

Abstract
Biophysical studies on amyloidogenic and aggregation-prone peptides often require large quantities of material. However, solid-phase synthesis, handling, and purification of peptides often present challenges on these scales. Recombinant expression is an attractive alternative because of its low cost, the ability to isotopically label the peptides, and access to sequences exceeding ∼50 residues. However, expression systems that seek to solubilize amyloidogenic peptides suffer from low yields, difficult optimizations, and isolation challenges. We present a general strategy for expressing and isolating amyloidogenic peptides in Escherichia coli by fusion to a polypeptide that drives the expression of attached peptides into bacterial inclusion bodies. This scheme minimizes toxicity during bacterial growth and enables the processing and handling of the peptides in denaturing solutions. Immobilized metal affinity chromatography, reverse phase HPLC, and cyanogen bromide cleavage are used to isolate the peptide, followed by further reverse phase HPLC, which yields milligram quantities of the purified peptide. We demonstrate that driving the peptides into inclusion bodies using fusion to BCL-XL-1/2 is a general strategy for their expression and isolation, as exemplified by the production of 11 peptides species.
Keywords: amyloid, peptide expression, BCL-XL, inclusion body
Introduction
Amyloidogenic peptides are the object of intense study because their misassembly into cross-beta sheet, or amyloid, structures is associated with a spectrum of aging-associated diseases, including Alzheimer's disease and type II diabetes.1,2 Understanding the mechanisms of amyloid assembly should provide insight into the etiology of these diseases and possibly enable interventional strategies. Amyloidogenic peptides are challenging to produce, purify, and handle3; and too often the scope of their study is limited by the quantities of the peptide that can be generated.
To date, all full-length amyloidogenic-peptide-expressing systems in E.coli have either expressed the peptides directly or expressed them by fusion to a solubilizing protein or protein domain. Direct expression is unreliable for short peptides. The solubilizing fusion partners used for expression of amyloidogenic peptides include maltose binding protein,4,5 glutathione S-transferase,6,7 thioredoxin,8,9 and poly(NANP).10 Because amyloidogenic peptides are naturally aggregation-prone, we designed a system where the peptide is attached to a fusion partner that directs the polypeptide into inclusion bodies in E.coli. This strategy takes advantage of the natural bacterial response for dealing with aggregating polypeptides, and relieves the cell of the potential toxicity conferred by soluble aggregates of these proteins.11,12 Indeed, it has recently been observed that bacterial inclusion bodies share biophysical properties with amyloid aggregates.13,14 Thus, inclusion-body-directing tags are a more natural choice for bacterial expression of amyloidogenic peptides than solubilizing tags. An amyloidogenic peptide fusion to ketosteroid isomerase (KSI), a sequence that prefers to form inclusion bodies, has been used for the expression of a short fragment of an amyloidogenic peptide, Aβ 11-2615; however, it is unclear whether this approach would efficiently afford inclusion bodies when fused to longer full-length amyloidogenic peptides. Fusion of KSI to the C-terminus of peptides of interest afford the unnatural C-terminal homoserine lactone after CNBr cleavage,16 which may be acceptable for model systems, but may not be acceptable for studies on native amyloidogenic peptides.
High-level expression of proteins as inclusion bodies has generally been eschewed in the structural biology community because of the difficulties associated with recovering native structure after resolubilization in chaotrope.17 Unlike many proteins that adopt well-defined structures, aggregation-prone amyloidogenic peptides are often weakly structured or intrinsically unstructured in solution.18,19 As preservation of structure is unnecessary, recovery of these peptides in a denatured state is preferable to prevent handling difficulties associated with aggregation.
Fusions to truncated BCL-XL proteins were previously used as a strategy for directing the expression of membrane-bound proteins to inclusion bodies. These inclusion bodies were reconstituted into SDS micelles and transferred to model bilayer-forming detergents for structural studies.20 These BCL-XL fusions yielded higher quantities of peptide relative to the typically used fusion partners, KSI and trpΔLE. We surmised that this strategy could be adapted for the expression of aggregation-prone amyloidogenic peptides, although we opted to use a different host plasmid, one that is arabinose inducible to gain enhanced control over the expression of these cytotoxic peptides. To our further advantage, BCL-XL was established as compatible with cyanogen bromide treatment; the cleavage method we chose given its low cost, its generation of a N-terminus with minimal restriction on the identity of the first amino acid, and the availability of this method to denaturing conditions.
Results
Vector design
In the work of Thai et al.,20 two constructs were tested: a 173-amino acid BCL-XL segment generated by truncating the C-terminus, and a 96-amino acid segment generated by truncating 77 additional amino acids from the N-terminus. For our purpose, we chose to use the shorter, 96-amino acid peptide as the basis for our fusion partner, which we refer to as BCL-XL-1/2. This choice was made because more of the mass of the inclusion is the amyloidogenic peptide of interest and because the shorter BCL-XL-1/2-amyloidogenic peptide fusions are more amenable to reverse phase HPLC purification.
The Thai et al.20 vector was altered in three important ways. First, our vector, distinct from the IPTG-inducible pET vector used by Thai et al., uses an arabinose-inducible promoter derived from the araBAD operon utilized in the Invitrogen pBAD vector, upon which the pBCA vector (see Fig. 1) used herein is based.21 This change was made to improve the control of amyloidogenic peptide expression to prevent amyloid-associated bacteriotoxicity before induction. Second, a downstream box (DSB, Fig. 1) was added N-terminal to the His7 purification tag to enhance ribosome binding,22 and a cationic spacer ([(K/R)(P/G)]5, Fig. 1) was inserted between the His7 purification tag and the BCL-XL-1/2 sequence to improve the yield and to aid in purification, respectively. Third, the Thai BCL-XL-1/2 sequence has been mutagenized for E. coli codon optimization and was modified by mutating an internal cysteine to serine (C35S) to prevent complications resulting from the potential generation of intermolecular disulfide bonds. A schematic of the pBCA plasmid and BCL-XL-1/2 construct is shown in Figure 1.
