

Abstract
Despite the critical importance of molecular specificity in bimolecular systems, in vitro display technologies have been applied extensively for affinity maturation of peptides and antibodies without explicitly measuring the specificity of the desired interaction. We devised a general strategy to measure, screen, and evolve specificity of protein ligand interactions analogous to widely used affinity maturation strategies. The specificity of binding to target and nontarget antibodies labeled with spectrally distinct fluorophores was measured simultaneously in protein mixtures via multiparameter flow cytometry, thereby enabling screening for high target antibody specificity. Isolated antibody specific ligands exhibited varying specificity, revealing critical amino acid determinants for target recognition and nontarget avoidance in complex mixtures. Molecular specificity in the mixture was further enhanced by quantitative directed evolution, yielding a family of epitopes exhibiting improved specificities equivalent, or superior to, the native peptide antigen to which the antibody was raised. Specificity screening simultaneously favored affinity, yielding ligands with three-fold improved affinity relative to the parent epitope. Quantitative specificity screening will be useful to screen, evolve, and characterize the specificity of protein and peptide interactions for molecular recognition applications.
Keywords: antibody, cell display, directed evolution, peptide epitope, specificity
Introduction
Polypeptide display technologies have been applied extensively for affinity maturation of peptides, antibodies, and other binding proteins, but are rarely exploited to engineer or evolve binding specificity. The improvement of ligand-binding affinity using display technologies is straightforward because the desired property (affinity) is directly linked to the mode of separation in affinity-based selection methods. Consequently, affinity maturation efforts using display technologies have proven enormously successful, generating antibodies and peptides with dramatically improved affinities for diagnostic and therapeutic applications.1–3 At the same time, because affinity and specificity are not generally correlated, high-affinity protein-binding ligands can be nonspecific.4,5
The purposeful manipulation and design of specificity has proven challenging. In most cases, libraries are screened for affinity, and specificity of the identified ligands is characterized by their extent of binding to other related or unrelated proteins.3,6 In other words, specificity is not typically measured during selection or screening. Common approaches to favor specificity to single targets involve selection in the presence of serum albumin (e.g., BSA) or blocking agents such as serum and milk.7,8 However, these approaches do not enable measurement of specificity during screening, and can yield ligands whose binding is unpredictably modulated by the blocking agent. Alternatively, the coupling of cell surface display systems with fluorescence-activated cell sorting (FACS) for quantitative library screening,1,3,9,10 provides an opportunity to screen for desired binding specificity. For example, yeast display has been applied to evolve a single-chain antibody to achieve desired crossreactivity with two botulinum neurotoxin subtypes.11 Methodologies to measure, screen for, and evolve binding specificity would be valuable to engineer polypeptide therapeutics and diagnostics that function more effectively in complex protein mixtures suited to their application, or enable recognition of two or more specified targets, while avoiding interactions with specified nontarget proteins.
Diagnostic reagents, including antibodies and peptides, typically require exceptional target specificity to function in biological fluids. In particular, peptides recognizing disease-associated serum antibodies12,13 hold potential for diagnostic development. Peptide display libraries have been used to identify epitopes recognized by serum antibodies from patients with Lyme disease,14 multiple sclerosis,15,16 and ovarian,13 breast,17 and prostate cancers.12,18 Such peptides must discriminate individual antibody species from a vast array of specificities present within the circulating repertoire. The specificity of identified ligands for the target disease antibodies ultimately determines their diagnostic utility. Consequently, general methods to evolve both affinity and specificity in the context of complex protein mixtures could be useful in creating more effective reagents for biomarker detection. Here, we present a general strategy to measure, screen for, and evolve ligand specificity in a complex mixture using bacterial display peptide libraries wherein the specificity of antibody binding is quantitatively measured via FACS. The ability to measure specificity enabled directed evolution to further enhance binding specificity of the identified antibody ligands.
Results
Subtractive screening for target-specific peptide mimotopes in serum IgG mixtures
In an effort to enhance the quality of affinity reagents obtained from serum antibody fingerprinting and biomarker discovery efforts, we sought to develop a general methodology to identify and then optimize peptide ligands with both high specificity and affinity for disease-specific antibodies amongst a background of unrelated serum antibodies. To accomplish this, a quantitative screen for highly specific target protein binding was developed using bacterial surface display libraries and FACS (see Fig. 1). Two distinct pools of IgG were prepared to simulate IgG samples isolated from the serum of patients with a known disease and appropriate control subjects for the purpose of serum antibody profiling. Specifically, a model serum antibody biomarker, anti-T7•tag IgG (1 nM), was spiked into an excess of pooled serum IgG (1 μM), thus mimicking a disease-associated IgG sample (disease IgG). Pooled IgG without the anti-T7•tag antibody comprised the simulated “control IgG.” Disease IgG and control IgG were labeled with red and green fluorophores, respectively (see Fig. 1). Antibodies that recognize native E. coli antigens were present in pooled human IgG but easily removed by a short incubation with E. coli that did not display peptides (see Fig. 1).
