

Abstract
It is difficult to properly validate algorithms that dock a small molecule ligand into its protein receptor using data from the public domain: the predictions are not blind because the correct binding mode is already known, and public test cases may not be representative of compounds of interest such as drug leads. Here, we use private data from a real drug discovery program to carry out a blind evaluation of the RosettaLigand docking methodology and find that its performance is on average comparable with that of the best commercially available current small molecule docking programs. The strength of RosettaLigand is the use of the Rosetta sampling methodology to simultaneously optimize protein sidechain, protein backbone and ligand degrees of freedom; the extensive benchmark test described here identifies shortcomings in other aspects of the protocol and suggests clear routes to improving the method.
Keywords: ligand docking, structure-based drug discovery, protein flexibility
Introduction
Almost all drugs used to treat human disease are small molecules, many of which bind to and modulate the activity of one or more enzymes or other proteins. Knowledge of the atomic structure of relevant protein—small molecule complexes greatly facilitates the rational design of new compounds and the improvement of potency in existing series of drugs. To this end, many pharmaceutical companies have created structure-based drug discovery programs. A central tool in these efforts is docking software, which predicts the binding mode of a protein—small molecule pair.
Starting from the work of Kuntz et al.,1 docking software has become more powerful, sophisticated, and successful. There are dozens of docking programs now available, which are compared and contrasted in recent reviews.2,3 One of the major remaining challenges for the field is to accurately model flexibility in the protein receptors.4 The recently developed RosettaLigand method offers one of the most extensive treatments of receptor flexibility to date.5,6
RosettaLigand uses the energy function and algorithms for stochastically exploring protein and small molecule conformations from the Rosetta molecular modeling software suite.7 The energy function consists of van der Waals, hydrogen bond, and implicit solvation terms, in addition to empirically derived torsional potentials. The docking algorithm combines stochastic Monte Carlo moves with gradient-based minimization. Major ligand conformations are pre-enumerated but are subjected to torsion space minimization during the simulation; rotamers for all receptor sidechains in the binding site are optimized simultaneously using a simulated annealing procedure; and the receptor backbone is allowed to minimize subject to restraints. The method and recent improvements have been described in recent publications.5,6
Many of the docking cases used to validate RosettaLigand were drawn from the Protein Data Bank,8 a public repository. However, as shown in Figure 1(A,B), public compounds are not necessarily representative of typical drug leads at private companies. The drug leads tend to be both larger and more flexible, making them more difficult to dock correctly. To create robust, useful docking programs, it is important to test against these “real world” cases.
Figure 1.
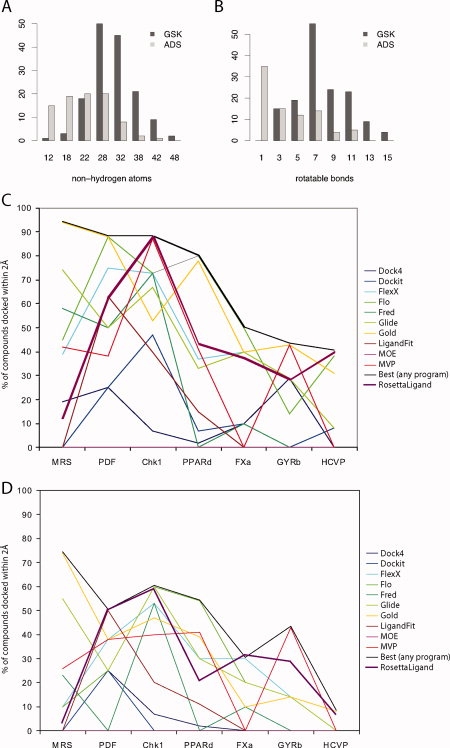
Comparison of RosettaLigand docking results. A: Comparison of the GlaxoSmithKline (GSK) compound collection used here to the Astex Diverse Set (ADS),11 a commonly used public-domain docking benchmark. GSK compounds tend to be larger than ADS compounds. B: GSK compounds tend to have more rotatable bonds than ADS compounds. C: Comparison of RosettaLigand docking to the programs assessed in Warren et al.9 Twenty pose predictions were evaluated from each program for each receptor/compound pair, and the percentage of compounds docked within 2 Å rmsd is recorded. RosettaLigand is shown in bold plum (MiniRosetta). D: The same comparison, but considering only one pose prediction per program/receptor/compound.
