

Abstract
Identification and characterization of recurrent supersecondary structural elements is central to understanding the rules governing protein tertiary structure. Here, we describe the GD box, a widespread noncontiguous supersecondary element, which we initially found in a group of topologically distinct but homologous β-barrels—the cradle-loop barrels. The GD box is similar both in sequence and structure and comprises two short unpaired β-strands connected by an orthogonal type-II β-turn and a noncontiguous β-strand forming hydrogen bonds with the β-turn. Using structure-based analysis, we have detected 518 instances of the GD box in a nonredundant subset of the SCOP database comprising 3771 domains. Apart from the cradle-loop barrels, this motif is also found in a diverse set of nonhomologous folds including other topologically related β-barrels. Since nonlocal interactions are fundamental in the formation of protein structure, systematic identification and characterization of other noncontiguous supersecondary structural elements is likely to prove valuable to protein structure modeling, validation, and prediction.
Keywords: supersecondary structural element, cradle-loop barrels, nonlocal interactions, locally similar protein motif
Introduction
Protein structures show a distinctive hierarchical order, in which amino acid chains (primary structure) form local, hydrogen-bonded elements (secondary structure) that assemble into specific, compact topologies (tertiary structure), and frequently also associate noncovalently into higher order assemblies (quaternary structure). Tertiary structures are often composed of multiple autonomously folding entities, named domains, which retain their overall fold and frequently also their function when they reoccur in different proteins, and thus represent units of evolution in today's proteins. However, domains are not the smallest unit of protein structure between the secondary and tertiary level. Unrelated domains frequently show recurrent local substructures, termed supersecondary structures, that comprise two or more secondary structure elements in specific geometric arrangements.1 Certain supersecondary structures are widespread in proteins, examples of which include β-hairpins, α-hairpins, β-meanders, and βαβ motifs,2 and all described so far are formed by adjacent segments in the polypeptide chain. However, given the central role of nonlocal interactions in the formation of protein structure (see for example Minor et al.3), it seems reasonable to assume that some widespread supersecondary structures should be formed by noncontiguous secondary structure elements.
We have identified one such structure, consisting of a recurrent βαβ-motif, in a group of topologically distinct but homologous β-barrels, which we called cradle-loop barrels for the shape of their putative substrate-binding loops. These barrels illustrate the evolution of folded proteins from simple oligomers of one fragment to the emergence of complex catalysis.4–7 To capture the relationship between such distinct folds originating from the same basic supersecondary structure, we proposed a new protein classification level, the metafold.8 The cradle-loop metafold comprises four distinct folds, the double-psi barrel [Fig. 1(A)], the swapped-hairpin barrel, the RIFT barrel, and the C-terminal barrel of bacterial fluorinating enzyme, each built from two copies of the conserved βαβ-element, either as monomers with internal sequence symmetry or as homodimers with one copy per subunit. A characteristic feature of the βαβ-element is a conserved 11 amino acid sequence motif, [h]-x-[h]-x(2)-G-[p]-x-[h]-x-[h], where [h] is hydrophobic and [p] polar. As the polar residue is frequently aspartate, we named this motif the GD box. In all cradle-loop barrels, this motif is structurally highly conserved and comprises two short unpaired β-strands connected by an orthogonal diverging type-II β-turn [Fig. 1(C)]. Diverging β-turns were first described in the I-sites library, which contains motifs that correlate both in sequence and structure.9 The glycine occupies the i + 2 position of the β-turn and the side chain of the polar residue in i + 3 accepts a hydrogen bond from the residue at position i. The backbone of the residues in i + 2 and i + 3 forms further hydrogen bonds to a noncontiguous β-strand from the symmetry-related half of the barrel [Fig. 1(D)]. Thus the GD box is an example of a noncontiguous supersecondary element; to our knowledge the first identified. We have analyzed the occurrence of the GD box in proteins of known structure. Our results show that it is widely represented in both homologous and nonhomologous contexts.
