


Abstract
G-quadruplex (or G4 DNA), a stable four-stranded structure found in guanine-rich regions, is implicated in the transcriptional regulation of genes involved in growth and development. Previous studies on the role of G4 DNA in gene regulation mostly focused on genomic regions proximal to transcription start sites (TSSs). To gain a more comprehensive understanding of the regulatory role of G4 DNA, we examined the landscape of potential G4 DNA (PG4Ms) motifs in the human genome and found that G4 motifs, not restricted to those found in the TSS-proximal regions, are bias toward gene-associated regions. Significantly, analyses of G4 motifs in seven types of well-known gene regulatory elements revealed a constitutive enrichment pattern and the clusters of G4 motifs tend to be colocalized with regulatory elements. Considering our analysis from a genome evolutionary perspective, we found evidence that the occurrence and accumulation of certain progenitors and canonical G4 DNA motifs within regulatory regions were progressively favored by natural selection. Our results suggest that G4 DNA motifs are ‘colonized’ in regulatory regions, supporting a likely genome-wide role of G4 DNA in gene regulation. We hypothesize that G4 DNA is a regulatory apparatus situated in regulatory elements, acting as a molecular switch that can modulate the role of the host functional regions, by transition in DNA structure.
INTRODUCTION
Certain types of guanine (G)-rich sequences can spontaneously fold into a stable four-stranded DNA structure, which is comprised of stacked G-quartets arranged from four Hoogsteen paired guanines, known as G-quadruplex or G4 DNA (1–4). A growing body of evidence indicates that G4 DNA structures are involved in various cellular functions, particularly in transcriptional regulation (2,5–7).
The formation of G4 DNA structures in the promoters of MYC, KRAS, PDGFA and INS genes has a remarkable influence on the level of gene transcription (8–16). An increasing number of G4 DNA-forming sequences have been identified and characterized in functional regions (e.g. promoters and enhancers) of many important cell growth-related genes including KIT, HIF1A, VEGFA, BCL2, RB1 and in various muscle-specific genes (17–25). In addition to these detailed studies on specific gene loci, several recent genome-wide analyses highlight multiple potential regulatory roles of G4 DNA structure.
First, G4 DNA structure forming sequences are prevalent throughout the human genome (26,27), raising the possibility that this DNA structural motif could act as a general regulatory signal. Second, the potential to form G4 DNA within the transcribed region of genes correlates with functional preferences, suggesting that genes with similar or related function could be coregulated based upon the presence of G4 DNA signal (28). Third, potential G4 DNA motifs (PG4Ms) in the transcription start site (TSS)-proximal region and promoter were associated with genome-wide gene expression, supporting a widespread role for G4 DNA in gene transcription (29–31). Lastly, bioinformatic studies revealed that PG4Ms were enriched in several functional regions including promoters, TSS-proximal regions, nuclease hypersensitive sites, RNA processing sites, 3′-untranslated regions and, most recently, in recombination hotspots (30,32–38), indicating multiple potential roles for G4 DNA in genome function.
In totality, these findings suggest that G4 DNA could be a common structural motif involved in gene regulation via various mechanisms. Since gene regulation is a systematic process controlled by many types of cis-regulatory elements and related trans-regulatory proteins (39), we hypothesized a functional association between G4 DNA and various gene regulatory elements. We asked whether a unique distribution of G4 motifs reflects their functional importance and whether the regulatory roles of G4 DNA are substantiated through pre-existing regulatory elements. Each of these possibilities are supported by the findings that potential G4 motifs are unevenly distributed in the human genome with a strong bias toward gene-associated regions, and that potential G4 motifs are constitutively enriched in seven types of well-known regulatory elements including TSS-proximal regions, nuclease hypersensitive sites, CpG islands, enhancers, insulators, conserved non-coding regulatory sequences and conserved transcription factor-binding sites. Herein, we demonstrate that this phenomenon was progressively favored by natural selection during the evolution of the human genome. Based on these findings we hypothesize that G4 DNA motifs are ‘colonized’ in regulatory elements where they may act as a structure-based regulatory apparatus for genome-wide gene regulation, and that this feature probably defines a general strategy for G4 DNA-mediated gene regulation.