Figure 1.
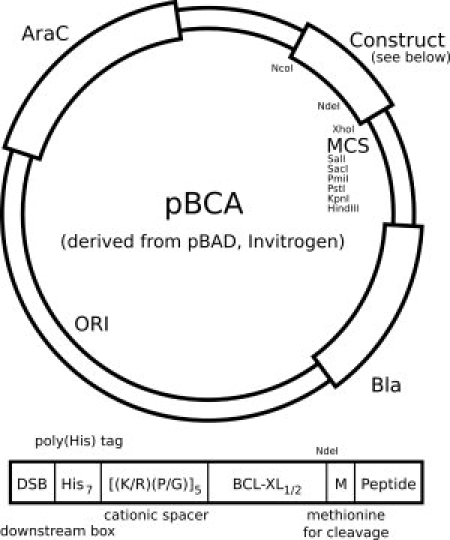
A schematic of the pBCA plasmid: the pBCA plasmid is derived from the pBAD plasmid (Invitrogen) and is modified to express the inclusion-body construct as indicated. A reconfigured multiple cloning site (MCS) has been engineered and a distal Nde1 site has been removed, affording a unique Nde1 site within the construct. The inclusion body construct includes a downstream box (DSB) for enhanced translation, a poly(His) tag for metal affinity chromatography, a cationic linker to improve purification, and the BCL-XL-1/2 sequence. This is followed by a methionine residue for cyanogen bromide cleavage provided by an in-frame Nde1 restriction site (CATATG).
Using standard molecular biology approaches (see Materials and Methods), we prepared plasmids for the expression of 11 peptides from four nonhomologous peptide families to demonstrate the versatility of the BCL-XL-1/2 expression system for the production of amyloidogenic peptides (Table I).23 These include peptides from the amylin family,18,21 the insulin family,24,25 the gelsolin amyloid fragment (AGel) family,26,27 and two peptides identified by limited proteolysis as the core of alpha synuclein fibrils.28
Table I.
A List of the Peptides Generated in the Course of this Study
| Peptide | Sequence | mw (kDa) |
|---|---|---|
| Amylins18,23 | ||
| Amylin (carboxylate) | KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY | 3904.3 |
| Amylin free acid | KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTYG | 3961.4 |
| Amylin + KR | KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTYGKR | 4245.7 |
| Amylin + CT | KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTYGKRNAVEVLKREPLNYPLY | 6145.9 |
| Proamylin | KATPIESHQVEKRKCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTYGKRNAVE VLKREPLNYPLY (see note added in Proof) | 7650.6 |
| Gelsolins26,27 | ||
| AGel 5 kDa | ATEVPVSWESFNNGNCFILDLGNNIHQWCGSNSNRYERLKATQVSKGIRDNER | 6067.6 |
| AGel 8 kDa | ATEVPVSWESFNNGNCFILDLGNNIHQWCGSNSNRYERLKATQVSKGIRDNER SGRARVHVSEEGTEPEA | 7860.5 |
| Insulins24,25 | ||
| Proinsulin | FVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGGGPGAGSL QPLALEGSLQKRGIVEQCCTSICSLYQLENYCN | 9388.7 |
| Proinsulin akita | FVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGGGPGAGSL QPLALEGSLQKRGIVEQCYTSICSLYQLENYCN | 9540.7 |
| α-Synucleins28 | ||
| α-Synuclein major core | GKTKEGVLYVGSKTKEGVVHGVATVAEKTKEQVTNVGGAVVTGVTAVAQKTV EGAGSIAAATGFVKKDQLGKN | 7256.3 |
| α-Synuclein minor core | GKTKEGVLYVGSKTKEGVVHGVATVAEKTKEQVTNVGGAVVTGVTAVAQKTV EGAGSIAAATGFVKKDQLGKNEEGAPQ | 7867.9 |
Expression and characterization
Generalized BCL-XL-1/2-peptide expression conditions are based upon the optimized conditions for the expression of the amylin free acid construct. Bacteria harboring the pBCA plasmid are grown to very high densities (OD600 ∼1.4). Expression was induced using arabinose, and the incubation temperature was increased to 40°C to promote inclusion body formation. The bacteria were harvested after a relatively short time (2 h). The araBAD-operon-containing BL21 strain performed marginally better than the recommended Top10 (modified DH10alpha) strain, possibly because of the extra copy of the araC gene for added expression control in spite of the innate metabolic consumption of arabinose found in this strain (data not shown). The BL21 strain was also preferable because of its faster generation time, enabling cultures to grow to desired high densities in a convenient timeframe.
Bacteria expressing the BCL-XL-1/2-amylin free acid fusion exhibited classical inclusion body morphology as revealed by electron microscopy imaging. Uninduced bacteria occasionally contained inclusion bodies, likely because of natural processes within bacteria, but these inclusion bodies did not stain with the polyhistidine antibody [Fig. 2(A)]. Inclusion bodies found in induced bacteria stained with secondary immunoconjugated gold particles bound to a monoclonal anti-polyhistidine primary antibody [Fig. 2(B)].