Figure 1.
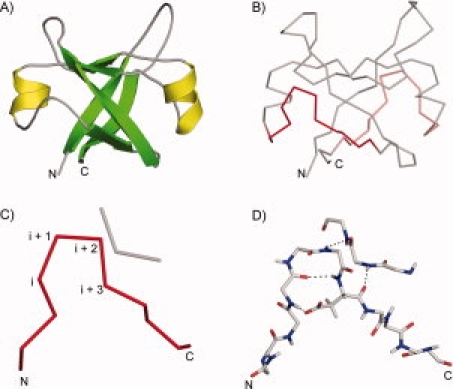
The GD box element. A: The double-psi barrel fold of VatN-N (PDB 1CZ4, residues 1–91) is shown in cartoon representation. α-helices are colored in yellow and β-strands in green. B: The double-psi barrel fold of VatN-N is shown in backbone representation. The unpaired hairpins (residues 34–44 and 77–87) from the two GD box elements are shown in red. C: The first GD box element from VatN-N (residues 34–44, 53–55) is shown. The positions corresponding to the type-II β-turn are marked. D: Detailed view of the hydrogen-bonding network of the first GD box element. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Results and Discussion
Searching for GD box elements in known structures
We used structure comparisons to detect GD boxes in SCOP25, a subset of the SCOP database filtered at 25% sequence identity. The GD box adopts a very similar structure in all cradle-loop barrels and we chose the first GD box from the N-terminal domain of the archaeal AAA chaperone VAT (PDB 1CZ4, residue 34–44) as the query structure. We were not aiming at detecting all GD box elements in an exhaustive manner, so we used a moderately generous root-mean-square deviation (RMSD) cutoff of 1.5 Å. Because one of the goals of this study was to establish whether the GD box forms the same noncontiguous interactions in all its embodiments, we did not consider the presence of hydrogen bonds between the residues at positions i + 2 and i + 3 of the β-turn and a noncontiguous β-strand in our searches.
We detected a total of 518 GD boxes in 420 distinct domains, which are classified into 134 folds in SCOP25 (Supporting Information Table S1); some domains contained multiple copies of the element. At 3771 domains total, this means that slightly >10% of all domains in SCOP25 contain at least one GD box. In all but 32 of the detected cases (6%), at least one of the residues at position i + 2 and i + 3 of the β-turn formed a backbone hydrogen bond with a residue in a noncontiguous β-strand. The noncontiguous interaction can thus be considered a general feature of GD boxes.
As a control, we wanted to assess the proportion to which type-II β-turns form GD boxes. We therefore detected all type-II β-turns in SCOP25 using Promotif10 and found 6327 instances, indicating that <10% of all type-II β-turns are part of GD box elements.
Apart from the cradle-loop barrels, the GD box is found in other topologically related, but nonhomologous barrels,8 as well as in a large number of folds that are not related either evolutionarily or topologically to the cradle-loop barrels [Fig. 2(A)]. The latter include the OB fold, the immunoglobulin-like fold [Fig. 3(A)], the NAD(P)-binding Rossmann fold, and the ubiquitin-like fold. Some folds contain multiple copies of the GD box element; for example, each half of the cradle-loop barrels contains its own copy of the GD box element and single-stranded [Fig. 3(B)] and double-stranded β-helices contain multiple overlapping copies. In most GD boxes, the diverging β-turn coordinates a β-strand that is distant in the linear polypeptide sequence and in some cases, this β-strand is contributed by a different subunit altogether. For example, in all homodimeric cases, such as in the transition state regulator AbrB from Bacillus subtilis (1YFB), the noncontiguous strand originates from the symmetry-related monomer. We propose that the GD box acts as a structural tether: on one hand, it provides structural specificity by allowing the polypeptide chain to snap into the final folded conformation once the hydrophobic collapse has produced a molten globule and brought the noncontiguous parts into approximate vicinity; on the other hand, it stabilizes the structure once folding is complete, by connecting elements which in many cases bracket the parts of the chain that form the hydrophobic core. Its geometric simplicity makes it compatible with a broad range of β topologies. These considerations may explain the widespread and frequently analogous representation of GD boxes in β folds.