Figure 1.
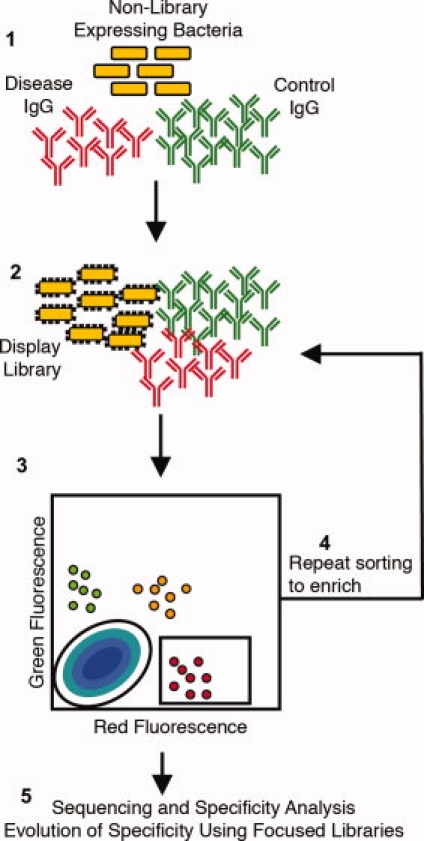
Quantitative specificity screening methodology. Two pools of serum IgG are labeled such that the disease IgG are red fluorescent and the control IgG are green fluorescent. Both pools are incubated separately with nonlibrary displaying bacteria to remove any IgG specific for bacteria (1). The two pools of IgG are then incubated simultaneously with the display library, with the control IgG added in excess (×10–100) of the disease IgG (2). Unbound IgG is removed by washing and cells are then sorted based on the levels of green and red fluorescence exhibited for the desired specificity for the disease IgG (3). Cells with high red and low green fluorescence are specific for the disease IgG. Collected cells are amplified and sorting is repeated for enrichment (4). After the final round of sorting, the specificity of clones is analyzed and their respective peptide sequences are determined and used to create focused libraries to further evolve their specificity (5).
In an effort to identify mimotopes that could specifically recognize only those antibody specificities unique to the disease IgG pool (i.e., the anti-T7•tag antibody), a bacterial display library comprised of random 15 amino acid insertions into an extracellular loop of outer membrane protein X (OmpX) was screened using FACS.20 The library was labeled simultaneously with the control and disease IgG mixtures, where the control IgG was present in a 10-fold excess to differentiate mimotopes binding to IgG shared among both pools. After labeling, roughly 0.3% of the library exhibited antibody binding, with varying degrees of specificities as assessed by the intensities of red and green fluorescence. The library was fractionated into three initial populations exhibiting differing ratios of red:green fluorescence. The majority of library members exhibiting binding fell into one of two categories; binding to both disease and control IgG (i.e., red+green+) or specific binding to disease IgG (red++green−) [Fig. 2(A)].
Figure 2.
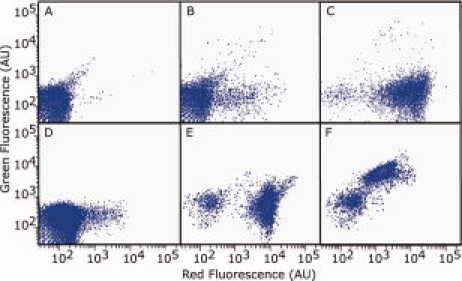
Enrichment of bacteria displaying peptides with varying specificity for the target antibody. Cells from the unsorted (A), cycle 1 sort (gate red++green−) (B), and cycle 4 sort (gate red++green−−) (C) populations of the random library. Cells from the unsorted (D) and round 4 sort for specificity (gate red++green−−) (E) and crossreactivity (gate red−green++) (F) of the linear library. Cells were incubated with 1 μM disease IgG and either ×10 (A, B, D) or ×100 (C, E, F) control IgG.
Sublibrary pools obtained from sorting the initial population were further sorted into populations with distinct specificity profiles as measured by flow cytometry. Individual isolated clones from these subpopulations also exhibited specificity measurements reflecting those of their parent populations [Fig. 3(A)]. Peptide sequences displayed by specific clones isolated from the red++green− and red++green−− included MX2QQ and MX3QQ motifs present in the wild-type T7•tag (MASMTGGQQMG) and related variants known to bind the anti-T7•tag (Supporting Information Table 2).3,20 In contrast, peptides identified from the nonspecific population (i.e., red+green+) exhibited a weak consensus unrelated to the wild-type T7•tag. Thus, two-color subtractive screening yielded ligands binding specifically to the target IgG in a 1000-fold background of unrelated IgG.
Figure 3.
