAC TAC CCA GTT GTG CCT CTG GGT-3′, with the introduced codon substitution italicized. The PCR-generated DNA was sequenced between the Blp I and Bam HI restriction enzyme sites to confirm the presence of the mutation. The product was then digested with Blp I and Bam HI and subcloned into pGF23 digested similarly. The pMAL2 expression plasmid, described earlier, encodes the full-length sequence for the murine, mature eALAS, with the arginine at position 85 mutated to leucine. The R85L/T430V double mutated variant was constructed by digesting the T430V-encoding plasmid (pTL30) with Xho I and Bam HI. This T430V mutation-encoding fragment was subcloned into pMAL2 digested similarly.
Protein purification, SDS-PAGE, protein determination, and steady-state analysis
Recombinant murine eALAS and the R85 and R85/T430 variants were purified from DH5α Escherichia coli bacterial cells containing the overexpressed protein as previously described.34 Sufficient expression of the T430V variant could not be obtained. Protein purity was estimated by SDS-PAGE37 and was always greater than 90%. Protein concentration was determined by the bicinchoninic acid method using bovine serum albumin as the standard.38 All protein concentrations are reported on the basis of a subunit molecular mass of 56 kDa. Enzymatic activity was determined by a continuous spectrophotometric assay at 30°C.39
Structural analyses
The protein data base files 2BWN, 2BWO, and 2BWP, corresponding to the R. capsulatus ALAS holoenzyme, succinyl-CoA bound, and glycine bound crystal structures were used as templates to model the murine eALAS Michaelis complex structure.4 Hydrogen bond determinations were accomplished using Deepview/Swiss-PdbViewer software.31,32
Circular dichroism spectroscopic measurements
Spectroscopic measurements were performed with enzyme that was dialyzed in 20 mM HEPES, pH 7.5 with 10% glycerol to remove free PLP. Circular dichroism (CD) spectra were obtained using an AVIV CD spectrometer calibrated for both wavelength maxima and signal intensity with an aqueous solution of D-10 camphorsulfonic acid.40 Protein concentrations were 100 μM for each enzyme tested. The final concentration of each CoA-derivative was 100 μM, giving a 1:1 molar ratio of enzyme to ligand. At least three CD spectra were collected per experiment and averaged using a 0.1 cm path length cuvette with a total volume of 300 μL. Blank CD spectra contained all components of the solution except enzyme. CD spectra containing the enzyme sample were collected immediately after adding the enzyme. The spectra of the samples containing enzyme were analyzed after subtracting the blank spectra.
Stopped-flow spectroscopy
All of the experiments were carried out at 30°C in 100 mM HEPES, pH 7.5, and 10% (v/v) glycerol. The concentration of reactants loaded into the two syringes was always two-fold greater than that present in the cell compartment after mixing, with glycine and the enzyme preincubated in one syringe and the CoA-derivative in another. Because of the difference in Km values for the CoA-derivatives among the two enzymes tested, different CoA-derivative concentrations were used to ensure the identification of a single enzyme catalyzed event. For the wild-type reaction, the final concentrations were: 120 μM wild-type ALAS and 130 mM glycine. The final concentrations of each independently examined CoA-derivative were: succinyl-CoA, 10 μM; octanoyl-CoA, 10 μM; butyryl-CoA, 20 μM; β-hydroxybutyryl-CoA, 30 μM; glutaryl-CoA, 30 μM. For the R85K-catalyzed reaction, the final concentrations were: 120 μM R85K and 130 mM glycine. The final concentrations of each independently examined CoA-derivative were: succinyl-CoA, 20 μM; octanoyl-CoA, 10 μM; butyryl-CoA, 10 μM; β-hydroxybutyryl-CoA, 10 μM; glutaryl-CoA, 20 μM. Rapid scanning stopped-flow kinetic measurements were conducted using an OLIS model RSM-1000 stopped-flow spectrophotometer. The dead time of this instrument is ∼2 ms, and the observation chamber optical path length is 4.0 mm. Scans covering the wavelength region 270–550 nm were acquired at a rate of 31, seven or three scans per second to condense the resulting data files to a tractable size for data fitting analysis. An external water bath was utilized to maintain constant temperature (30°C) of the syringes and observation chamber. Observed rate constants were determined by global fitting of the acquired spectral data sets, using the single value decomposition software provided by OLIS.41 The quality of fits were judged by visual analysis of the calculated residuals in conjunction with the Durbin–Watson statistic.42 Single turnover data were interpreted using a three kinetic step mechanism as described by Eq. (1).
| (1) |
The observed rate constants were determined from at least three replicate experiments, and the reported values represent the average and standard error of measurement for each experimental condition.