GlaxoSmithKline (GSK) has carefully assembled an extensive set of such real-world test cases, taken from their in-house drug discovery program. In 2005, GSK used this set to rigorously evaluate 10 widely used docking programs (often in collaboration with the program's creators) on their ability to generate and identify near-native ligand poses.9 From the perspective of algorithm developers, a valuable feature of this data set is that the structures are not publicly known, so evaluations can be conducted “blind.” This provides a more stringent and believable assessment of the strengths and weaknesses of a docking algorithm than does a typical retrospective benchmark. In light of this unique opportunity, GSK has continued to collaborate with research groups to assess additional docking methods.
This article presents the results of RosettaLigand docking on the GSK test set in the context of results from other widely used programs. The docking test set consisted of eight receptors and 136 ligands spanning 21 classes. The receptors were Chk1 kinase (Chk1), factor Xa (FXA), gyrase B (GyrB), methionyl tRNA synthetase (MRS), hepatitis C RNA polymerase (HCVP), peroxisome proliferator-activated receptor-δ (PPARD), and polypeptide deformylase from E. coli and S. pneumococcus (PDF); descriptions of these test systems along with exemplars of the 21 classes of ligands were previously published.9 For incorrectly predicted cases, we consider the causes of failure, and suggest potential remedies to overcome specific difficulties.
Methods
The RosettaLigand protocol was substantially the same as described in Davis and Baker.5 To reduce the computation time, we used a 6 Å diameter binding site and 1000 trajectories instead of 10 Å and 5000 trajectories. Total time was ∼4 days on 100 processors, or about 35 processor-hours per compound. Some cases would likely have benefited from additional sampling. Evaluation of the results and comparison with the other methods was based both on the lowest energy structure and the 20 lowest energy structures that differed from one another by at least 1 Å rmsd.
Docking was conducted with the structures, binding site definitions, and small molecules provided by GSK, without foreknowledge of the experimentally determined binding modes or conformations (although we had some prior experience with public structures of PPARD, PDF, and FXA).
Results
For each receptor, we tallied the fraction of compounds docked within 2 Å rmsd, evaluating both the single lowest energy pose and the 20 lowest energy unique poses for each case. (It is clearly preferable for the lowest energy structure to be correct; when this is not the case small molecule docking can still be informative if the correct solution is among a relatively small set of top models). The results for RosettaLigand are listed in Table I. Considering the best rmsd for any pose in the top 20, RosettaLigand was very successful with the Chk1 target, generating poses within 2 Å for 88% of the 15 Chk1 ligands. For an additional three targets (HCVP, PDF, and PPARδ), RosettaLigand generated good poses for at least 40% of the ligands for each target. MRS was an exceptionally difficult target for RosettaLigand; several of the issues discussed later affected these predictions. The few MRS successes were chemically very similar to failed cases, suggesting insufficient sampling was the primary culprit for the failures. In fact, we found that five-fold greater sampling (i.e., additional trajectories) converted MRS failures to successes in two of five test cases; 20- to 50-fold greater sampling for MRS might be needed to eliminate all failures due to sampling. Considering only the top single pose, the trends remain the same while the absolute success rates decline across the board.
Table I.
RosettaLigand Docking Results
| Any pose |
Best scoring pose |
|||
|---|---|---|---|---|
| Percent within 2 Å | Number within 2 Å | Percent within 2 Å | Number within 2 Å | |
| Chk1 | 88% | 15 (17) | 59% | 10 (17) |
| FXa | 38% | 6 (16) | 31% | 5 (16) |
| Gyrb | 29% | 2 (7) | 29% | 2 (7) |
| HCVP | 40% | 6 (15) | 7% | 1 (15) |
| MRS | 12% | 4 (33) | 3% | 1 (33) |
| 63% | 5 (8) | 50% | 4 (8) | |
| PPARδ | 43% | 23 (53) | 21% | 11 (53) |
“Any Pose” refers to the top 20 best-scoring predictions.