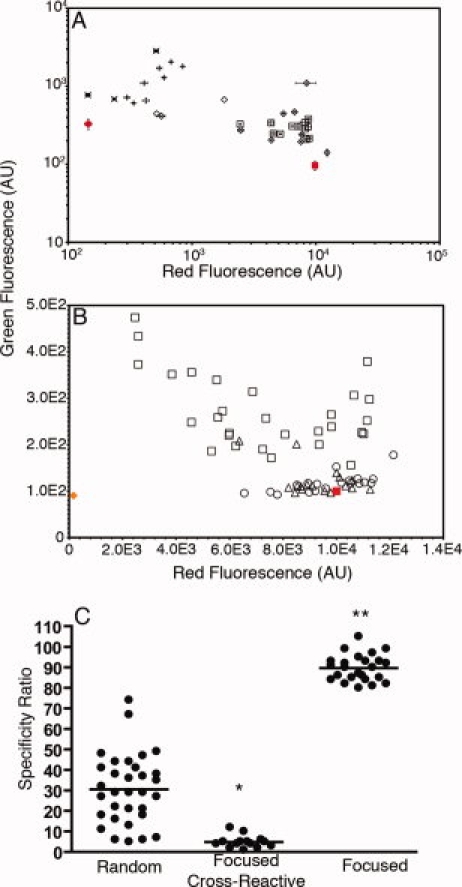
Quantitative specificity measurements of clones obtained using flow cytometry. (A) Quantitative measurements of clonal red and green fluorescence after labeling with 10 μM control and 1 μM disease IgG isolated from red−green+ (×), red+green+ (+), red++green− (open diamonds), and red++green−− (open squares) gates after three rounds of sorting from the random library. A clone overexpressing the cell surface display scaffold (without an insert) was used as a negative control (red circle). Error bars represent the standard deviation about the mean of at least three experiments. (B) The red and green fluorescence of clones isolated after four and five rounds of sorting from the random library in the red++green−− gate (open squares) and after three and four rounds of sorting from the linear (open triangles) and constrained libraries (open circles) after incubation with 100 μM control IgG and 1 μM disease IgG. A clone overexpressing the library scaffold with no peptide insert was used as a negative control (orange diamond). Error bars representing the standard deviation of at least three experiments were omitted for clarity from panel B. All values in (A) and (B) were normalized to the fluorescence of cells displaying the wt T7 tag (red square). (C) The specificity ratios of clones sorted from the random library using the red++green−− gate (random) are plotted along with the ratios of clones isolated by screening for crossreactivity (focused crossreactive) and specificity (focused) from both the linear and constrained libraries. Lines show the mean value in each group. *Indicates a statistical difference from the random and focused groups (P < 0.0001). **Indicates a statistical difference from the random and focused crossreactive groups (P < 0.0001).
Sorting for improved specificity for the target antibody
To identify peptide ligands with improved specificity for the target antibody, two additional rounds of sorting were performed with enriched library pools from the red++green− and red++green−− populations wherein the control IgG concentration (100 μM) was 100-fold higher than that of the disease IgG (1 μM) [Fig. 2(C)]. Individual clones were isolated and their apparent specificities were quantitatively ranked using the ratio of red- to green-fluorescence, or specificity ratio [Fig. 3(B)]. Specificity ratios exhibited by antibody ligands ranged from 5 to 74, when that of the wild-type T7•tag was normalized to a ratio of 100. All binders from the random library exhibited an apparent specificity less than that of the peptide to which the antibody was raised [Fig. 3(B)].
Directed evolution of the peptide mimotope specificity
Since it is unlikely that mimotopes identified from random peptide libraries will have optimal affinity and specificity for antibody biomarker detection, we anticipated that mutagenesis and screening would enable directed evolution of mimotope specificity, to enhance diagnostic utility. Toward this end, two focused libraries were designed based on two consensus motifs among clones having a red:green ratio greater than 35 (MGXQQ, CQQ/MM/LC) (Table I), yielding libraries of the form X5MGXQQX5 (linear) and X5CQQ/MM/LCX5 (constrained). To enable screening for both high affinity and specificity, focused libraries were constructed as N-terminal fusions using the eCPX display scaffold.21 Each library was screened for improved specificity with the control IgG present at a 100-fold higher concentration than the disease IgG. Even in the presence of the higher concentration of control IgG, 3% of the linear library members and 1% of the constrained library members exhibited antibody binding, a 3 to 10-fold increase relative to the random library. Thus, as expected, the focused second-generation libraries contained a higher frequency of highly target specific binding peptides.
Table I.
Peptide Sequences and Specificity Ratios of Clones Isolated from Random and Designed Libraries
 |
Individual clones isolated from the focused libraries exhibited substantially higher specificities than did clones isolated from the random library [Fig. 3(B), Table I]. Clones from the focused libraries exhibited reduced crossreactivity and increased specificity for the anti-T7•tag IgG [Fig. 3(B)], when compared with peptides isolated from the random library using flow cytometry. The specificity ratios of several clones from the focused libraries were equivalent to or exceeded that of the wild-type T7•tag used to raise this monoclonal antibody (Table I). The improvements in the specificities of clones from both focused libraries were significantly higher (P < 0.0001) than those of the most specific clones from the random library [Fig. 3(C)]. Of course, we cannot rule out the possibility that a reduced specificity ratio could be observed in the context of a new set of normal donor IgG. For comparison, the focused libraries were also screened for nonspecific or crossreactive binders to the control IgG [Fig. 2(F)], and clones from the crossreactive populations exhibited specificity ratios between 1 and 12, indicating significantly lower specificities for the disease IgG relative to random library clones (P < 0.0001) and specific clones from the focused libraries (P < 0.0001) [Fig. 3(C)].