Figure 2.
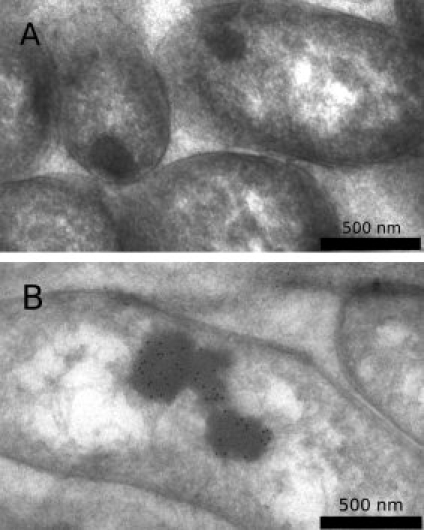
(A) Uninduced bacteria harboring a pBCA plasmid with an amylin free acid insert generate inclusion bodies, but they are not very prevalent and fail to label when stained using an anti-polyhistidine primary antibody and an immunogold-conjugated secondary antibody. (B) Induced bacteria generally generate inclusion bodies, which exhibit immunogold staining using the same protocol.
Processing of inclusion bodies
After harvesting and overnight freezing, bacterial cells were lysed using sonication and the insoluble components were subjected to repeated cycles of washing by sonication in fresh buffers and pelleting by centrifugation. SDS-PAGE monitoring of this process indicated increasing enrichment of the desired peptide away from endogenous E. coli proteins [Fig. 3(A)]. As designed, throughout the wash steps the insoluble pellets contained the expressed peptide fusion, evidenced by the presence of reactivity to an anti-polyhistidine antibody [Fig. 3(B)] only in the pellet samples.
Figure 3.
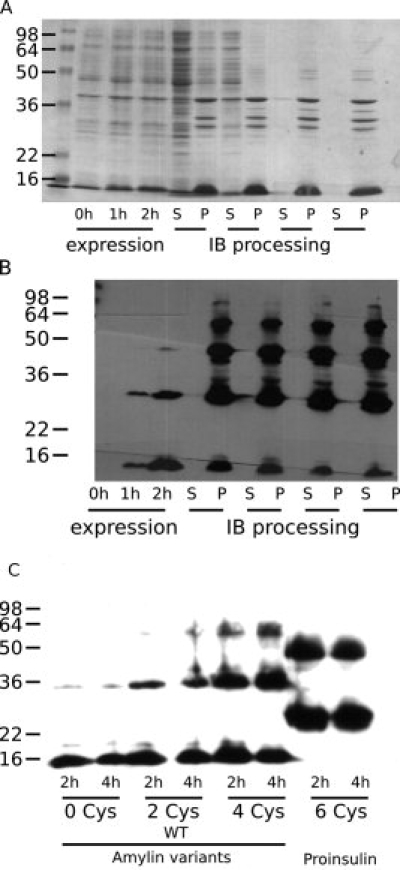
A typical bacterial expression run is exemplified by the amylin free acid fusion construct. (A) Coomassie-stained gel of whole cell lysates 0, 1, and 2 h postinduction illustrates the increasing titer of the engineered peptide; samples of supernatant (S) and pellet (P) after successive rounds of centrifugation illustrate retention of the engineered peptide in the pellets and increasing levels of purity. (B) Anti-polyhistidine Western blot of the same gel confirms the identity of the engineered peptide and illustrates the presence of oligomeric species. Comparison of the Coomassie gel and the Western blot suggests that the oligomers have a higher affinity for the primary antibody, presumably because of polyvalency. (C) The presence of increasing numbers of cysteine residues contributes to an increased population of the oligomeric states; however, as is illustrated by the differences between the amylin constructs and the proinsulin construct, peptide identity also contributes significantly to the oligomer distribution.
Complications in inclusion body processing
The induced BCL-XL-1/2 fusion polypeptides migrated upon SDS-PAGE as SDS denaturation-resistant oligomeric structures. These oligomers appear shortly after induction in E. coli and persist and intensify over the course of inclusion body washing [Fig. 3(B)]. To determine if intermolecular disulfide bonding could be responsible for this phenomenon, an amylin mutant with no cysteines and an amylin mutant with an increased cysteine count were generated and inserted in the fusion polypeptide construct, and the postinduction oligomer content was probed by Western blotting.
The amylin analog containing no cysteines yielded oligomeric species, albeit in very low quantities; increasing the cysteine count resulted in progressively increased dimer and higher order oligomer content, suggesting that disulfide oxidation can trap oligomers as covalently bonded species [Fig. 3(C)]. It is clear that oligomerization does not correlate solely with the presence of cysteines, as proinsulin, which contains three times as many cysteines as amylin, exhibits a prominent dimer band, but higher order oligomer accumulation is less pronounced than that of the amylin-derived species containing fewer cysteine residues [Fig. 3(C)]. Thus, both peptide sequence and cysteine count determine oligomerization propensity. The increase in oligomerization with cysteine count underscores the need to keep cysteine-containing peptides reduced when using this expression system and vindicates the choice to mutate the wild-type BCL-XL cysteine residue to serine in the BCL-XL-1/2 sequence.
The BCL-XL-1/2-alpha synuclein minor core fragment fusion exhibited noteworthy solubility and required a modification of the standard process described earlier for effective inclusion body production. Without modification, some of the fusion peptide was sequestered as inclusion bodies, but most of the product persisted in the soluble supernatant after centrifugation [Fig. 4(A)]. Inclusion body sequestration could be enhanced by increasing the time or temperature of induction, or increasing both [Fig. 4(B)]. The alpha synuclein fusion polypeptide could be nearly completely forced into the insoluble fraction by shifting the postinduction temperature to 42°C instead of 40°C and harvesting the bacteria 8 h postinduction, instead of 2 h.