MATERIALS AND METHODS
Identification of PG4Ms
PG4Ms were identified using a previously described program, Quadparser, developed by Huppert et al. Detailed instructions for using this program have been fully described elsewhere (27). Briefly, Quadparser recognizes DNA sequences that have four or more G-runs which contain three or more continuous Gs as a potential G4 DNA motif (G≥3N1–7G≥3N1–7G≥3N1–7G≥3; where N refers to any base). Because genomic DNA is presented as a single strand, both G- and C-patterns (C≥3N1–7C≥3N1–7C≥3N1–7C≥3) were applied to identify PG4Ms on both strands. Two points should be noted here. (i) PG4Ms mentioned in this study and identified by Quadparser refer to distinctive G4 DNA-forming sequences that do not overlap with others. For example, if a DNA sequence contains six G-runs (sequence denoted in box brackets) each of which has at least three continuous Gs: [GGG]TAT[GGG]TAT[GGG]TAT[GGG]TAT[GGG]TAT[GGG], then three overlapping G4 structures might theoretically be formed using the first (1–4), the middle (2–5) or the last four (3–6) G-runs. However, Quadparser outputs this sequence as only one distinctive PG4M to avoid predicting multiple G4-forming sites from a single region of DNA. (ii) In many cases, each identified single PG4M could form diverse types of G4 structure with different topological arrangements by differentially assigning a Guanine into the G-quartet or loop regions. Again, the Quadparser recognizes it as a single distinctive G4-forming site. For example, the DNA sequence GGGGACGGGCACGGGTAAGGG can potentially form two types of G4 DNA by assigning the forth G either into the G-quartet (G[GGG]ACT[GGG]CAC[GGG]TAA[GGG]) or the first loop region ([GGG]GACT[GGG]CAC[GGG]TAA[GGG]). The default rule for PG4M identification was ‘GC 3417’ in which GC represents G- and C- pattern, ‘3’ represents the minimal length of G-run, ‘4’ represents the minimal number of G-run, and ‘17’ represents the range of the length of the loop. The prediction rule can be revised to identify other G4-related motifs. Here, we modified the 3417 rule to ‘GC 2417’, ‘GC 3317’, etc. to identify progenitor or mock PG4Ms according to the specific purpose (see main text for detail). Since mRNA is single-stranded only G-pattern was applied to predict PG4Ms and G4-related motifs in mRNA sequence (e.g. ‘G 3417’, ‘G 2417’, etc.).
DNA sequences and GREs
DNA sequences for the human genome (Hg18) were downloaded directly from the UCSC genome browser (40). Only assembled sequences were used to analyze the genome-wide location of PG4Ms. Genomic coordinates of the TPR (from −500 to +500) were obtained from ‘RefSeq Gene’ track of UCSC genome browser (Hg18); CGIs were obtained from ‘CpG Island’ track of UCSC (Hg 18) and NHSs were obtained from ‘Duck DNase Sites’ track of UCSC (Hg 17) (41). The profile of PG4Ms in NHSs (5158 sites) was analyzed in a previous study (32), but in the study presented here we used a much more comprehensive dataset (95 723 sites); cTFBS were obtained from ‘TFBS conserved’ track of UCSC (Hg18). cTFBSs were predicted on the basis of sequence similarity to the consensus sequence of known TFs and on sequence conservation across human/mouse/rat genomes. To ensure reliability of the data, only cTFBSs that have a Z score ≥2.33 (corresponding to a P value of 0.01) and that do not overlap with exonic sequences were used. Since cTFBSs are short (typically between 6–12 bp in length) discrete sequences which make it unfeasible to directly calculate the frequency of PG4Ms, we clustered the cTFBSs based on density (cTFBS cluster). cTFBSs that occurred within a distance of 500 bp were combined and the clusters that (i) contain five or more cTFBSs and (ii) are longer than 100 bp, were used in the following analysis. The DNA sequences for the above-described UCSC deposited regulatory elements that were extracted directly from the UCSC browser. For other literature-reported GREs, a bed file was created and uploaded into the UCSC custom track to download the sequences from corresponding genome assemble. Specifically, coordinates of the human insulator were obtained from the CTCF-binding site database (CTCFBSDB) (42), where only those insulators identified experimentally by ChIP-chip (43) or ChIP-seq (44) were used for analysis. Predicted cis-regulatory modules identified by evolutionary and sequence pattern extraction through reduced representation (ESPERR) were obtained from Taylor et al. (45). Regions with a regulatory potential (RP) score of at least 0.05 for at least 200 bp and that do not overlap with known exons [referred to as noncoding regulatory sequences (NCRS)] were used for further analysis (downloaded from http://www.bx.psu.edu/∼ross/dataset/DatasetHome.html). Human enhancers predicted from 79 tissues were obtained from Pennacchio et al. (46); human pseudogenes were obtained from the Pseudogene.org database (47). The information for PG4M-positive GREs and nearby genes (within 10 kb) can be found in Supplementary Table S1.
Data analysis
Genome coordinates among different assemblies (e.g. Hg16, Hg17) were converted to Hg18 using lift over tool (http://genome.ucsc.edu/cgi-bin/hgLiftOver). Galaxy genome analysis tools were used for performing conversion, intersection, subtraction and combination of genomic coordinates (48). The conservation score (by phastCons) for the human genome was obtained from (49) and assigned to GREs. All GREs were pooled and sorted according to their average conservation scores (phastCons score, based on 17-species alignment), where GREs with a score lower than 0.1 were excluded. Sequences are simulated through a first-order Markov chain model using sliding windows, which may more accurately reflect the nucleotide arrangement than sequence permutation. The Markov chain is a probabilistic model describing the state transition that future states, which will be reached through a probabilistic process, depend only upon the present state. For each window, the Markov transition probabilities for bases A, T, G and C were computed from the real sequence. The simulation sequence for this window was then generated based on the 4 × 4 transition matrix with the first base generated according to the overall base frequencies. Beginning from the 5′-end, the window moved along the DNA sequence; the whole chromosome was simulated in this manner. Since the window size affects simulation output, we tested window sizes of various lengths (from 50 to 200 bp). The ratio of observed to expected (O/E) number of PG4M was found to be equal to 1 in the real genome when the size was 120 bp. We, thus, reported results using the window size of 120 bp, unless otherwise specified.