Figure 2.
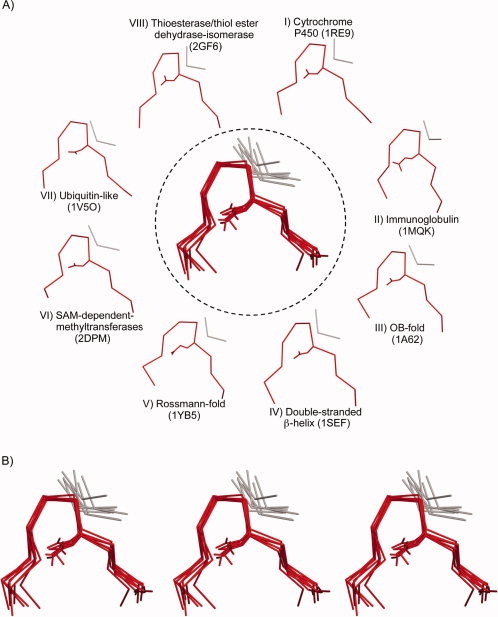
A gallery of GD box elements. A: GD box elements from eight different folds are shown. The unpaired hairpin is shown in red and the noncontiguous segment in gray. A structural superposition of the GD box elements is shown in the center. The structures shown are (I) 1RE9: residues 300–310, 290–292, (II) 1MQK: chain L, 11–21, 77–79, (III) 1A62: 89–99, 54–56, (IV) 1SEF: 223–233, 208–210, (V) 1YB5: 143–153, 173–175, (VI) 2DPM: 183–193, 232–235, (VII) 1V5O: 71–81, 9–11, and (VIII) 2GF6: 68–78, 11–13. B: Wall-eye and cross-eye stereo view of the superposition shown in panel A. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Figure 3.
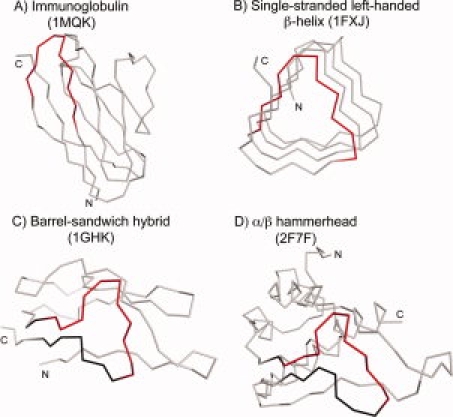
A gallery of GD box-containing folds. The contiguous segment of the GD box element is shown in red in all the structures. The βββ-motif containing the GD box element is shown in black in (C) and (D). The structures shown are (A) 1MQK: chain L, (B) 1FXJ: chain A, 252–329, (C) 1GHK, and (D) 2F7F: chain A, 4–140.
Sequence features of the GD box motif
Although the searches for the GD box elements were made by structural comparison, their sequences (considering only the unpaired hairpin) follow the same characteristic pattern as in the cradle-loop barrels: [h]-x-[h]-x(2)-G-[p]-x-[h]-x-[h] (Table I, Fig. 4). Hydrophobic residues are strongly favored at positions 3, 9, and 11, and to lesser extent at position 1. Positions 4, 5, 6, and 7 correspond to the four positions (i, i + 1, i + 2, and i + 3) of the type-II β-turn; at these positions hydrophilic residues are favored. Position 6 is dominated by glycine and to lesser extent by asparagine. Type-II β-turns favor cysteine, serine, and lysine at position i + 311; however, in GD box elements the i + 3 position is predominantly occupied by aspartic acid and to lesser extent by glutamine and glutamic acid.
Table I.