Intrinsic protein fluorescence quenching and product binding
The presteady state kinetics of the product binding reaction of ALAS and the R85 and T430 variants were examined by measuring changes in the intrinsic protein fluorescence intensity. An OLIS RSM-1000F rapid mixing spectrofluorimeter, equipped with a high-intensity xenon arc lamp, was used to follow the reaction. The enzyme and ligand in 20 mM HEPES (pH 7.5) and 10% glycerol were maintained at 30°C in separate syringes before their mixing in the reaction chamber. The concentrations of enzyme and ligand in the reaction chamber were 1/2 of those in the syringes. The intrinsic protein fluorescence, as measured with 5 μM enzyme, was evaluated in the presence of increasing concentrations of the product, ALA. The excitation wavelength and the slit width were 280 and 5 mm, respectively. Scheme 1 illustrates the relationship between wavelength maximum and the dynamic process being monitored. The emitted light was filtered using a cutoff filter (WG 320; 80% transmittance at 320 nm; Edmund Optics, Barrington, NJ). Typically, 500 time points were collected for varying lengths of time, and three or more experiments were averaged. Each averaged data set was then fitted to Eq. (2), using the global fitting software provided with the instrument.
| (2) |
where Fobs(t) is the observed fluorescence change (in arbitrary units) at time t, kobs1 is the observed first-order rate constant, A1 is the pre-exponential factor and A0 is the offset. The observed rate constants were then plotted against ligand concentration and the data were fitted to Eq. (3) by nonlinear regression. The rates of dissociation (koff) and association (kon) as well as the ligand binding constants (KD) were calculated from the asymptotic maximal observed rate, the ordinate intercept, and the ligand concentration (x) in Eq. (3).
| (3) |
Glossary
Abbreviations:
- AAT
aspartate aminotransferase
- ALAS
5-aminolevulinate synthase
- ALA
5-aminolevulinate
- AON
8-amino-7-oxononanoate
- AONS
8-amino-7-oxononanoate synthase
- CD
circular dichroism
- CoA
coenzyme A
- HEPES
(N-[2-hydroxyethyl] piperazine-N′-[2-ethane sulfonic acid])
- KBL
2-amino-3-ketobutyrate-CoA ligase
- meALAS
murine erythroid ALAS
- MOPS
4-morpholinepropanesulfonic acid
- NAD+
β-nicotinamide adenine dinucleotide
- PLP
pyridoxal 5′-phosphate
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SPT
serine palmitoyl transferase.
References
- 1.Webster SP, Alexeev D, Campopiano DJ, Watt RM, Alexeeva M, Sawyer L, Baxter RL. Mechanism of 8-amino-7-oxononanoate synthase: spectroscopic, kinetic, and crystallographic studies. Biochemistry. 2000;39:516–528. doi: 10.1021/bi991620j. [DOI] [PubMed] [Google Scholar]
- 2.Alexeev D, Alexeeva M, Baxter RL, Campopiano DJ, Webster SP, Sawyer L. The crystal structure of 8-amino-7-oxononanoate synthase: a bacterial PLP-dependent, acyl-CoA-condensing enzyme. J Mol Biol. 1998;284:401–419. doi: 10.1006/jmbi.1998.2086. [DOI] [PubMed] [Google Scholar]
- 3.Ikushiro H, Hayashi H, Kagamiyama H. Reactions of serine palmitoyltransferase with serine and molecular mechanisms of the actions of serine derivatives as inhibitors. Biochemistry. 2004;43:1082–1092. doi: 10.1021/bi035706v. [DOI] [PubMed] [Google Scholar]
- 4.Astner I, Schulze JO, van den Heuvel J, Jahn D, Schubert WD, Heinz DW. Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J. 2005;24:3166–3177. doi: 10.1038/sj.emboj.7600792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eliot AC, Kirsch JF. Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 6.Yard BA, Carter LG, Johnson KA, Overton IM, Dorward M, Liu H, McMahon SA, Oke M, Puech D, Barton GJ, Naismith JH, Campopiano DJ. The structure of serine palmitoyltransferase; gateway to sphingolipid biosynthesis. J Mol Biol. 2007;370:870–886. doi: 10.1016/j.jmb.2007.04.086. [DOI] [PubMed] [Google Scholar]