RESULTS
Landscape of PG4Ms in the human genome
PG4Ms, G≥3N1–7G≥3N1–7G≥3N1–7G≥3 and C≥3N1–7C≥3N1–7C≥3N1–7C≥3 (where the C pattern represents PG4Ms in the negative DNA strand) were prevalent; while at the same time, they were shown to be unevenly distributed in the human genome (Supplementary Figure S1). Basically, if the PG4Ms were randomly distributed then the number of PG4Ms counted on a given genomic length could be modeled as a Poisson process. For a large value of λ (>10), the Poisson distribution could be approximated by the Normal distribution N (λ,λ) (where λ is the mean of the Poisson distribution). Here, we calculated the number of PG4Ms in 100 kb genomic windows (mean = 12.515). Because the calculation of PG4Ms was affected by regional GC composition, we corrected the effects of heterogeneity of chromosome and GC content by transforming the numbers to normalized values within each of the chromosomes and the categories of similar GC content as 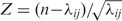 (where n is the observed number for each window and λij is the mean for all windows belonging to chromosome category i and GC-content category j). Under the null hypothesis that PG4Ms were evenly distributed, we expected that the normalized values would follow the Standard Normal distribution. However, the observed distribution was significantly different to the expectation (Kolmogorov–Smirnov test, D = 0.082, P < 0.001, Figure 1A), which was found to be biased toward high or low values with the standard division of 1.362, indicating a genome-wide nonuniformity.
(where n is the observed number for each window and λij is the mean for all windows belonging to chromosome category i and GC-content category j). Under the null hypothesis that PG4Ms were evenly distributed, we expected that the normalized values would follow the Standard Normal distribution. However, the observed distribution was significantly different to the expectation (Kolmogorov–Smirnov test, D = 0.082, P < 0.001, Figure 1A), which was found to be biased toward high or low values with the standard division of 1.362, indicating a genome-wide nonuniformity.
Figure 1.

PG4Ms are unevenly distributed with a strong preference for genes-associated regions in the human genome. (A) Histogram of the frequencies of PG4Ms (blue) corrected for chromosome and GC content. Red curve shows the expected standard normal distribution and the broken black curve is a fit curve for the real distribution. (B) Distribution of PG4Ms correlates with gene regions (GRs). Frequency of PG4Ms (red) and GRs (blue) are plotted across the human chromosomes (showing chromosome 1) in 500-kb window size with a 50-kb step size. Only the GRs that fall completely within a window or that occupy an entire window were counted.
By examining the distribution of PG4Ms in the genome and corresponding genomic features, we observed that PG4Ms were more likely to occur within gene regions (GRs, defined as the transcribed sequence of a gene and the 5-kb flanking region on both sides, so as to include splice variants). A total of 239 901 PG4Ms were identified in 1369.08 Mb GRs, producing a frequency of 0.175, significantly higher than the genome average of 0.126 (360 438 PG4Ms in 2858.01 Mb). Correlation analysis showed that the distributions of PG4Ms and GRs were coupled across the human genome (Spearman ρ = 0.588, P < 0.001) and this observation was valid for all human chromosomes (Supplementary Figure S1). As a representative example shown in Figure 1B for chromosome 1, PG4Ms-rich regions are generally colocalized with gene-rich regions and vice versa.
Because we and others have previously reported that PG4Ms were enriched in TSS-proximal regions, we tested whether the positive correlation was exclusively due to the PG4Ms in this region by recalculating the correlation coefficient after masking all of the PG4Ms located in TSS-proximal regions (from −500 to +500). The correlation between PG4Ms and GRs remained highly significant (Spearman ρ = 0.541, P < 0.001), suggesting that the co-existence of PG4Ms and genes could be extended outside the TSS-proximal region. These results are consistent with the emerging hypothesis that G4 DNA is a regulatory motif involved in transcriptional regulation. Recently, great attention has been paid toward investigating or modeling the regulatory role of PG4Ms in the region proximal to the TSS (e.g. promoter and specific TFBSs). The unexplained co-existence of PG4Ms and genes inspired us to examine the profile and potential role of PG4Ms in many other regulatory elements.
Constitutive enrichment of PG4Ms in various gene regulatory elements
Gene expression is a highly integrated process controlled by various cis-regulatory elements and corresponding proteins, whereby their combinations and interactions set the basis for gene regulation and result in the spatiotemporal patterns of gene expression (Figure 2A). Under the assumption that the regulatory role of G4 DNA is substantiated through pre-existing gene regulatory elements (GREs), seven types of well-known GREs (both specific GREs and those regions with high frequencies of GREs and regulatory potential) were collected based on genome-wide experimental and computational studies (Figure 2B) to examine the possible relationship between PG4Ms and gene regulation.
Figure 2.