Since improvements in apparent specificity could be due to either reduced affinities to nontarget antibodies, or increased affinity for the target antibody the dissociation constants of representative first and second-generation epitopes were measured using surface plasmon resononance. Interestingly, the highly specific epitope identified by directed evolution (GSEGLMGPQQKWVGGKK) exhibited roughly two-fold higher affinity (KD = 1.7 (±0.3) × 10−7 M) than the wild-type T7•tag (KD = 2.8 (±0.5) × 10−7 M), and more than three-fold improved affinity relative to the first generation epitope RCRGMMGPQQSTRDTKK (KD = 5.3 (±0.2) × 10−7 M). These results suggest that the higher specificity of this peptide was in part achieved by improved affinity for the target antibody without concomitant increases in affinity for nontarget antibodies. Experiments to assess binding affinity to the pooled IgG were also performed. However, affinity could not be determined because of high background signals obtained from nonspecific binding of IgG in the reference flow cell which exceeded the signals obtained in the flow cells with immobilized peptides.
Highly specific peptides obtained by directed evolution exhibited a preference for proline in the eighth position and for glycine in the fourth position (xxxGxMGPQQxxxxx). In contrast, low-specificity ligands from the same library yielded highly crossreactive clones with a distinct consensus of xxxPYMGYQQxxxxx, and none exhibited proline at the eighth position. Similarly, specific sequences from a disulfide-constrained library exhibited a preference for glutamine over methionine in the eighth position while crossreactive sequences preferred methionine or lysine. These results demonstrate that key determinants of specificity can be identified by analysis of a panel of specificity-evolved ligands.
Discussion
The function of engineered proteins, including peptides and antibodies, in practical applications is critically dependent on their target recognition specificity. Even so, experimental methods to measure and manipulate specificity in complex mixtures are seldom used in practice. The term “specificity” is frequently used to describe qualitatively the molecular selectivity or discrimination capabilities in protein interactions and is a complex function of contact area/pair-wise interactions, amino acid content, surface topology, backbone and side chain mobility, concentration, and other parameters.22 Consequently, specificity is not generally defined explicitly. However, specificity is potentially quantifiable. For instance, consider species Xi binding to Xj to form a complex XiXj in a mixture of n noncovalently interacting species at equilibrium,
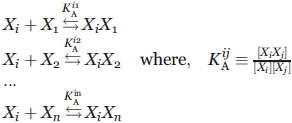 |
Here, a systemic, or extrinsic specificity for interaction i,j φij can be defined as a concentration dependent variable,
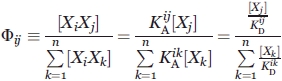 |
(1) |
where KAij and KDij are the equilibrium association and dissociation constants respectively for the i,j complex [XiXj]. As defined, φij ranges from zero, when i and j do not interact, to infinity for a hypothetical infinitely specific complex between Xi and Xj. However, while KDij is typically measured in vitro using purified proteins, seldom are the complete set of KDij and [Xk] values (the denominator) known. For this reason, the “specificity” of a given interaction is usually investigated by determining the extent of binding between species i and a small, arbitrary subset of all possible n proteins present in the sample.23,24 Here, multiparameter flow cytometry instrumentation was used to quantify the magnitude of the numerator and the aggregate denominator of Eq. (1), thus enabling specificity-based library screening, directed evolution, and clone characterization.
Although molecular specificity has been enhanced by affinity maturation strategies, it is clear from 1 that affinity and specificity need not be correlated. Indeed, experimental studies have demonstrated that high-affinity ligands can be nonspecific. GTP-binding aptamers with affinities ranging from 9 nM to 8 μM were identified, and their specificity was measured by measuring affinities toward a set of related GTP analogs. High-affinity aptamers were not more specific for GTP. The authors concluded that high specificity arises only when direct selection pressure for specificity is applied.25 Quantitative affinity measurements of a panel of small molecule kinase antagonists for a subset of the kinome further demonstrate that high-affinity antagonists can be nonspecific leading to unacceptable side-effect profiles.19 Even high-affinity antibody fragments selected from phage display libraries can be nonspecific.26 These studies suggest that when specificity constraints are important for the intended application, specificity should be considered explicitly in library screening and directed evolution processes.