To place these results within the context of the previous GSK evaluation, comparison with results obtained using other docking programs are shown in Figure 1(C,D). Like the other programs, we did relatively well for some receptors and poor for others. Although no program was consistently the top performer, RosettaLigand showed better than average performance for all targets except MRS. It also seems to enjoy a small comparative advantage in selecting native-like poses, as its performance relative to other programs is slightly stronger when considering only a single top pose per ligand [Fig. 1(D)].
Discussion
RosettaLigand seems to perform comparably with the most widely used current docking programs. Also like the other programs, the quality of results varies significantly from target to target: no one program is consistently the best performer across all targets. Although there are an encouraging number of successful predictions, there are also plenty of incorrect ones. Incorrect dockings arise either from failing to generate a native-like pose (search), or from failing to recognize it as such (scoring). Search problems seem to be more significant for RosettaLigand, although the GSK set also illustrates challenges in scoring. We describe search issues first, followed by scoring issues; but in general we refrain from identifying these with specific receptors or ligands to avoid compromising the “blind” status of this set for other researchers.
Deep pockets
A clear search problem is presented by one receptor, which features multiple deep, tight-fitting pockets. On one hand, it is difficult to find the precise ligand geometry that fits all pockets simultaneously. On the other hand, trying to fill the pockets in succession may create insurmountable energy barriers for the simulation. We confirmed that this is a search problem by showing that the minimized native structures were significantly lower in energy than the best docked poses. The difficulty is compounded by RosettaLigand's use of a pre-enumerated conformer library, which is unlikely to contain precisely the native binding geometry. One possible solution would be to dock ligand fragments in their pockets separately and rebuild the linkers afterward. An alternative solution is to divide the ligand into logically distinct segments, analogous to the amino acid residues of a protein, but to maintain chemical connectivity. In proteins, Rosetta's simulated annealing rotamer packing method can simultaneously optimize hundreds of sidechains with thousands of rotamers each. Although a monolithic ligand is limited to a few thousand conformers, dividing the same ligand into two parts and enumerating a thousand finer-grained conformers for each would allow consideration of one million overall ligand conformations without requiring significant additional compute time or memory.
Spurious receptor flexibility
A second search problem is self-inflicted: RosettaLigand allows all sidechains in the binding pocket to be flexible. Although this is important for binding sites that show real flexibility, in some cases, it allows the binding site to adopt non-native configurations with false binding pockets. For example, an aromatic residue in the MRS binding site often flipped to a conformation not seen in the crystal structures, which blocked the true pocket but allowed the ligand to make favorable interactions with that sidechain. Sometimes this is a true search problem, in that the native pose is lower in energy but is never sampled; in other cases, the non-native pose is favored and this becomes a scoring failure. A solution to this problem could be to give an even larger energy bonus to native sidechain rotamer conformations because there is experimental evidence (the crystal structure) that in the absence of ligand they are the lowest energy conformations for the sidechain.
Multiple hydrogen bonds
The most pervasive problem for this set involves directional polar interactions. For a number of the receptors, the best RosettaLigand pose was close to native, but missed the opportunity to make additional hydrogen bonds that were obvious on inspection. Missing these interactions makes it difficult to distinguish native-like poses from non-native poses based on energy. One common case concerns ligand H-bond donors or acceptors separated by two or three bonds, which interact with adjacent sites on the receptor. A 180° rotation about one of the intervening bonds makes the difference between those groups pointing in the same direction or opposite directions, but may change the overall shape of the conformer relatively little. In several cases, our conformer library contained only the incorrect relative orientation, and only one of the two possible H-bonds was ever sampled. Currently, RosettaLigand is unable to access the correct conformer if it is not close to one present in the input conformer library, because minimization of the ligand torsions very rarely crosses torsional barriers. This lack of hopping between torsional minima may be limiting even for moderately flexible compounds: the radius of convergence seems to be 1 Å rmsd or a bit less. Obtaining a sufficient coverage of conformation space might require segmenting the ligand as proposed earlier. Alternatively, finer-grained conformer libraries could be generated around low-energy poses from a first round of docking, allowing refinement of those poses. Finally, one might devise a sampling strategy that explicitly focuses on exploring such 180° flips, or one that notices adjacent unsatisfied donor-acceptor pairs and tries to match them. The challenge is to make such changes while maintaining the overall position of the ligand and its existing favorable interactions.