PG4Ms are constitutively enriched in various GREs. (A) Overview of transcriptional regulation and regulatory elements. The interactions between regulatory elements and their binding proteins and interactions between different types of regulators (both genetic and epigenetic) result in a complex regulatory network which provides the basis of gene regulation and regulates the loading of the transcription initiation complex. Gene regulatory elements analyzed in this study are represented by blue text. (B) General information on the seven types of GREs. (C) Frequency of PG4Ms in seven types of GREs and controls including genome sequence, intergenic regions and pseudogenes. TPRs were used as a positive control. The GREs are arranged according to the frequency of PG4Ms (from higher to lower). All proceeding figures adhere to this order. (D) Frequency of PG4Ms in GREs after masking those overlapping with the TPRs. The percentages of GREs that overlapped with TPRs are shown above the bar graph. *P < 0.001 (Mann–Whitney tests).
The specific GREs analyzed here include: (i) Nuclease hypersensitive sites (NHSs), an indicator of open chromatin and highly accessible DNA sequences (41) and proven reliable guides to identify regulatory elements (41,50). (ii) Enhancers, cis-regulatory sequences that elevates the transcription level of an adjacent gene (51); enhancers can reside within the flanking regions, introns, UTRs or can be a considerable distance from the target gene they regulate. (iii) Insulators, DNA elements that prevent the regulatory effects passing from one chromatin domain to another, establishing and maintaining the boundaries of chromatin domains (52,53). (iv) Conserved transcription factor-binding sites (cTFBSs), important cis-regulatory sequences that primarily regulate initiation of gene transcription either directly or indirectly, through the binding of cognate transcription factors. In addition, those regions with high frequency of GREs and high regulatory potentials also include (v) CpG islands (CGI) and (vi) predicted regulatory sequences. CGIs are typically associated with promoter and TSS-proximal regions of genes in vertebrate genomes, especially with housekeeping genes, and are regulatory targets of DNA methylation (54). The predicted regulatory sequences are elements predicted from multi-species alignments using the ESPERR method (45). We used such predicted functional elements, located in noncoding regions (NCRSs), in this study. Finally, (vii) the TSS-proximal region (TPR) is of interest, as well; many regulatory elements, such as the core promoter and a number of cis-regulatory elements, tend to be located in the region proximal to the TSS and play an essential role in gene regulation (39). The enrichment and selection of PG4Ms in the TPR (from −500 to +500) have been previously well studied (32,34), hence we used it as a positive control in this study.
We found a significant discrepancy in the frequency of PG4Ms between GREs and control regions, including the bulk genome, intergenic regions and pseudogenes (Figure 2C). A total of 92 316 PG4Ms (25.6%) were observed to overlap with known GREs analyzed herein, and the percentage was great than 4-fold compared to the assumption that PG4Ms were evenly distributed throughout the human genome. As shown in Figure 2C, the frequencies of PG4Ms ranged from 0.32 to 1.23 in GREs, which were significantly higher than that for the total genome, intergenic regions and pseudogenes (from 0.10 to 0.13) (Mann–Whitney tests, P < 0.001 in all cases; Bonferroni-corrected). Considering that CGIs are characterized by a high density of CG dinucleotides and a high GC content which would lead to overestimation, the frequency of PG4Ms in CGI was calculated after masking all CpG dinucleotides (CGI_CpG–).
Many GREs tend to be located in the TPR, where a high frequency of PG4Ms has been reported in previous studies (32,34). We questioned whether the high frequency of PG4Ms observed in other GREs was due to those subsets which overlapped with the TPR. Therefore, we masked all of the GREs that overlap with the TPR (at least 100 bp) in each dataset and recalculated the frequency of PG4Ms. Results showed that the frequency of PG4Ms remained significantly higher than the controls, albeit relatively decreased (Figure 2D, Mann–Whitney tests, P < 0.001 in all cases; Bonferroni-corrected). These data indicate that the enrichment of PG4Ms in GREs is a consequence of a large-scale adoption of this sequence motif.
Under the neutral model, the accumulation of PG4Ms could be achieved by an elevation of the GC content in regions of the genome, led by biased AT to GC mutation or/and gene conversion that favors the fixation of AT to GC mutations (55,56). To test this, DNA sequences (1 kb in length) from GREs and the bulk genome were randomly selected and classified into groups according to the GC content with a uniform scale range (1%). We compared the frequency of PG4Ms in GREs and the bulk genome for each group with a similar GC content. If the higher frequency of PG4Ms observed in GREs is simply caused by the difference of GC composition, we would expect a comparable frequency of PG4Ms between GREs and bulk genome. As shown in Figure 3A, however, the frequencies in GREs were significantly higher than that in the bulk genome, when GC content was considered.
Figure 3.