Positional Propensities for Each of the 11 Positions of the GD Box
| Residue | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ILE | 1.3 | 0.8 | 3.2 | 0.2 | 0.6 | 0 | 0.2 | 1.4 | 3 | 1.2 | 2.7 |
| PHE | 1.3 | 1.2 | 1.7 | 0.2 | 0.2 | 0.2 | 0.1 | 0.7 | 1.4 | 0.8 | 2.1 |
| VAL | 1.2 | 1 | 2.9 | 0.4 | 1.1 | 0 | 0.8 | 1.7 | 4.8 | 1.2 | 3 |
| LEU | 1.2 | 0.6 | 3.1 | 0.3 | 0.3 | 0 | 0.1 | 0.8 | 2.2 | 1.2 | 1.7 |
| TRP | 0.8 | 0.3 | 0.9 | 0.3 | 0 | 0 | 0 | 0.9 | 0.9 | 0.8 | 1.2 |
| MET | 1.1 | 0.4 | 2.1 | 0.4 | 0.4 | 0.4 | 2 | 0.7 | 0.5 | 1 | 0.7 |
| ALA | 1.3 | 0.5 | 0.7 | 0.8 | 1.3 | 0.1 | 0.9 | 0.7 | 0.3 | 0.9 | 1 |
| GLY | 0.7 | 1.1 | 0.1 | 1 | 0.3 | 10.4 | 0.6 | 0.2 | 0.3 | 0.9 | 0.5 |
| CYS | 1.5 | 0.6 | 0.7 | 0 | 0.6 | 0 | 0.6 | 0.1 | 1.3 | 0.6 | 1.3 |
| TYR | 0.8 | 0.9 | 1.1 | 0.6 | 0.3 | 0.3 | 0.3 | 0.8 | 1.5 | 1.6 | 1 |
| PRO | 1.2 | 1.4 | 0.7 | 2.4 | 4.5 | 0 | 0 | 1 | 0 | 0.5 | 0.7 |
| THR | 1.3 | 1.6 | 0.2 | 1.1 | 0.6 | 0.1 | 1.3 | 2.2 | 0.2 | 1.8 | 0.4 |
| SER | 0.6 | 1.4 | 0.2 | 0.9 | 0.7 | 0.2 | 1.3 | 0.9 | 0.1 | 1 | 0.4 |
| HIS | 0.9 | 1.7 | 0.3 | 1.2 | 1.2 | 0.3 | 0.5 | 1.1 | 1.3 | 1 | 0.2 |
| GLU | 0.7 | 1.1 | 0.1 | 1.7 | 1.6 | 0.3 | 1.9 | 1.2 | 0 | 1 | 0.2 |
| ASN | 0.8 | 1 | 0 | 0.8 | 0.9 | 1.7 | 0.4 | 0.9 | 0 | 0.4 | 0.5 |
| GLN | 0.8 | 1 | 0.1 | 1.7 | 1 | 0.3 | 2.2 | 0.9 | 0.2 | 1 | 0.3 |
| ASP | 0.5 | 1 | 0 | 0.5 | 1.2 | 0.5 | 5.4 | 0.2 | 0.1 | 0.5 | 0.7 |
| LYS | 0.9 | 1.2 | 0.1 | 2.9 | 1.4 | 0.1 | 0.5 | 1.2 | 0.1 | 0.7 | 0.2 |
| ARG | 1.1 | 0.7 | 0.2 | 1.8 | 1 | 0.3 | 0.1 | 1.6 | 0.3 | 1.2 | 0.3 |
Amino acids are ranked in the order of decreasing hydrophobicity. Positions 4, 5, 6, and 7 correspond to the four positions of the type-II β-turn.
Figure 4.