Figure 4.
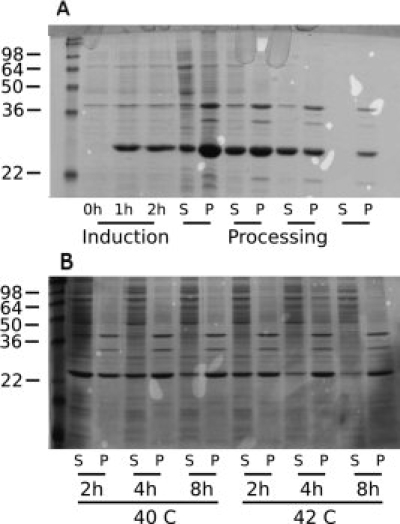
Expression of alpha synuclein (minor core fragment) exhibited altered inclusion body dynamics. (A) A Coomassie-stained gel of the expression and processing steps shows large quantities of the engineered peptide in the soluble fraction post-lysis. Persistence of the peptide in the second and third soluble fractions is likely due to the presence of unlysed bacteria or pellet disassembly. (B) A Coomassie-stained gel of a reoptimization of the expression conditions varying on time and induction temperature. Despite its propensity to resolubilize the BCL-XL-1/2 construct, the minor core fragment can be driven into inclusion bodies by elevating the expression temperature and harvesting after extended periods of time.
Under the standard conditions described earlier, the BCL-XL-1/2-alpha synuclein major core fragment fusion peptide also exhibited some solubility. However, the relative ratio of soluble versus insoluble for this species was not as dramatic (see Supporting Information). This sequence differs from the minor core fragment sequence only by the lack of the C-terminal six-residue sequence EEGAPQ. Hence, the usually predictable inclusion body formation can be disrupted by subtle sequence-specific effects; although it seems that the factors preventing sequestration into inclusion bodies can be overcome by altering the environmental conditions, in particular the temperature.
Postprocessing purification
Once isolated from most of the soluble E. coli components, the inclusion bodies are subjected to a 5- to 7-day resolubilization procedure using highly denaturing and reducing conditions: 7.2M guanidine-HCl, tris(carboxyethyl) phosphine. The peptides are then subjected to ultracentrifugation to clear the remaining insoluble material, including lipids, nucleic acids, and possibly protein aggregates. The remaining supernatant contains the fusion polypeptide and is subjected to immobilized metal affinity chromatography, which affords a highly purified sample. The eluted peptides appear to comprise monomeric BCL-XL-1/2 fusion peptides, persistent dimers, and occasionally, proteolyzed fragments. These are then subjected to RP-HPLC (C4) to further purify and remove the guanidine denaturant. The RP-HPLC solvents (acetonitrile or isopropanol) were removed by lyophilization, affording a dry sample of the fusion peptide.
The lyophilate was subjected to cyanogen bromide cleavage in 7.2M guanidine-HCl and separated from the fusion polypeptide by a second round of RP-HPLC, again using a C4 column. Additional purification could be achieved by subjecting the peptide to a final round of RP-HPLC using a C18 chromatography column.
Yields of peptides
Upon cleavage and repurification, all constructs yielded between 5 and 10 mg of peptide as measured by the dry mass of the lyophilate, including TFA counterion salts, which can be nonstoichiometric (see Table II). These values corresponded to a yield of ∼15–20% for the cyanolysis reaction. Although typical cyanolysis reactions can exhibit yields approaching 50%,29,30 our reaction exhibited lower yields. We ascribe this to the self-association tendencies of these peptides and their propensity to form oligomers and aggregates. Any persistence of such aggregates upon resolubilization of the lyophilate may create steric occlusion or accessibility problems near the critical methionine residue. Overnight treatment in denaturant was critical for the success of the reaction.
Table II.
Yields of Fusion Polypeptide and Cleaved Peptide from a 4 L of Shake-Flask Culture
| Family | Construct | Fusion protein yield (4 L) | Final yield (4 L) |
|---|---|---|---|
| Amylin | Amylin (carboxylate) | 130 mg | 9 mg |
| Amylin free acid | 250 mg | 10 mg | |
| Amylin + KR | 180 mg | 6 mg | |
| Amylin + CT | 170 mg | 7 mg | |
| Proamylin | 150 mg | 4 mg | |
| Gelsolin | 5 kDa aGel | 210 mg | 9 mg |
| 8 kDa aGel | 170 mg | 6 mg | |
| α-Synuclein | Major core | 220 mg | 12 mg |
| Minor core | 330 mg | 7 mg | |
| Insulin | Proinsulin | 206 mg | 10 mg |
| Akita proinsulin | 140 mg | 8 mg |
Discussion
The solid-phase synthesis of aggregation-prone amyloidogenic peptides, while capable of producing large quantities, is often complicated by on-resin aggregation. Furthermore, impurities found in this process are often truncations or deletions, which may be difficult to separate by HPLC. By contrast, expression gives access to longer polypeptides, enables isotopic labeling, and facilitates orthogonal purification using tags. Final HPLC steps on expressed peptides ultimately separate peptides from chemically distinct species, improving overall purity.