PG4Ms in GREs are favored by natural selection. (A) The relationship between GC content and frequency of PG4Ms were examined in bulk genome and GREs, respectively. Frequencies of PG4Ms were calculated in 1-kb windows, randomly selected from the bulk genome (red) and GREs (blue) and grouped by GC content with a uniform scale range (1%). Only GC content within the normal range of the human genome (40–60%) are shown. (B) Frequency of PG4Ms in GREs contrasted with their genomic environment. Frequencies of PG4Ms in GREs (red bars), in the GRE-upstream and GRE-downstream regions (with equal length to the GREs) are plotted for each type of GRE. (C) Influence of different window sizes of the Markov simulation (50–200 bp) on the ratio of observed number of PG4Ms to that of expected (O/E) was modeled for the bulk genome (100 times). (D) PG4Ms in GREs are favored by natural selection. O/E values were calculated for each type of GRE and controls. The standard variations for the O/E values (Markov simulations, 100 repeats) are too low to be visualized in the figure.
With the observation of the uneven distribution of PG4Ms in the human genome and their constitutive enrichment in GREs, we began further tests to determine whether PG4M in GREs were driven by a neutral mechanism or natural selection.
PG4Ms in GREs are favored by natural selection
Since the GREs investigated in this study were generally short (median length 634 bp), it is reasonable to expect that those GRE sequences share a similar mutation rate and base composition with their flanking sequences if no selection force is acting upon that sequence. Thus, we compared the frequency of PG4Ms in GREs to that of the flanking sequences. The genomic regions, both upstream and downstream, with an equal length as that of the corresponding GRE, were analyzed. In general, we found that the frequency of PG4Ms in GREs, with the exception of enhancer regions, contrasted to their genomic environment with statistical significance (Figure 3B, Kruskal–Wallis test, P < 0.001). Again, this observation is not consistent with the neutral model expectation.
To detect the selection on the accumulation of PG4Ms in GREs in a more straightforward manner, we compared the number of expected PG4Ms in GREs to the observed number. Instead of using a permutation approach, we chose a more sophisticated Markov chain simulation which has the advantage of keeping not only the base composition (e.g. GC content), but also other features of the original sequences such as the frequency of dinucleotides. Since the window size affects the output of sequence simulation, various window sizes (from 50 to 200 bp) were analyzed for 100 repeats. We computed the bulk genome and found when the window size was 120 bp, the observed number of PG4Ms is similar to the simulated ones (Figure 3C). We, therefore, used this window size to assess the GREs. We compared the observed and expected number of PG4Ms in all types of GREs and controls. The 1 kb TSS-proximal regions (from –500 to +500) were used as positive controls because PG4Ms in this region have been previously shown to be under selection. Nonregulatory sequences were chosen as negative controls and included randomly selected genomic sequences (100 000 representative sequences range from 100 bp to 5 kb in length), intergenic regions (10 000 random sequences), transcribed regions of pseudogenes and the coding strand of exonic regions (mRNA sequences). The results were striking in that we found that the O/E ratio of the number of PG4Ms was significantly higher in all of the GREs compared to those observed for the controls (Figure 3D, t-tests, P < 0.001 for all cases, Bonferroni-corrected). It suggests that the PG4Ms in the GREs were favored by natural selection. In addition, our results were consistent with previous findings that PG4Ms were significantly repressed in mRNA sequences (27), as we observed a low O/E value (<1) for PG4Ms in mature mRNA sequences.
Progenitors of PG4Ms in GREs are favored by natural selection
Typically, a DNA sequence that contains four stretches of G-runs with at least three continuous Gs and interrupted by one to seven bases is identified as a PG4M, expressed by the rule of G≥3N1–7G≥3N1–7G≥3N1–7G≥3 (G4-3417). The number of G-runs (at least four) is equivalent to the four Gs in the G-quartet, and the number of Gs (at least three) in each G-run is equivalent to the number of G-quartet in G4 DNA (Figure 4A). This is a complex motif, hence, the gain and accumulation of PG4Ms in GREs would not be expected to be accomplished very quickly. In order to track the process of evolution, the prediction rule of PG4M was first modified to predict the ‘PG4M progenitor’ by changing the minimal number of G-runs (Figure 4B). We compared the O/E values of PG4Ms progenitors between GREs and controls (progenitor–O/E) under the Markov model using a 120-bp window with 100 replicates. We sequentially reduced the number of G-runs to three, two and one leading to the rules of G4-3317, G4-3217 and G4-3117, respectively. Since a portion of higher-order progenitors could be special cases of the lower-order progenitor, for example G4-3317 contains some G4-3417 and G4-3217 contains some G4-3317 and G4-3417, we excluded all of those lower-order progenitor motifs that have any overlap with the higher-order G4-3417 for above analyses to eliminate the influence of canonical PG4Ms on our observation. When the number of G-runs is one, the values of O/E were almost equal for all GREs and controls (Figure 4C), suggesting that the basic motif of G4-3117 is not favored by natural selection. However, starting from when the number of G-run reached two, a disparity of O/E values occurred between GREs and controls (Mann–Whitney tests, P < 0.001). This disparity slightly increased when the number of G-runs increased from two to three as shown in Figure 4D–E, indicating that the selection forces increased steadily towards the canonical G4 DNA structure. We suggest that G4-3217 and G4-3317 would be progenitor sequences during the selection process since they could readily produce PG4Ms by the conjunction of the progenitor sequences, via duplication or recombination. However, the possibility that G4-3217 and G4-3317 had already gained other functions, besides the formation of G4 DNA, could not completely ruled out.