- 7.Eisenberg M. Biosynthesis of biotin and lipoic acid. In: Neidhardt FC, Ingraham JL, Low KB Jr, Magasanik B, Schaechter M, Umbarger M, editors. Escherichia coli and Salmonella typhimurium. Washington, DC: American Soc. Microbiol.; 1987. pp. 544–550. [Google Scholar]
- 8.Hanada K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim Biophys Acta. 2003;1632:16–30. doi: 10.1016/s1388-1981(03)00059-3. [DOI] [PubMed] [Google Scholar]
- 9.Bell SC, Turner JM. Bacterial catabolism of threonine. Threonine degradation initiated by l-threonine-NAD+ oxidoreductase. Biochem J. 1976;156:449–458. doi: 10.1042/bj1560449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merrill AH., Jr Characterization of serine palmitoyltransferase activity in Chinese hamster ovary cells. Biochim Biophys Acta. 1983;754:284–291. doi: 10.1016/0005-2760(83)90144-3. [DOI] [PubMed] [Google Scholar]
- 11.Han G, Gable K, Yan L, Allen MJ, Wilson WH, Moitra P, Harmon JM, Dunn TM. Expression of a novel marine viral single-chain serine palmitoyltransferase and construction of yeast and mammalian single-chain chimera. J Biol Chem. 2006;281:39935–39942. doi: 10.1074/jbc.M609365200. [DOI] [PubMed] [Google Scholar]
- 12.Shoolingin-Jordan PM, LeLean JE, Lloyd AJ. Continuous coupled assay for 5-aminolevulinate synthase. Methods Enzymol. 1997;281:309–316. doi: 10.1016/s0076-6879(97)81037-3. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi A, Fujisawa S. Effect of L-carnitine on mitochondrial acyl CoA esters in the ischemic dog heart. J Mol Cell Cardiol. 1994;26:499–508. doi: 10.1006/jmcc.1994.1060. [DOI] [PubMed] [Google Scholar]
- 14.Hunter GA, Rivera E, Ferreira GC. Supraphysiological concentrations of 5-aminolevulinic acid dimerize in solution to produce superoxide radical anions via a protonated dihydropyrazine intermediate. Arch Biochem Biophys. 2005;437:128–137. doi: 10.1016/j.abb.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 15.Onuki J, Teixeira PC, Medeiros MH, Dornemann D, Douki T, Cadet J, Di Mascio P. Is 5-aminolevulinic acid involved in the hepatocellular carcinogenesis of acute intermittent porphyria? Cell Mol Biol (Noisy-le-grand) 2002;48:17–26. [PubMed] [Google Scholar]
- 16.Shoolingin-Jordan PM, Al-Daihan S, Alexeev D, Baxter RL, Bottomley SS, Kahari ID, Roy I, Sarwar M, Sawyer L, Wang SF. 5-Aminolevulinic acid synthase: mechanism, mutations and medicine. Biochim Biophys Acta. 2003;1647:361–366. doi: 10.1016/s1570-9639(03)00095-5. [DOI] [PubMed] [Google Scholar]
- 17.Hunter GA, Zhang J, Ferreira GC. Transient kinetic studies support refinements to the chemical and kinetic mechanisms of aminolevulinate synthase. J Biol Chem. 2007;282:23025–23035. doi: 10.1074/jbc.M609330200. [DOI] [PubMed] [Google Scholar]
- 18.Schnackerz KD, Tai CH, Potsch RK, Cook PF. Substitution of pyridoxal 5′-phosphate in D-serine dehydratase from Escherichia coli by cofactor analogues provides information on cofactor binding and catalysis. J Biol Chem. 1999;274:36935–36943. doi: 10.1074/jbc.274.52.36935. [DOI] [PubMed] [Google Scholar]
- 19.Moscowitz A. Some applications of the kronig-kramers theorem to optical activity. Tetrahedron. 1961;13:48–54. [Google Scholar]
- 20.Hunter GA, Ferreira GC. Pre-steady-state reaction of 5-aminolevulinate synthase. Evidence for a rate-determining product release. J Biol Chem. 1999;274:12222–12228. doi: 10.1074/jbc.274.18.12222. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Ferreira GC. Transient state kinetic investigation of 5-aminolevulinate synthase reaction mechanism. J Biol Chem. 2002;277:44660–44669. doi: 10.1074/jbc.M203584200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt A, Sivaraman J, Li Y, Larocque R, Barbosa JA, Smith C, Matte A, Schrag JD, Cygler M. Three-dimensional structure of 2-amino-3-ketobutyrate CoA ligase from Escherichia coli complexed with a PLP-substrate intermediate: inferred reaction mechanism. Biochemistry. 2001;40:5151–5160. doi: 10.1021/bi002204y. [DOI] [PubMed] [Google Scholar]