Although no one system is an ideal solution for the expression of all peptides, a careful choice of expression vectors can afford high yields of products. Not all peptides are well suited for our expression system—the Alzheimer's A-beta peptides (both 1-40 and 1-42), for example, contain an internal methionine which would be subject to fragmentation during the cyanogen bromide cleavage step. This internal methionine can affect the aggregation characteristics of the A-beta peptide when oxidized,31 suggesting that even a conservative mutation, such as mutation to leucine,10,32 could result in nontrivial effects. It may be possible to express methionine-bearing peptides by using N-chloro-succinimide to selectively cleave at the C-terminus of a tryptophan residue, but this was not attempted for our system.33 It may also be possible to engineer sites enabling enzymatic cleavages to be conducted in denaturing solutions by specialized, kinetically stable proteases.34
Native amylin contains a C-terminus that is amidated. The chemical transformation to convert the C-terminal carboxylic acid (amylin (carboxylate), Table I), resulting from our recombinant expression, to a primary amide was not explored in this study. However, we believe that it should be possible to achieve this using EDC/NHS activation of the amylin C-terminal carboxylate followed by treatment with aqueous ammonia, as amylin itself contains no carboxylate side chains. Williamson and Miranker35 have recombinantly made the nonamyloidogenic, rodent C-terminally amidated amylin sequence, taking advantage of ammonia treatment of an intein-derived thioester. Considering that the intein has to be folded in order to function, this strategy could be problematic for amyloidogenic peptide expression.
Considering these limitations, our strategy resulted in multimilligram quantities of amyloidogenic peptides from laboratory-scale bacterial expression using our constructs. Moreover, the strategy is general, applicable to the expression of 11 different peptides from four different families of amyloidogenic peptides, including epitope-tagged peptides, and truncations. Some of the constructs generated enabled the production of peptide variants for biophysical study.18 In one construct, a greater than fivefold improvement in expression yield was achieved over our previous efforts,26 notwithstanding improved purity conferred by our revised purification strategy. We envision that this platform will be useful for the expression of other amyloidogenic peptides, as well as for the expression of isotopically labeled samples, which are critical for modern solid-state NMR studies on amyloid structures.10,36,37 Overall, the versatility of this system demonstrates that using the internal system for handling misfolded peptides is a sound and general strategy for the expression of naturally aggregation-prone peptides in E. coli.
Materials and Methods
Preparation of plasmid vectors and inserts
pBCA was prepared by modification of the pBAD (Invitrogen) plasmid vector. The multiple cloning site of the pBAD vector was altered by insertion of a synthesized double-stranded oligonucleotide and ligation of the oligonucleotide to the plasmid digested with the XhoI and HindIII restriction enzymes. An NdeI restriction site on the opposite side of the plasmid was eliminated by the insertion of a sticky-end double-stranded oligonucleotide generated by annealing two complementary, single-stranded oligonucleotides. The modified BCL-XL-1/2 open reading frame was prepared by repeated cycles of assembly PCR38 to contain an optimized codon set. The following changes in peptide sequence were made from the original human BCL-XL gene (accession: Z23115): only residues 117–212 were used, M159 and M170 were mutated to leucines,20 and C151 was mutated to a serine.
Individual fusion constructs were generated by extension PCR to generate a 5′ NdeI and a 3′ double stop codon (TAGTAA) and XhoI restriction site. The PCR products and the pBCA plasmid were then subjected to digestion by the respective restriction enzymes, isolation of fragments using agarose gel electrophoresis, and ligation using T4 ligase (New England Biolabs). Some ligations failed, likely owing to primer design and overhang requirements of the NdeI enzyme. These required insertion of the PCR product into the pCR 2.1 plasmid using a commercial TA cloning kit (Invitrogen), followed by bacterial production of the shuttle plasmid and excision of the insert using NdeI and XhoI restriction enzymes, followed by insertion into a pBCA plasmid by ligation using T4 ligase as described earlier.
Peptide production and characterization (general)
Plasmids were transformed into chemically competent E. coli strain BL21 (DE3). A 6-mL starter culture containing 50 μg/mL carbenicillin was inoculated with a colony of the transformed bacteria; this starter culture was harvested by centrifugation (4000g, 10 min) after overnight growth. The resulting pellet was twice subjected to a wash step, where the culture was resuspended in fresh Luria-Bertani (LB) broth, and centrifuged on a benchtop centrifuge (14,000g, 1 min) to separate the bacteria from the supernatant broth.
This culture was reinoculated into a 100-mL second starter culture containing 50 μg/mL carbenicillin. After 2–3 h, this starter was harvested, washed twice as mentioned earlier, and reinoculated into 4 L of LB broth culture (in 1 L Fernbach flasks) containing 25 mM sodium phosphate (pH 7.4) and 25 μg/mL ampicillin. Bacterial growth was monitored by UV; typically the desired induction density (OD600 = 1.4 AU) was reached after 2 h of culture growth.
Peptide expression was induced by the addition of 20 mL of 20% (w/v) arabinose in water (5 mL per liter). Samples for analysis were taken before induction and after 1 and 2 h; after 2 h, the bacteria were harvested by centrifugation (20 min, 16,000g) and frozen overnight at −80°C.