Figure 4.

Progenitor PG4Ms are favored by selection in GREs. To analyze the generation and selection of PG4Ms during evolution, we modified the prediction rule of PG4Ms (3417) based on the structural property of canonical G4 DNA structure (A). ‘3’ represents the minimal number of continuous Gs (length of G-run); ‘4’ represents minimal number of G-runs; ‘17’ represents the range of the length of arbitrary bases between G-runs (loop length, between one and seven bases). Minimal number of G-runs for PG4M prediction (four) was changed to 3/2/1 (B and C–F) and the minimal length of G-run (three) was changed to 2/1 (B and G–I). The ratio of observed to expected (O/E) number of PG4Ms and modified PG4Ms was calculated for each type of GRE and control, respectively. The broken blue lines highlight the O/E value for the genome in each case. The standard variations for Markov simulations (100 repeats) are too low to be visualized in the figure.
We next investigated the influence of the minimal number of continuous Gs in each G-run (length of G-run) on the enrichment of PG4Ms. This was carried out by reducing the minimal length of each G-run to two and one base (G4-2417 and G4-1417, respectively; mock PG4Ms) while other parameters remained constant. As shown in Figure 4G–I, the O/E values significantly differed between GREs and controls only when the length of the G-runs reached three guanines (G4-3417). No obvious differences in the O/E values were found between GREs and controls for the reduced length of G-runs to two or one. Thus, our results suggested that G-runs with three continuous guanines were functionally important, consistent with the structural property of G4 DNA that contributes to formation and stabilization of G4 DNA typically requiring three or more G-quartets.
Progressive selection of canonical and progenitor PG4Ms in GREs during genome evolution
In addition to the detection of the type of selection status, we further traced the progressive selection of PG4Ms in GREs on a vertical time axis. Since only canonical G4-3417 and the progenitor motifs G4-3317 and G4-3217 were favored by selection, here we focused on these three motifs for further analysis. In general, relatively more conserved GREs (with a higher phastCons score) tend to be older than less conserved ones (with a lower phastCons score) that might create more recently during evolution. All GREs were pooled and sorted according to the average phastCons scores (based on 17-species alignment). We calculated the average O/E values of canonical PG4Ms (G4-3417) as well as the values of those progenitor PG4Ms in windows containing 1000 GREs. Interestingly, we found a strong negative correlation between the phastCons scores and the average O/E value for all of the motifs (Figure 5, Pearson correlation r < −0.9, P < 0.001 for each case). Thus, the progenitors G4-3217 (Figure 5C), G4-3317 (Figure 5B) and canonical G4-3417 (Figure 5A) were under gradually increasing selection pressure. The pressure acts toward both canonical structure and PG4M progenitors in newer GREs.
Figure 5.

Canonical PG4Ms and PG4M progenitors are progressively selected in GREs during evolution. To analyze the selection for canonical PG4Ms (G4-3417) and PG4M-progenitors during evolution, GREs were pooled and ranked based on conservation score (phastCons) from 17-species alignment. The average O/E value of PG4Ms (A) and corresponding PG4M progenitors (B and C) was calculated for every 1000 GREs. Sequence simulations were performed using 120-bp window sizes for 100 repeats.
In summary, we provided evidence that PG4Ms are under evolutionary selection resulting in a strong nonuniform and GRE-preferred pattern in the human genome. The selection process for the occurrence of PG4Ms in GREs acted on both the length (at least three continuous Gs) and the number of G-runs (at least two), and bring the ‘mature’ PG4Ms into GREs step-by-step.
Genome-wide colocalization of selected PG4M-enriched regions with GREs
It is realistic that not all predicted PG4Ms will necessarily fold into G4 DNA in vivo, the selected PG4M-enriched regions would be more biologically relevant compare to individual PG4Ms. Based on the genome-wide density of PG4Ms, we identified 18 283 PG4M-clustered regions (G4 clusters, Supplementary Table S2) according to the following criteria: (i) there were at least three PG4Ms and (ii) the maximum distances between the neighboring PG4Ms in each cluster were no longer than 500 bp. Although PG4Ms were highly prevalent in the human genome, G4 clusters were relatively rare and the identified number of G4 clusters was significantly higher than expected (Markov simulation, 120 bp), though they exhibit a similar number of individual PG4Ms. G4 clusters not only showed a high level of G4 frequency (over 50-fold), but importantly, the frequency was 2.1-fold as compared to that which would be expected (Markov simulation, 120-bp window). The G4 cluster represents the hotspots of PG4Ms selection in the genome. We examined whether the G4 cluster is more associated with GREs than individual PG4Ms.
We first asked whether there is a colocalization tendency of G4 clusters with GREs. When all overlapping GREs were merged into a single entity, we found that the distribution of G4 clusters was generally coupled with GREs (Supplementary Figure S2). We calculated the distances between each G4 cluster and its nearest GREs (defined as the difference between the coordinates of the two midpoints) and then determined the median value. Five thousand sets of random clusters, of which the number and cluster length were kept identical to actual G4 clusters, were generated to calculate the null distribution of this value. We found that the value obtained from the real genome (482 bp) was significantly smaller than that obtained from the randomized data (mean = 4212 bp) (P < 0.001). This result showed that G4 clusters were generally close to GREs, indicating a strong genome-wide colocalization tendency.