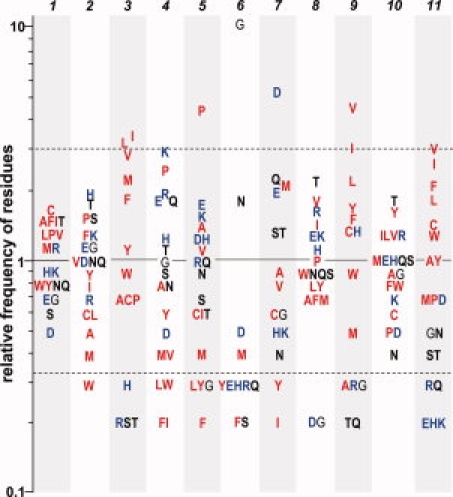
The relative preference of occurrence for the 20 amino acids at the 11 positions of the GD box. Dotted lines indicate the position of residues that are three times more frequent and three times less frequent than expected, respectively. Hydrophobic residues are colored in red, charged residues in blue, and uncharged hydrophobic residues in bold black. The 11 positions of the GD box are indicated. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Evolutionary consequences
Because structure space is finite, unrelated proteins tend to converge on similar local structures. This is reflected in the fact that over half of the residues in the most highly populated folds are found in one of the three most common supersecondary structures, that is αα-hairpins, ββ-hairpins, and βαβ-elements.2 In contrast, sequence space is essentially infinite and many sequences are compatible with a particular local structure. Sequence convergence should thus be highly unlikely. For this reason, statistically significant sequence similarity is considered the best marker for homology. However, short, structurally constrained parts of the polypeptide chain do converge on specific sequence motifs, as described here for the GD box. In such cases, and particularly where these structurally constrained elements form a substantial part of the entire polypeptide chain, it seems possible that the convergent sequence motifs would lead to a level of overall sequence similarity that could be interpreted (erroneously) as indicative of homology.
We therefore wanted to evaluate to what extent sensitive sequence comparison methods, such as those based on the comparison of profile hidden Markov models (HMMs), would return scores for GD box-containing proteins that are normally seen between homologous proteins. To this end, we made pairwise comparisons of profile HMMs for all GD box-containing domains and clustered them by a force-directed procedure, using the statistical significance of the pairwise comparisons to assign attractive and repulsive forces to each profile pair in a three-dimensional map (see Methods section). At settings at which we recover the cradle-loop barrels as a cluster, whose homologous origin we have documented in a series of studies, most other GD box-containing folds did not exhibit connections to the cradle-loop barrels or to each other. The convergent similarity of GD boxes is thus not sufficient to suggest homology between evolutionarily unrelated domains, even when these are being compared by methods calibrated for the detection of very distant relationships.
Two other clusters formed in the three-dimensional map. One contained the Rossmann folds, whose connections relied however on a different supersecondary structure, a dinucleotide-binding βαβ-element whose homologous origin has been discussed previously.12 The other contained the barrel-sandwich hybrid and the α/β-hammerhead folds. The similarity between these two folds relies on a GD box-containing βββ-element, which resembles a hammerhead [Fig. 3(C,D)]. The barrel-sandwich hybrid fold is pseudo-symmetric and contains two homologous copies of the βββ-element. The α/β-hammerhead fold contains one copy of the βββ-element. We conclude that these two folds most likely have arisen from an ancestral βββ-element—the barrel-sandwich hybrid fold by duplication and the α/β-hammerhead fold by accretion.
Application to tertiary structure prediction
Protein folding is still an unsolved problem.13,14 One encouraging approach has been through methods, such as ROSETTA,15 which use fragment libraries to predict tertiary structure by assembling local structural features. However, these methods are mainly successful for domains with less than about 100 residues. One reason for their poor scalability may lie in the fact that they do not consider nonlocal interactions, which become progressively more important with the size of the fold. Enriching these fragment libraries with widespread noncontiguous supersecondary structures that have clear sequence-structure patterns, such as the GD box, should make it possible to include knowledge of nonlocal interactions into this approach.