Frozen cultures were thawed at room temperature for ∼10 min, resuspended in 50 mL lysis buffer (50 mM Tris, 100 mM NaCl, 0.1% (w/v) NaN3, 5 mM EDTA, 0.1% v/v Triton X-100, pH 8.4), and sonicated until a homogenous suspension was obtained. The suspension was centrifuged (40 min, 20,000g) to pellet the insoluble matter, and the pellet resuspended by sonication in a further 50 mL of lysis buffer including 10 ng of DNAse 1. The suspension was centrifuged to pellet the insoluble matter, and the pellet resuspended by sonication in 50 mL of wash buffer (50 mM Tris, 100 mM NaCl, 0.1% (w/v) NaN3 5 mM EDTA, pH 8.4) including 10 ng of DNAse 1. This pellet was subjected to two further rounds of resuspension and centrifugation in 50 mL of low pH buffer (50 mM Tris, 50 mM MES, 100 mM NaCl, 0.1% (w/v) NaN3, pH 5.0) each. The final pellet was resuspended in 50 mL of IMAC buffer A (50 mM Tris, ∼7.2M GdnHCl, pH 8.4), with the addition of 24 mg TCEP, and incubated at 4°C with rotary agitation (10 rpm) for 7 days.
The resuspended peptide pellet was centrifuged once at medium speed (38,000g, 10 h) and twice at high speed (100,000g, 2 h each) to remove insoluble matter and filtered using a 0.22-μm PVDF filtration membrane before loading using FPLC (Akta, GE Healthsciences) onto the TALON cobalt IMAC resin (Clontech) loaded in an HR-16 column housing (GE Healthsciences). The TALON resin was pre-equilibrated in IMAC buffer A, the flow rate was dropped to 0.1 mL/min, and the resolubilized inclusion bodies were passed over the resin to immobilize the peptide of interest. After loading the resolubilized inclusion bodies, the flow rate was increased to 0.5 mL/min and briefly (10 mL) washed in more IMAC buffer A. This wash was followed by an exchange into IMAC buffer B (50 mM Tris, 50 mM MES, ∼7.2M GdnHCl, pH 5.0), which effected elution.
Fractions containing the fusion polypeptide as evidenced by higher UV absorbance at 280 nm were pooled, a further 24 mg TCEP was added and the peptide was incubated overnight, and subjected to HPLC purification (C4, Vydac). HPLC purification was performed using a 30–60% gradient of 95% acetonitrile with 0.5% (v/v) TFA with the balance being water containing 0.5% (v/v) TFA. A mixture of 70% 2-propanol, 25% methanol, 5% deionized water with 0.5% (v/v) TFA was used when acetonitrile was unavailable. Fractions containing the fusion peptide were pooled and lyophilized.
Lyophilized peptide was resuspended (10 mg/mL) in cleavage buffer (7.2M GdnHCl with one molar excess HCl: prepared by adding 50 mL of a 5M HCl stock solution to 171 g GdnHCl and the volume made up to 250 mL), sonicated for 30 min and incubated overnight to disaggregate the peptide, and then subjected to an equal volume of 5 mg/mL (∼100-fold molar excess) cyanogen bromide in the same buffer for cleavage over 36 h at 25°C. The peptide was then subjected to separation from the fusion peptide by HPLC purification using a gradient tailored to each peptide (C4, Vydac), and lyophilization.
Further purification was achieved by resuspending the peptide in 7.2M GdnHCl, 50 mM Tris, pH 8.0 with or without 12 mg TCEP, depending on the oxidation state desired, overnight incubation, followed by lowering the pH by dilution (1:1) in 7.2M GdnHCl, 50 mM MES, pH 4.0, and performing a third round of HPLC purification (C18, prep, Phenomenex; or C18, semi-prep, Vydac), again on gradients tailored to each peptide of interest.
Electron microscopy
Immunoelectron microscopy on ultrathin sections was performed as described with minor variations.39 Induced and noninduced bacterial cells were fixed in 4% paraformaldehyde, 0.1% glutaraldehyde in 0.1M phosphate buffer, embedded in gelatin, and cryoprotected in 2.3M sucrose before freezing. Thin cryosections (∼100 nm) were cut on a Leica FC6 freezing ultratome and mounted on nickel mesh grids. Each grid was processed on individual droplets of the following solutions on Parafilm at room temperature: 50 mM glycine, 10% fetal calf serum (FCS) in PBS, primary antibody (anti-polyhistidine antibody produced in mouse: clone HIS-1: Sigma) diluted 1:50 in 5% FCS (control grids were prepared from noninduced cells). Subsequently, the grids were washed in 0.2% FCS, incubated in goat anti-mouse tagged with 12 nm gold (Jackson ImmunoResearch, West Grove, PA) diluted 1:20 in 5% FCS in PBS. Grids were then washed in PBS alone, fixed in 1% glutaraldehyde in PBS, washed in double deionized H2O, and contrasted in uranyl oxalate (pH 7). Each individual grid was then picked up in ice-cold uranyl acetate/methyl cellulose (pH 4) mixture using copper wire loops. Once dry, the grids were examined on a Philips CM100 TEM (FEI, Hillsbrough, OR) at 80 kV. Images were documented using a Megaview III CCD camera (Soft Imaging Systems, Lakewood, CO).
PAGE analysis of peptide expression
Whole cell-containing media (undiluted) and supernatants (1:10 dilution in wash buffer) and pellets (1:10 dilution in wash buffer) from the first four stages of inclusion body preparation were subject to SDS-PAGE in the tris-glycine system with 15% polyacrylamide (w/v); SeeBlue 2 (Invitrogen; Carlsbad, CA) ladder was used to estimate molecular weights. Aliquots of the IMAC process were diluted (1:9) in 10 mM Tris, 8M urea, 0.1% (w/v) SDS before loading onto the gel; SeeBlue 2 ladder was diluted (1:1) in the same buffer. For the IMAC process gels, two extra lanes on either side of the sample lanes were loaded with 10 mM Tris, 8M urea, 0.1% (w/v) SDS. 6× Laemmeli sample buffer (375 mM Tris, 9% (w/v) SDS, 50% (v/v) glycerol, 0.03% (w/v) bromophenol blue, pH 6.8) was added to all samples for all gels and the samples were boiled ∼5 min before loading.