We next qualified the portion of G4 clusters that was located within each type of GRE, respectively. We found that the number of G4 clusters overlapping with GREs was significantly higher than would be expected using the random cluster as a control. The overlap between G4 clusters and GREs was from 4- to 20-fold as compared to that observed for the control. Similar results were obtained for the PG4Ms located in the G4 clusters (Figure 6A). Using the same approach we next assessed the portion of individual PG4Ms overlapping with GREs. Results showed that although the portion of individual PG4Ms located in GREs was fom 3- to 11-fold in comparison to the control (number and length were kept identical to actual PG4Ms), more G4 clusters tended to be located within regulatory regions for all types of GREs. Significantly, over 60% of the G4 clusters overlapped with at least one GRE, which was ∼2.3-fold higher than individual PG4Ms and was ∼5-fold as compared to the random cluster, making the G4 cluster (selected PG4Ms-enriched regions) a decent feature for predicting regulatory sequences. Two representative examples are shown in Figure 6, wherein genomic regions rich in GREs tend to have a high number of G4 clusters (red, Figure 6B) and vice-versa (Figure 6C). Specifically, many of the G4 clusters are colocalized with GREs compared to that observed for the control (black, Figure 6B and C). Since the cTFBS clusters analyzed herein contained a collection of the binding sites of different transcription factors (TFs), we further examined which TFBSs were specifically enriched in the G4 cluster. We analyzed the presence of known TFBSs in the G4 clusters and found 71 TFBSs were significantly enriched in the G4 cluster (Figure 6D and Supplementary Table S3). This result raises the possibility that G4 DNA would potentially modulate the binding and regulatory role of those TFs in specific cellular conditions.
Figure 6.

Genome-wide colocalization of G4 clusters with GREs. (A) Percentage of individual PG4Ms (red), G4 clusters (blue) and PG4Ms in the G4 clusters (grey) that overlapped with GREs was calculated for each type of GRE. The percentage was normalized to randomized datasets in which the number and length of elements were kept identical to original data for individual PG4Ms, G4 cluster and PG4Ms in the G4 clusters, respectively. (B and C) UCSC genome browser view of two representative genomic regions (500 kb in length) in chromosome 1, showing a general colocalization of G4 clusters and GREs. Vertical lines in different colors represent the locations of G4 clusters, GREs and random cluster (control); the name for each element is indicated above the lines. 6A shows that genomic region rich in genes and GREs tend to have a high number of G4 clusters, and the G4 clusters are generally colocalized with GREs; 6B shows the opposite situation. (D) Enriched TFBSs in G4 clusters. Enriched sequence motifs for known eukaryotic TFBSs were searched for in G4 clusters using a cis-regulatory element annotation system (CEAS, http://ceas.cbi.pku.edu.cn) (83). Top 10 TFBSs with the highest number of hits and enrichment scores are listed. The complete list of 71-enriched TFBSs in G4 cluster (at least 1.5-fold) can be found in Supplementary Table S3.
DISCUSSION
G4 DNA-mediated transcriptional regulation has been investigated and described in some specific genes. These findings highlighted a regulatory role of this unusual four-stranded DNA structure and suggest a new therapeutic approach for cancer therapy using G4 DNA-based gene modulation via DNA structure specific ligands (10,22,57,58). In addition to wet-lab experiments, current bioinformatic-based studies of potential G4 motifs in the TSS-flanking region also demonstrated an association between PG4M and gene regulation, function and expression levels. Due to the prevalence of PG4Ms in the human genome, it is interesting to test whether G4 motifs are involved in genome-wide gene regulation to a greater degree and in a more ubiquitous manner.
Potential implications for the distribution features of PG4Ms in gene regulation
To date, a large number of regulatory elements encoded in the human genome have been identified and characterized (59). Some of the GREs analyzed herein were specific regulatory elements (e.g. insulators and enhancers), some were regions with a high potential to harbor regulatory elements (such as NCRSs and NHSs) and some were identified in specific tissues or cell types (e.g. enhancer and insulator). Our results showed that PG4Ms were prevalent in all types of GREs examined, and the frequency of PG4Ms was significantly higher than expected when compared to a number of genomic controls, indicating that PG4Ms in GREs are favored during genome evolution.
By examining the current distribution and tracing the process of evolution for potential G4 DNA motifs, our results suggest a genome-wide ‘colonization’ of the regulatory region by PG4Ms in the human genome. These findings have led to a hypothesis that G4 DNA is a regulatory apparatus situated within or near other gene regulatory elements and the transitions of DNA structure in PG4Ms might function as molecular switches that modulate the role of the functional regions wherein they are situated. This is a biologically ‘intelligent’ strategy since it takes advantage of the established functions, regulations and interactions of GREs, and of the ability of PG4Ms to form alternative structures that can potentially influence the activity and behavior of GREs, hence providing an extensive regulatory role in gene regulation.