Affinity maturation using in vitro display technologies is generally accomplished by randomization and affinity-based selection with limiting concentrations of antigen or screening after a prolonged dissociation phase.1–3 Our results and consideration of Eq. (1) suggest an alternative approach to simultaneously optimize affinity and specificity, wherein measurements of nontarget protein binding to competing interaction partners enables quantitative screening for optimal affinity and specificity. By increasing the concentrations of nontarget interaction partners in the system, one can increase the magnitude of the denominator of 1, thereby requiring increased target binding to maintain or improve the value of the specificity parameter φij. Interestingly, screening for protein binding peptides within the bacterial cytoplasm (where nontarget protein concentrations are very high) only yielded peptide ligands with KD values <1 μM,27 despite the fact that ligands with KDs greater than 10 μM also should have been recovered since intracellular receptor/ligand concentrations were greater than 100 μM. This unexpected result can be understood considering 1, since high target affinity could overcome the presence of high nontarget protein concentrations (e.g., in E. coli) approaching 1 mM to maintain a high specificity value φij.
Previous efforts to evolve the specificity of protein ligand interactions have relied on sequential positive and negative affinity selections of protein display libraries. Consequently, the desired specificity characteristics of library members are not measured during the screening process. For example, phage and mRNA display approaches have used sequential selection, rather than quantitative measurements, to favor specificity between a small set of related targets. mRNA display has been applied to identify peptides exhibiting a roughly 100-fold selectivity for G-protein subunits Gαi1 and Gαs(s), by first depleting the library with a column matrix of immobilized nontarget, and then performing mRNA library panning with a molar excess of the target Gα as a competitor.28 Similarly, anticortisol antibodies with improved fine specificity were identified previously by combining computational prediction of optimal substitution sites and a phage display selection in the presence of an excess of steroid anologs prednisolone and dexamethasone.29 Nevertheless, these strategies do not enable measurement of interactions with nontargets during screening thereby rendering identification of selection conditions that maximize specificity difficult. For instance, one cannot readily determine whether all library members capable of interacting with the target have been depleted from the library population.
In this study, we simulated a serum antibody sample from a subject with disease by spiking an IgG (1 nM) of known affinity and specificity into pooled control IgG (1 μM), such that target “biomarker” represents 0.1% of the circulating antibody repertoire. These concentrations were chosen to mimic a 10-fold dilution of serum antibodies, while enabling counter screening with a 10- to 100-fold excess of control antibody, and are comparable with those in previous antibody fingerprinting studies.18,30 Initially, competing control Ig was used in a 10-fold excess to favor a high level of clone recovery, and then in 100-fold excess in later rounds to ensure disease specificity. We have neither determined the minimum target antibody concentration nor apparent antibody affinity necessary for mimotope identification. In our experience, antibody specific, peptide-displaying bacterial clones frequently exhibit high apparent affinity that can be attributed to bivalent binding to adjacent epitopes on the cell surface. The high apparent affinities afforded by multivalent display systems should be useful for identifying binders to low abundance serum biomarkers. Finally, the specificity-based screening approach presented here would, in principle, enable screening for absent biomarkers, present in control sera but not in disease sera.
Specificity-based screening of bacterial or yeast cell display libraries via cytometry is anticipated to enable engineering of proteins with novel specificity properties including broadened or multispecificity—the ability of one ligand to specifically recognize two or more distinct targets. Phage display has been applied to evolve the anti-HER-2 therapeutic Traztuzumab to exhibit dual specificity for HER-2 and VEGF, which resulted in increased efficacy in animal tumor models.31 Specificity screening using FACS may be especially useful for engineering antibody and nonantibody protein therapeutics with desired multispecificity and/or reduced off-target binding. Using FACS, multispecificity could be achieved by screening with two target ligands, and all relevant nontargets labeled with three spectrally distinct fluorophores. Phage display has also been applied to identify antibodies that crossreact with human and murine homologs of BAFF-R to facilitate rapid transitioning from preclinical to clinical studies,32 and to broaden the specificity of an antisulfonamide antibody for antibiotic detection. Similarly, yeast display enabled evolution of scFv antibodies that crossreact with distinct subtypes of Botulinum neurotoxin,11 though interactions with relevant background nontargets were not quantified. The present approach to evolve specificity of protein and peptide interactions analogous to affinity maturation approaches will enable the development of proteins and peptides with improved specificity characteristics for a variety of molecular recognition applications.
Materials and Methods
Reagents and strains
All experiments were performed with E. coli strain MC1061 (F-araD139 Δ(ara-leu)7696 galE15 galK16 Δ(lac)X74 rpsL (StrR) hsdR2 (rK− mK+) mcrA mcrB1).33 A bacterial display library comprised of random 15-mer insertions in extracellular loop 2 of outer membrane protein X (OmpX)34 was used in the initial screens to identify peptide epitopes for the target antibody. Bacterial cultures were grown at 37°C with vigorous shaking in LB media supplemented with chloramphenicol (34 μg/mL) for expansion and additionally supplemented with arabinose (0.2% for random library and 0.04% for the focused libraries) to induce peptide display. Reagents were supplied as follows: Primers and oligonucleotides (Operon Biotechnologies), restriction enzymes (New England Biolabs), streptavidin-R-phycoerythrin conjugate (SA-PE) (Invitrogen), anti-biotin mAb R-phycoerythrin (Miltenyi Biotec), biotinylated anti-T7•tag mAb (Novagen), and human serum IgG (Sigma). Biotinylation of IgG was carried out using the FluoReporter® mini-biotin-XX protein labeling kit (Invitrogen), and IgG was also labeled with Alexa Fluor® 488 carboxylic acid (Invitrogen) according to the manufacturer's instructions.