After electrophoresis, gels were treated (min. 5 h incubation after heating to boiling with microwave irradiation) with Coomassie Brilliant Blue G-250 (1% w/v in 50% acetic acid, 20% methanol), and destained overnight in 50% acetic acid, 20% methanol to visualize total peptide content. Alternatively, the separated proteins were transferred onto a nitrocellulose membrane and subjected to Western blot using a mouse monoclonal anti-polyhistidine antibody (Sigma) (1:5000 dilution in 20 mM Tris, 100 mM NaCl, 0.1% (v/v) Tween, 4% (w/v) condensed milk, pH 7.6) followed by a goat anti-mouse-HRP secondary antibody (1:5000 dilution in the same buffer) to visualize specific peptide content.
Note added in Proof:
The underlined KA in the proamylin sequence was originally annotated by PubMed as part of the proamylin sequence, but is now annotated as part of the signal peptide. The sequence, as given, is what was expressed, however the underlined KA should not have been included.
Acknowledgments
The authors thank Theresa Fassel for her assistance in the preparation of electron micrographs and Jeremy Mills for useful advice in the optimization of bacterial expression.
References
- 1.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 2.Carrell RW, Gooptu B. Conformational changes and disease—serpins, prions and Alzheimer's. Curr Opin Struct Biol. 1998;8:799–809. doi: 10.1016/s0959-440x(98)80101-2. [DOI] [PubMed] [Google Scholar]
- 3.Abedini A, Singh G, Raleigh DP. Recovery and purification of highly aggregation-prone disulfide-containing peptides: application to islet amyloid polypeptide. Anal Biochem. 2006;351:181–186. doi: 10.1016/j.ab.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 4.Hortschansky P, Schroeckh V, Christopeit T, Zandomeneghi G, Fändrich M. The aggregation kinetics of Alzheimer's beta-amyloid peptide is controlled by stochastic nucleation. Protein Sci. 2005;14:1753–1759. doi: 10.1110/ps.041266605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazor Y, Gilead S, Benhar I, Gazit E. Identification and characterization of a novel molecular-recognition and self-assembly domain within the islet amyloid polypeptide. J Mol Biol. 2002;322:1013–1024. doi: 10.1016/s0022-2836(02)00887-2. [DOI] [PubMed] [Google Scholar]
- 6.Paulsson JF, Schultz SW, Köhler M, Leibiger I, Berggren P, Westermark GT. Real-time monitoring of apoptosis by caspase-3-like protease induced FRET reduction triggered by amyloid aggregation. Exp Diabetes Res. 2008;2008:865850. doi: 10.1155/2008/865850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ratnaswamy G, Koepf E, Bekele H, Yin H, Kelly JW. The amyloidogenicity of gelsolin is controlled by proteolysis and pH. Chem Biol. 1999;6:293–304. doi: 10.1016/S1074-5521(99)80075-1. [DOI] [PubMed] [Google Scholar]
- 8.Krampert M, Bernhagen J, Schmucker J, Horn A, Schmauder A, Brunner H, Voelter W, Kapurniotu A. Amyloidogenicity of recombinant human pro-islet amyloid polypeptide (ProIAPP) Chem Biol. 2000;7:855–871. doi: 10.1016/s1074-5521(00)00034-x. [DOI] [PubMed] [Google Scholar]
- 9.Lopes DHJ, Colin C, Degaki TL, de Sousa ACV, Vieira MNN, Sebollela A, Martinez AMB, Bloch CJ, Ferreira ST, Sogayar MC. Amyloidogenicity and cytotoxicity of recombinant mature human islet amyloid polypeptide (rhIAPP) J Biol Chem. 2004;279:42803–42810. doi: 10.1074/jbc.M406108200. [DOI] [PubMed] [Google Scholar]
- 10.Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Döbeli H, Schubert D, Riek R. 3D structure of Alzheimer's amyloid-beta(1–42) fibrils. Proc Natl Acad Sci USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haataja L, Gurlo T, Huang CJ, Butler PC. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev. 2008;29:303–316. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, Finch CE, Krafft GA, Klein WL. Self-assembly of Abeta(1–42) into globular neurotoxins. Biochemistry. 2003;42:12749–12760. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- 13.Carrió M, González-Montalbán N, Vera A, Villaverde A, Ventura S. Amyloid-like properties of bacterial inclusion bodies. J Mol Biol. 2005;347:1025–1037. doi: 10.1016/j.jmb.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 14.Wang L, Maji SK, Sawaya MR, Eisenberg D, Riek R. Bacterial inclusion bodies contain amyloid-like structure. PLoS Biol. 2008;6:e195. doi: 10.1371/journal.pbio.0060195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharpe S, Yau W-M, Tycko R. Expression and purification of a recombinant peptide from the Alzheimer's β-amyloid protein for solid-state NMR. Protein Expr Purif. 2005;42:200–210. doi: 10.1016/j.pep.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 16.Kuliopulos A, Walsh CT. Production, purification, and cleavage of tandem repeats of recombinant peptides. J Am Chem Soc. 1994;116:4599–4607. [Google Scholar]