A problem with using nonlocal interactions as restraints is that, while the contiguous part of the element will be recognizable based on its sequence pattern, the nonlocal interaction partner will be difficult to identify. In NMR structure calculation from ambiguously assigned cross-peaks, as well as in protein–protein docking, one faces similar problems. Indeed, fragment assembly could be viewed as a protein–protein docking task. One can deal with the ambiguities by using ambiguous distance restraints16,17: the restraint is defined on the whole set of possible distances; the one that is most compatible with the overall structure is then picked automatically during the structure calculation process. Such a restraint-based approach would significantly reduce the computational complexity, and thus, would allow modeling and prediction of larger proteins as well.
Methods
For this study, we used the SCOP18 database (version 1.73) filtered for a maximum of 25% sequence identity. After filtering out all NMR structures and all X-ray structures with a resolution of worse than 2 Å, we obtained a subset comprising 3771 domains.
For structure comparisons, we used an implementation of the rigid-body superposition algorithm described by Challis et al.19 Only Cα atoms were considered for superposition. The GD box motif from the N-terminal domain of the archaeal AAA chaperone VAT (PDB code 1CZ4, residues 34–44) was compared with all 11 residue fragments from the SCOP25 dataset. Fragments with an RMSD <1.5 Å with respect to the probe fragment were pooled together. All fragments without a type-II β-turn were removed from this set. We classified as canonical GD boxes all fragments in which at least one of the residues at position i + 2 and i + 3 of the type-II β-turn was involved in hydrogen-bonding interactions with a non-neighboring residue; the remaining fragments were classified as noncanonical. The programs Promotif10 and HBPlus20 were used to detect β-turns and to calculate hydrogen bonds, respectively. The noncanonical fragments were further analyzed in the context of the full protein: if the residues in the β-turn formed hydrogen bonds to a non-neighboring residue, the fragment was also classified as canonical.
The positional propensities for each position of the GD box was calculated as Pi(a) = Fi(a)/F(a), where Pi(a) is the positional propensity of amino acid “a” at the position “i”, Fi(a) is the frequency of “a” at position “i”, and F(a) is the background frequency of “a” in the dataset.
For sequence comparisons, we used HHsearch,21 which is a highly sensitive homology search method based on the pairwise comparison of HMMs. We built multiple sequence alignments for all GD box-containing domains using the buildali.pl script from the HHsearch package. This script is a modified PSI-BLAST procedure, which suppresses the corruption of alignments by preventing the inclusion of nonhomologous sequence segments at the ends of PSI-BLAST high-scoring pairs. Profile HMMs were calculated from the alignments using hhmake (from the HHsearch package) with default settings. We then performed all possible pairwise comparisons between them using HHsearch with default settings and clustered them by their pairwise P-values using CLANS,22 an implementation of the Fruchterman-Reingold algorithm that scales log-P-values into attractive forces in a force field. Clustering was done to equilibrium in 2D at a P-value cutoff of 1.0e-03 using default settings.
References
- 1.Rao ST, Rossmann MG. Comparison of super-secondary structures in proteins. J Mol Biol. 1973;76:241–256. doi: 10.1016/0022-2836(73)90388-4. [DOI] [PubMed] [Google Scholar]
- 2.Salem GM, Hutchinson EG, Orengo CA, Thornton JM. Correlation of observed fold frequency with the occurrence of local structural motifs. J Mol Biol. 1999;287:969–981. doi: 10.1006/jmbi.1999.2642. [DOI] [PubMed] [Google Scholar]
- 3.Minor DL, Jr, Kim PS. Context-dependent secondary structure formation of a designed protein sequence. Nature. 1996;380:730–734. doi: 10.1038/380730a0. [DOI] [PubMed] [Google Scholar]