- 23.Dunathan HC. Conformation and reaction specificity in pyridoxal phosphate enzymes. Proc Natl Acad Sci USA. 1966;55:712–716. doi: 10.1073/pnas.55.4.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunt MC, Solaas K, Kase BF, Alexson SE. Characterization of an acyl-coA thioesterase that functions as a major regulator of peroxisomal lipid metabolism. J Biol Chem. 2002;277:1128–1138. doi: 10.1074/jbc.M106458200. [DOI] [PubMed] [Google Scholar]
- 25.Hunt MC, Alexson SE. Novel functions of acyl-CoA thioesterases and acyltransferases as auxiliary enzymes in peroxisomal lipid metabolism. Prog Lipid Res. 2008;47:405–421. doi: 10.1016/j.plipres.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 26.Engel CK, Kiema TR, Hiltunen JK, Wierenga RK. The crystal structure of enoyl-CoA hydratase complexed with octanoyl-CoA reveals the structural adaptations required for binding of a long chain fatty acid-CoA molecule. J Mol Biol. 1998;275:859–847. [PubMed] [Google Scholar]
- 27.Engel CK, Mathieu M, Zeelen JP, Hiltunen JK, Wierenga RK. Crystal structure of enoyl-coenzyme A (CoA) hydratase at 2.5 angstroms resolution: a spiral fold defines the CoA-binding pocket. EMBO J. 1996;15:5135–5145. [PMC free article] [PubMed] [Google Scholar]
- 28.Abo-Hashema KA, Cake MH, Lukas MA, Knudsen J. The interaction of acyl-CoA with acyl-CoA binding protein and carnitine palmitoyltransferase I. Int J Biochem Cell Biol. 2001;33:807–815. doi: 10.1016/s1357-2725(01)00049-8. [DOI] [PubMed] [Google Scholar]
- 29.Furuyama K, Sassa S. Interaction between succinyl CoA synthetase and the heme-biosynthetic enzyme ALAS-E is disrupted in sideroblastic anemia. J Clin Invest. 2000;105:757–764. doi: 10.1172/JCI6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gong J, Kay CJ, Barber MJ, Ferreira GC. Mutations at a glycine loop in aminolevulinate synthase affect pyridoxal phosphate cofactor binding and catalysis. Biochemistry. 1996;35:14109–14117. doi: 10.1021/bi961296h. [DOI] [PubMed] [Google Scholar]
- 31.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 33.Creighton TR. Proteins, structures and molecular properties. New York: W.H. Freeman; 1983. [Google Scholar]
- 34.Ferreira GC, Dailey HA. Expression of mammalian 5-aminolevulinate synthase in Escherichia coli. Overproduction, purification, and characterization. J Biol Chem. 1993;268:584–590. [PubMed] [Google Scholar]
- 35.Gong J, Hunter GA, Ferreira GC. Aspartate-279 in aminolevulinate synthase affects enzyme catalysis through enhancing the function of the pyridoxal 5′-phosphate cofactor. Biochemistry. 1998;37:3509–3517. doi: 10.1021/bi9719298. [DOI] [PubMed] [Google Scholar]
- 36.Miyazaki K, Takenouchi M. Creating random mutagenesis libraries using megaprimer PCR of whole plasmid. Biotechniques. 2002;33:1033–1034. 1036–1038. doi: 10.2144/02335st03. [DOI] [PubMed] [Google Scholar]
- 37.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 38.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 39.Hunter GA, Ferreira GC. A continuous spectrophotometric assay for 5-aminolevulinate synthase that utilizes substrate cycling. Anal Biochem. 1995;226:221–224. doi: 10.1006/abio.1995.1217. [DOI] [PubMed] [Google Scholar]
- 40.Chen GC, Yang JT. Two-point calibration of cicular dichrometer with D-10-camphosulphonic acid. Anal Lett. 1977;10:1195–1207. [Google Scholar]
- 41.Tsai MD, Weintraub HJ, Byrn SR, Chang C, Floss HG. Conformation-reactivity relationship for pyridoxal Schiff's bases. Rates of racemization and alpha-hydrogen exchange of the pyridoxal Schiff's bases of amino acids. Biochemistry. 1978;17:3183–3188. doi: 10.1021/bi00609a002. [DOI] [PubMed] [Google Scholar]
- 42.Durbin J, Watson GS. Testing for serial correlation in least squares regression. Biometrika. 1970;37:409–414. [PubMed] [Google Scholar]