Library construction
The focused libraries were constructed to be displayed at the N-terminus of a variant of circularly permuted OmpX (eCPX).10 Oligonucleotide primers 1 and 3 (Supporting Information Table 2) were used in a PCR with plasmid pB33 eCPX21 as the template to construct the library with the form X5MGXQQX5, where X is any amino acid. PCR products were digested with SfiI and subsequently ligated into similarly digested (HincII/SfiI) pB33eCPX. Ligated DNA was transformed into strain MC1061 by electroporation and resulted in 1.2 × 108 CFU. The focused library with the form X5CQM/QM/LCX5 was constructed similarly using primers 2 and 3 and resulted in 3.3 × 108 CFU.
Depletion of E. coli specific IgG before screening
An overnight culture of cells overexpressing the library scaffold (either OmpX or eCPX) without a displayed peptide were diluted (1:50) in fresh media and grown until the optical density at 600 nm (OD600) was between 0.5 and 0.7 (1.5–2 hr). Cells were induced for 30 min to 1 hr at 37°C. The OD600 of the culture was measured and an appropriate volume of cells were centrifuged at 3000 rcf for 5 min. Cells were resuspended in either disease or control IgG at a concentration of 109 cells/mL. Samples were incubated at room temperature on a LabQuake rotary shaker for 1.5 to 2 hr. Samples were then centrifuged at 3000 rcf for 5 min using a swinging bucket rotor. The IgG in the supernatant was recovered, filtered using a syringe fitted with a 0.22 μM filter, and aliquots of each sample were stored at −20°C.
Library screening and clonal analysis using flow cytometry
For screening and analysis, overnight cultures were diluted (1:50) in fresh media and grown until the OD600 was between 0.5 and 0.7 (1.5–2 hr). Cells from the random library were induced for 30 min and cells from the focused library were induced for 1 hr. A volume of culture corresponding to the desired number of cells (5 × 108 cells for the first round of sorting or for subsequent rounds, a 5-fold oversampling of the number of cells retained from the previous round) was washed once or twice in 1 mL total volume PBS by centrifugation at 3000 rcf for 5 min. The final cell pellet was resuspended in PBS and then added to a mixture of either 10 μM control IgG and 1 μM disease IgG or 100 μM control IgG and 1 μM disease IgG. Samples were incubated for 30 min at 37°C on a LabQuake rotary shaker. Samples were washed two times in 1 mL ice-cold PBS by centrifugation at 4°C and then labeled with 5–30 nM SA-PE or anti-biotin R-phycoerythrin conjugate on ice for 45 min. Cells were washed in 1 mL ice-cold PBS by centrifugation at 4°C, resuspended in ice-cold PBS to a concentration between 107 and 108 cells/mL, and analyzed or sorted on a FACSAria™ cell sorter (Becton Dickinson). Sorted cells were amplified by overnight growth and plated directly on agar for isolation of single clones.
Peptide affinity measurement using surface plasmon resonance
Mass spectrometry was performed on the synthetic peptides used in the binding experiments by the manufacturer. Binding experiments were performed at 25°C using a Biacore 3000 instrument equipped with research-grade CM5 sensor chips (GE Healthcare). Synthetic peptides pepT7 (MASMTGGQQMGKK), pep27 (RCRGMMGPQQSTRDTKK), and pepM3.7 (GSEGLMGPQQKWVGGKK) were diluted in 10 mM acetate, pH 5.0 and coupled to the surface of individual flow cells on the sensor chip with amine-coupling reagents (EDC, NHS; and sodium ethanolamine HCl, pH 8.5) (GE Healthcare) using the immobilization application wizard in the Biacore 3000 control software. This resulted in an immobilization of 36 RUs for pepT7, 196 RUs for pep27, and 190 RUs for pepM3.7. An additional flow cell on the chip was activated and capped without any peptide coupled to the surface and used as a reference for all binding experiments.
HBS-EP (0.01M HEPES pH 7.4, 0.15M NaCl, 3 mM EDTA, 0.005% v/v surfactant P2, GE Healthcare) was the running buffer for all experiments. The anti-T7•tag antibody was diluted in the running buffer and concentrations ranging from 1 nM to 1 μM were injected in the order of increasing concentration over the sensor chip at 30 μL/min. Association and dissociation of the antibody were both monitored for 5 min. A regeneration step consisting of a 5 s injection of 10 mM glycine-HCl, pH 1.5 (GE Healthcare) was performed after each dissociation period. Kinetic data was analyzed using the bivalent analyte model in the BIAevaluation software.