- 4.Coles M, Diercks T, Liermann J, Groger A, Rockel B, Baumeister W, Koretke KK, Lupas A, Peters J, Kessler H. The solution structure of VAT-N reveals a ‘missing link’ in the evolution of complex enzymes from a simple betaalphabetabeta element. Curr Biol. 1999;9:1158–1168. doi: 10.1016/S0960-9822(00)80017-2. [DOI] [PubMed] [Google Scholar]
- 5.Coles M, Djuranovic S, Soding J, Frickey T, Koretke K, Truffault V, Martin J, Lupas AN. AbrB-like transcription factors assume a swapped hairpin fold that is evolutionarily related to double-psi beta barrels. Structure. 2005;13:919–928. doi: 10.1016/j.str.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 6.Coles M, Hulko M, Djuranovic S, Truffault V, Koretke K, Martin J, Lupas AN. Common evolutionary origin of swapped-hairpin and double-psi beta barrels. Structure. 2006;14:1489–1498. doi: 10.1016/j.str.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 7.Ammelburg M, Hartmann MD, Djuranovic S, Alva V, Koretke KK, Martin J, Sauer G, Truffault V, Zeth K, Lupas AN, Coles M. A CTP-dependent archaeal riboflavin kinase forms a bridge in the evolution of cradle-loop barrels. Structure. 2007;15:1577–1590. doi: 10.1016/j.str.2007.09.027. [DOI] [PubMed] [Google Scholar]
- 8.Alva V, Koretke KK, Coles M, Lupas AN. Cradle-loop barrels and the concept of metafolds in protein classification by natural descent. Curr Opin Struct Biol. 2008;18:358–365. doi: 10.1016/j.sbi.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 9.Bystroff C, Baker D. Prediction of local structure in proteins using a library of sequence-structure motifs. J Mol Biol. 1998;281:565–577. doi: 10.1006/jmbi.1998.1943. [DOI] [PubMed] [Google Scholar]
- 10.Hutchinson EG, Thornton JM. PROMOTIF—a program to identify and analyze structural motifs in proteins. Protein Sci. 1996;5:212–220. doi: 10.1002/pro.5560050204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hutchinson EG, Thornton JM. A revised set of potentials for beta-turn formation in proteins. Protein Sci. 1994;3:2207–2216. doi: 10.1002/pro.5560031206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lupas AN, Ponting CP, Russell RB. On the evolution of protein folds: are similar motifs in different protein folds the result of convergence, insertion, or relics of an ancient peptide world? J Struct Biol. 2001;134:191–203. doi: 10.1006/jsbi.2001.4393. [DOI] [PubMed] [Google Scholar]
- 13.Kryshtafovych A, Venclovas C, Fidelis K, Moult J. Progress over the first decade of CASP experiments. Proteins. 2005;61(Suppl 7):225–236. doi: 10.1002/prot.20740. [DOI] [PubMed] [Google Scholar]
- 14.Lupas AN. The long coming of computational structural biology. J Struct Biol. 2008;163:254–257. doi: 10.1016/j.jsb.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 15.Simons KT, Bonneau R, Ruczinski I, Baker D. Ab initio protein structure prediction of CASP III targets using ROSETTA. Proteins. 1999;37(Suppl 3):171–176. doi: 10.1002/(sici)1097-0134(1999)37:3+<171::aid-prot21>3.3.co;2-q. [DOI] [PubMed] [Google Scholar]
- 16.Nilges M. Calculation of protein structures with ambiguous distance restraints. Automated assignment of ambiguous NOE crosspeaks and disulphide connectivities. J Mol Biol. 1995;245:645–660. doi: 10.1006/jmbi.1994.0053. [DOI] [PubMed] [Google Scholar]
- 17.Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 18.Lo Conte L, Ailey B, Hubbard TJ, Brenner SE, Murzin AG, Chothia C. SCOP: a structural classification of proteins database. Nucleic Acids Res. 2000;28:257–259. doi: 10.1093/nar/28.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Challis JH. A procedure for determining rigid body transformation parameters. J Biomech. 1995;28:733–737. doi: 10.1016/0021-9290(94)00116-l. [DOI] [PubMed] [Google Scholar]
- 20.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J Mol Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 21.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 22.Frickey T, Lupas A. CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics. 2004;20:3702–3704. doi: 10.1093/bioinformatics/bth444. [DOI] [PubMed] [Google Scholar]