- 17.Sørensen HP, Mortensen KK. Advanced genetic strategies for recombinant protein expression in Escherichia coli. J Biotechnol. 2005;115:113–128. doi: 10.1016/j.jbiotec.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 18.Yonemoto IT, Kroon GJA, Dyson HJ, Balch WE, Kelly JW. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry. 2008;47:9900–9910. doi: 10.1021/bi800828u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chandra S, Chen X, Rizo J, Jahn R, Südhof TC. A broken alpha-helix in folded alpha-synuclein. J Biol Chem. 2003;278:15313–15318. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
- 20.Thai K, Choi J, Franzin CM, Marassi FM. Bcl-XL as a fusion protein for the high-level expression of membrane-associated proteins. Protein Sci. 2005;14:948–955. doi: 10.1110/ps.041244305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rush GJ, Steyn LM. Translation enhancement by optimized downstream box sequences in Escherichia coli and Mycobacterium smegmatis. Biotechnol Lett. 2005;27:173–179. doi: 10.1007/s10529-004-7664-0. [DOI] [PubMed] [Google Scholar]
- 23.Paulsson JF, Andersson A, Westermark P, Westermark GT. Intracellular amyloid-like deposits contain unprocessed pro-islet amyloid polypeptide (proIAPP) in beta cells of transgenic mice overexpressing the gene for human IAPP and transplanted human islets. Diabetologia. 2006;49:1237–1246. doi: 10.1007/s00125-006-0206-7. [DOI] [PubMed] [Google Scholar]
- 24.Huang K, Dong J, Phillips NB, Carey PR, Weiss MA. Proinsulin is refractory to protein fibrillation: topological protection of a precursor protein from cross-beta assembly. J Biol Chem. 2005;280:42345–42355. doi: 10.1074/jbc.M507110200. [DOI] [PubMed] [Google Scholar]
- 25.Ron D. Proteotoxicity in the endoplasmic reticulum: lessons from the Akita diabetic mouse. J Clin Invest. 2002;109:443–445. doi: 10.1172/JCI15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huff ME, Balch WE, Kelly JW. Pathological and functional amyloid formation orchestrated by the secretory pathway. Curr Opin Struct Biol. 2003;13:674–682. doi: 10.1016/j.sbi.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 27.Maury CP. Gelsolin-related amyloidosis. Identification of the amyloid protein in Finnish hereditary amyloidosis as a fragment of variant gelsolin. J Clin Invest. 1991;87:1195–1199. doi: 10.1172/JCI115118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miake H, Mizusawa H, Iwatsubo T, Hasegawa M. Biochemical characterization of the core structure of alpha-synuclein filaments. J Biol Chem. 2002;277:19213–19219. doi: 10.1074/jbc.M110551200. [DOI] [PubMed] [Google Scholar]
- 29.Schmoldt H, Wentzel A, Becker S, Kolmar H. A fusion protein system for the recombinant production of short disulfide bond rich cystine knot peptides using barnase as a purification handle. Protein Expr Purif. 2005;39:82–89. doi: 10.1016/j.pep.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 30.Assadi-Porter FM, Patry S, Markley JL. Efficient and rapid protein expression and purification of small high disulfide containing sweet protein brazzein in E. coli. Protein Expr Purif. 2008;58:263–268. doi: 10.1016/j.pep.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hou L, Shao H, Zhang Y, Li H, Menon NK, Neuhaus EB, Brewer JM, Byeon IL, Ray DG, Vitek MP, Iwashita T, Makula RA, Przybyla AB, Zagorski MG. Solution NMR studies of the A beta(1–40) and A beta(1–42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J Am Chem Soc. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 32.Döbeli H, Draeger N, Huber G, Jakob P, Schmidt D, Seilheimer B, Stüber D, Wipf B, Zulauf M. A biotechnological method provides access to aggregation competent monomeric Alzheimer's 1–42 residue amyloid peptide. Biotechnology (NY) 1995;13:988–993. doi: 10.1038/nbt0995-988. [DOI] [PubMed] [Google Scholar]
- 33.Lischwe MA, Sung MT. Use of N-chlorosuccinimide/urea for the selective cleavage of tryptophanyl peptide bonds in proteins. Cytochrome c. J Biol Chem. 1977;252:4976–4980. [PubMed] [Google Scholar]
- 34.Jaswal SS, Sohl JL, Davis JH, Agard DA. Energetic landscape of alpha-lytic protease optimizes longevity through kinetic stability. Nature. 2002;415:343–346. doi: 10.1038/415343a. [DOI] [PubMed] [Google Scholar]
- 35.Williamson JA, Miranker AD. Direct detection of transient alpha-helical states in islet amyloid polypeptide. Protein Sci. 2007;16:110–117. doi: 10.1110/ps.062486907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luca S, Yau W, Leapman R, Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siemer AB, Ritter C, Steinmetz MO, Ernst M, Riek R, Meier BH. 13C, 15N resonance assignment of parts of the HET-s prion protein in its amyloid form. J Biomol NMR. 2006;34:75–87. doi: 10.1007/s10858-005-5582-7. [DOI] [PubMed] [Google Scholar]
- 38.Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene. 1995;164:49–53. doi: 10.1016/0378-1119(95)00511-4. [DOI] [PubMed] [Google Scholar]
- 39.Raposo G, Kleijmeer M, Postuma G, Slot J. Immunogold labeling of ultrathin cryosections: application in immunology. In: Herzenberg LA, Weir D, Blackwell C, Geuze H, editors. 5th Ed. Vol. 4. Cambridge, MA: Blackwell Science; 1996. pp. 1–11. Weir's handbook of experimental immunology. [Google Scholar]