Glossary
Abbreviations:
- FACS
fluorescence-activated cell sorting
- eCPX
enhanced circularly permuted OmpX.
References
- 1.Daugherty PS, Chen G, Olsen MJ, Iverson BL, Georgiou G. Antibody affinity maturation using bacterial surface display. Protein Eng. 1998;11:825–832. doi: 10.1093/protein/11.9.825. [DOI] [PubMed] [Google Scholar]
- 2.Boder ET, Midelfort KS, Wittrup KD. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc Natl Acad Sci USA. 2000;97:10701–10705. doi: 10.1073/pnas.170297297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bessette PH, Rice JJ, Daugherty PS. Rapid isolation of high-affinity protein binding peptides using bacterial display. Protein Eng Des Sel. 2004;17:731–739. doi: 10.1093/protein/gzh084. [DOI] [PubMed] [Google Scholar]
- 4.Clackson T, Wells JA. In vitro selection from protein and peptide libraries. Trends Biotechnol. 1994;12:173–184. doi: 10.1016/0167-7799(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 5.Szwajkajzer D, Carey J. Molecular and biological constraints on ligand-binding affinity and specificity. Biopolymers. 1997;44:181–198. doi: 10.1002/(SICI)1097-0282(1997)44:2<181::AID-BIP5>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 6.Levin AM, Weiss GA. Optimizing the affinity and specificity of proteins with molecular display. Mol Biosyst. 2006;2:49–57. doi: 10.1039/b511782h. [DOI] [PubMed] [Google Scholar]
- 7.Brown CK, Modzelewski RA, Johnson CS, Wong MK. A novel approach for the identification of unique tumor vasculature binding peptides using an E. coli peptide display library. Ann Surg Oncol. 2000;7:743–749. doi: 10.1007/s10434-000-0743-0. [DOI] [PubMed] [Google Scholar]
- 8.Lunder M, Bratkovic T, Doljak B, Kreft S, Urleb U, Strukelj B, Plazar N. Comparison of bacterial and phage display peptide libraries in search of target-binding motif. Appl Biochem Biotechnol. 2005;127:125–131. doi: 10.1385/abab:127:2:125. [DOI] [PubMed] [Google Scholar]
- 9.Harvey BR, Georgiou G, Hayhurst A, Jeong KJ, Iverson BL, Rogers GK. Anchored periplasmic expression, a versatile technology for the isolation of high-affinity antibodies from Escherichia coli-expressed libraries. Proc Natl Acad Sci USA. 2004;101:9193–9198. doi: 10.1073/pnas.0400187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rice JJ, Schohn A, Bessette PH, Boulware KT, Daugherty PS. Bacterial display using circularly permuted outer membrane protein OmpX yields high affinity peptide ligands. Protein Sci. 2006;15:825–836. doi: 10.1110/ps.051897806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Rodriguez C, Levy R, Arndt JW, Forsyth CM, Razai A, Lou J, Geren I, Stevens RC, Marks JD. Molecular evolution of antibody cross-reactivity for two subtypes of type A botulinum neurotoxin. Nat Biotechnol. 2007;25:107–116. doi: 10.1038/nbt1269. [DOI] [PubMed] [Google Scholar]
- 12.Mintz PJ, Kim J, Do KA, Wang X, Zinner RG, Cristofanilli M, Arap MA, Hong WK, Troncoso P, Logothetis CJ, Pasqualini R, Arap W. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat Biotechnol. 2003;21:57–63. doi: 10.1038/nbt774. [DOI] [PubMed] [Google Scholar]
- 13.Vidal CI, Mintz PJ, Lu K, Ellis LM, Manenti L, Giavazzi R, Gershenson DM, Broaddus R, Liu J, Arap W, Pasqualini R. An HSP90-mimic peptide revealed by fingerprinting the pool of antibodies from ovarian cancer patients. Oncogene. 2004;23:8859–8867. doi: 10.1038/sj.onc.1208082. [DOI] [PubMed] [Google Scholar]
- 14.Kouzmitcheva GA, Petrenko VA, Smith GP. Identifying diagnostic peptides for lyme disease through epitope discovery. Clin Diagn Lab Immunol. 2001;8:150–160. doi: 10.1128/CDLI.8.1.150-160.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Somers V, Govarts C, Hellings N, Hupperts R, Stinissen P. Profiling the autoantibody repertoire by serological antigen selection. J Autoimmun. 2005;25:223–228. doi: 10.1016/j.jaut.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 16.Govarts C, Somers K, Hupperts R, Stinissen P, Somers V. Exploring cDNA phage display for autoantibody profiling in the serum of multiple sclerosis patients: optimization of the selection procedure. Ann NY Acad Sci. 2007;1109:372–384. doi: 10.1196/annals.1398.043. [DOI] [PubMed] [Google Scholar]