Tautomers and protonation states
The final search problem is in sampling tautomer and protonation states, as the bound form may not be the dominant form in solution. Because of time constraints in this experiment (which was carried out over a single 1-week period at GSK), we used only a single physically reasonable tautomer and protonation state, but these differed in a number of cases from the actual bound states in the crystal, which makes accurate energy evaluation impossible. It is possible to enumerate tautomers and/or protonation states (and their relative energies) in the input to RosettaLigand; adding and removing protons on the ligand is treated by the same Rosetta machinery that substitutes amino acids during protein design. However, if there are many states to consider and many possible conformations, the total number of ligand “rotamers” becomes prohibitively large. Again, one possible solution is to segment the ligand, in cases where tautomer and protonation decisions for each segment are independent of the others. In some cases, though, it is likely there will still be too many possible states to explore effectively. Another approach, albeit untested, would be to score hydrogen bonds without explicitly modeling polar hydrogen (i.e., using heavy atom coordinates only). Each group could act as either a donor or acceptor, with an appropriate energy penalty for the high-energy state (depending on pH). The results of this coarse grained search could be used to direct hydrogen placement for a subsequent more accurate all atom hydrogen bond energy evaluation.
Hydrophobic scoring and solvation
The clearest scoring problem concerns hydrophobic interactions. For example, PPARD ligands feature multiple bulky, hydrophobic groups that are similar in shape and size. It is often difficult to determine which group belongs in which pocket of the receptor, because the various possible poses score as roughly isoenergetic. Although a native-like pose is often contained within the top 20 poses, it is often not the best scoring one (Fig. 1). It is possible that better conformational search would improve the detailed fit to the point that our existing energy function could identify the native pose, but we can also learn from the scoring approach of programs that perform particularly well on PPARD, such as GOLD, which uses a softened van der Waals potential that has been parameterized to favor hydrophobic contacts.10 For other targets, some compound classes bind with hydrophobic groups quite solvent exposed, sometimes eschewing an “obvious” hydrophobic pocket nearby. In many cases, these binding structures are also surprising to human experts, underscoring the subtlety of solvent interactions. We may be able to resolve these issues by refining the parameters and atom types in our implicit solvation model, or these cases may require a more sophisticated model of solvation.
Despite these shortcomings, we are encouraged to find that the performance of RosettaLigand is similar to other, more established programs, although their specific strengths and weaknesses vary. On the other hand, even the best programs fail to generate a native-like docked pose 70% of the time for at least one receptor, a humbling result. Testing with the unique set of compounds provided by GSK has revealed shortcomings that were not obvious from publicly available data sets, and this will enable further progress in the development of RosettaLigand.
References
- 1.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 2.Cozzini P, Kellogg GE, Spyrakis F, Abraham DJ, Costantino G, Emerson A, Fanelli F, Gohlke H, Kuhn LA, Morris GM, et al. Target flexibility: an emerging consideration in drug discovery and design. J Med Chem. 2008;51:6237–6255. doi: 10.1021/jm800562d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: current status and future challenges. Proteins. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 4.Leach AR, Shoichet BK, Peishoff CE. Prediction of protein-ligand interactions. Docking and scoring: successes and gaps. J Med Chem. 2006;49:5851–5855. doi: 10.1021/jm060999m. [DOI] [PubMed] [Google Scholar]
- 5.Davis IW, Baker D. RosettaLigand docking with full ligand and receptor flexibility. J Mol Biol. 2009;385:381–392. doi: 10.1016/j.jmb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 6.Meiler J, Baker D. RosettaLigand: protein-small molecule docking with full side-chain flexibility. Proteins. 2006;65:538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- 7.Das R, Baker D. Macromolecular modeling with Rosetta. Annu Rev Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 8.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucl Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 10.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 11.Hartshorn MJ, Verdonk ML, Chessari G, Brewerton SC, Mooij WT, Mortenson PN, Murray CW. Diverse, high-quality test set for the validation of protein-ligand docking performance. J Med Chem. 2007;50:726–741. doi: 10.1021/jm061277y. [DOI] [PubMed] [Google Scholar]