- 17.Hansen MH, Ostenstad B, Sioud M. Antigen-specific IgG antibodies in stage IV long-time survival breast cancer patients. Mol Med. 2001;7:230–239. [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Yu J, Sreekumar A, Varambally S, Shen R, Giacherio D, Mehra R, Montie JE, Pienta KJ, Sanda MG, Kantoff PW, Rubin MA, Wei JT, Ghosh D, Chinnaiyan AM. Autoantibody signatures in prostate cancer. N Engl J Med. 2005;353:1224–1235. doi: 10.1056/NEJMoa051931. [DOI] [PubMed] [Google Scholar]
- 19.Fabian MA, Biggs WH, III, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 20.Bessette PH, Hu X, Soh HT, Daugherty PS. Microfluidic library screening for mapping antibody epitopes. Anal Chem. 2007;79:2174–2178. doi: 10.1021/ac0616916. [DOI] [PubMed] [Google Scholar]
- 21.Rice JJ, Daugherty PS. Directed evolution of a biterminal bacterial display scaffold enhances the display of diverse peptides. Protein Eng Des Sel. 2008;21:435–442. doi: 10.1093/protein/gzn020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silverman J, Liu Q, Bakker A, To W, Duguay A, Alba BM, Smith R, Rivas A, Li P, Le H, Whitehorn E, Moore KW, Swimmer C, Perlroth V, Vogt M, Kolkman J, Stemmer WP. Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains. Nat Biotechnol. 2005;23:1556–1561. doi: 10.1038/nbt1166. [DOI] [PubMed] [Google Scholar]
- 24.Sparks AB, Rider JE, Hoffman NG, Fowlkes DM, Quillam LA, Kay BK. Distinct ligand preferences of Src homology 3 domains from Src, Yes, Abl, Cortactin, p53bp2, PLCgamma, Crk, and Grb2. Proc Natl Acad Sci USA. 1996;93:1540–1544. doi: 10.1073/pnas.93.4.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carothers JM, Oestreich SC, Szostak JW. Aptamers selected for higher-affinity binding are not more specific for the target ligand. J Am Chem Soc. 2006;128:7929–7937. doi: 10.1021/ja060952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Haard HJ, van Neer N, Reurs A, Hufton SE, Roovers RC, Henderikx P, de Bruine AP, Arends JW, Hoogenboom HR. A large non-immunized human Fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J Biol Chem. 1999;274:18218–18230. doi: 10.1074/jbc.274.26.18218. [DOI] [PubMed] [Google Scholar]
- 27.You X, Nguyen AW, Jabaiah A, Sheff MA, Thorn KS, Daugherty PS. Intracellular protein interaction mapping with FRET hybrids. Proc Natl Acad Sci USA. 2006;103:18458–18463. doi: 10.1073/pnas.0605422103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Austin RJ, Ja WW, Roberts RW. Evolution of class-specific peptides targeting a hot spot of the Galphas subunit. J Mol Biol. 2008;377:1406–1418. doi: 10.1016/j.jmb.2008.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chames P, Coulon S, Baty D. Improving the affinity and the fine specificity of an anti-cortisol antibody by parsimonious mutagenesis and phage display. J Immunol. 1998;161:5421–5429. [PubMed] [Google Scholar]
- 30.Zanoni G, Navone R, Lunardi C, Tridente G, Bason C, Sivori S, Beri R, Dolcino M, Valletta E, Corrocher R, Puccetti A. In celiac disease, a subset of autoantibodies against transglutaminase binds toll-like receptor 4 and induces activation of monocytes. PLoS Med. 2006;3:1637–1653. doi: 10.1371/journal.pmed.0030358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bostrom J, Yu SF, Kan D, Appleton BA, Lee CV, Billeci K, Man W, Peale F, Ross S, Wiesmann C, Fuh G. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science. 2009;323:1610–1614. doi: 10.1126/science.1165480. [DOI] [PubMed] [Google Scholar]
- 32.Lee CV, Hymowitz SG, Wallweber HJ, Gordon NC, Billeci KL, Tsai SP, Compaan DM, Yin J, Gong Q, Kelley RF, DeForge LE, Martin F, Starovasnik MA, Fuh G. Synthetic anti-BR3 antibodies that mimic BAFF binding and target both human and murine B cells. Blood. 2006;108:3103–3111. doi: 10.1182/blood-2006-03-011031. [DOI] [PubMed] [Google Scholar]
- 33.Casadaban MJ, Cohen SN. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol. 1980;138:179–207. doi: 10.1016/0022-2836(80)90283-1. [DOI] [PubMed] [Google Scholar]
- 34.Hu X, Bessette PH, Qian J, Meinhart CD, Daugherty PS, Soh HT. Marker-specific sorting of rare cells using dielectrophoresis. Proc Natl Acad Sci USA. 2005;102:15757–15761. doi: 10.1073/pnas.0507719102. [DOI] [PMC free article] [PubMed] [Google Scholar]