PG4Ms may provide a molecular switch to regulate the function of GREs
The detailed molecular mechanisms by which G4 DNA structures regulate gene expression remain largely unknown although accumulating direct and indirect evidence obtained both in vivo and in vitro supports the bona fide existence of functional G4 DNA in vivo (60,61). Several previous studies proposed specific potential models to explain G4 DNA-mediated gene regulation (7,8,15,25). Having found that potential G4 DNA motifs are selected for enrichment within or near GREs in the human genome, we hypothesize that G4 structure-mediated gene regulation is a common mechanism of gene regulation (Figure 6).
-
PG4Ms may regulate the binding of regulatory proteins in GREs by switching DNA structure (Figure 7A–C). The interactions between cis-regulatory elements and corresponding regulatory proteins are essential for gene regulation. The PG4Ms in GREs may regulate the recognition and binding of cognate regulatory proteins, hence modulating gene expression. Specifically, the formation of G4 DNA in PG4Ms would (a) recruit regulatory proteins that have G4 DNA-specific-binding activity (Figure 7A); (b) inhibit the binding of regulatory proteins (Figure 7B) and (c) result in distinct protein-binding patterns (Figure 7C) with different regulatory consequences. In this model, the transitions in local DNA structure in GREs could provide regulatory signals which could then be sensed and read by the interacting proteins.
Many G4 DNA-specific-binding proteins have been identified and characterized, some of them have been further shown to regulate gene expression (61). For example, a biochemical study suggests that Pur-1 can stimulate insulin gene transcription by recognizing G4 DNA formed in the promoter region (15). Some regulatory proteins preferred binding at double-stranded or single-stranded DNA targets, such as Sp1 and single-strand binding proteins CNBP and hnRNPK associated with transcriptional regulation of the MYC gene (62,63). The formation of G4 DNA in the recognition sites may thus interfere with the recognition or binding process and, in turn, influence gene expression. It has been hypothesized that formation of G4 DNA in the NHEIII region of the MYC promoter would block the binding of specific transcription factors so as to inhibit gene transcription (7,8). In addition, DNA sequences with different structures may recruit different regulatory proteins. In vitro studies demonstrated that proteins show significantly different affinities to DNA molecules with different topologies. For example, MyoD homodimers preferentially bind to bimolecular G4 DNA, while MyoD-E47 heterodimers bind more tightly to double-stranded E-box DNA (7,64). The transitions of local DNA structures would therefore create different binding profiles of regulatory proteins to regulate gene expression.
Two recently conducted analyses indicate that PG4Ms (especially those upstream of transcription start sites) overlap with many G-rich TFBSs including Sp1, KLF, EKLF, MAZ, EGR-1 and Ap-2 (65,66). Eddy et al. commented that the colocalization of PG4Ms with TFBSs would challenge the notion that the PG4M upstream of the TSS were involved in gene regulation (66). However, as mentioned above, we suggest that, on the contrary, this phenomenon might support the hypothesis that G4 DNA plays a role in gene regulation. First, formation of G4 and binding of TFs could function independently under different situations or in different cell types. Second, although occupancy of G-rich TFBSs by TFs could hamper the formation of G4 DNA, we could not rule out the opposite action. Third, being a mutually exclusive dynamic process for binding of regulatory protein and formation of G4 DNA, the competition itself could provide a regulatory mechanism during the course of gene regulation. Actually, significant overlapping of PG4Ms with many functional elements and the enrichment of TFBSs in G4 clusters (Figure 6D and Supplementary Table S3) indicate a potential interaction. To further investigate this possibility, it would be interesting to analyze the binding properties of those regulatory proteins whose reorganization sites are located in the PG4M-enriched regions, or to identify the dynamics of in vivo binding patterns of those proteins by ChIP-chip or ChIP-seq following the treatment of G4 DNA-specific stabilizing or destabilizing ligands.
G4 DNA may regulate the role of host GREs by its unique biophysical property (Figure 7D). The topology of DNA, such as negative supercoiling, regulates gene expression remarkably (67,68). Hence, it is possible that G4 structure per se can regulate cellular function through its unique biophysical properties. For example, formation of G4 DNA during DNA replication would hamper the reading and copying of genetic information (69,70). Transcription of several G-rich sequences produces a unique G4 DNA-containing G-loop structure that contributes to recombination and hypermutation (71,72). Furthermore, our previous study hypothesized that G4 DNA stimulates gene transcription by stabilizing DNA in an open conformation and rendering the template strand unpaired for a high rate of transcription (29).
While DNA sequence elements significantly influence gene activity, epigenetic mechanisms also regulate gene function considerably without changing the DNA sequence (73). It is also possible that G4 DNA regulates gene expression through epigenetic mechanisms. CpG islands are targets of DNA methylation; cytosine methylation in the CpG context plays an important role in gene silencing (74). Previous in vitro studies demonstrated that DNA methyltransferase recognizes and catalyzes DNA substrates with certain secondary structures (e.g. hairpins and G4 DNA) at a remarkable high efficiency, suggesting that DNA conformation would be involved in regulating DNA methylation (75,76). In this study the CpG islands are found to be rich in PG4Ms, we thus propose that PG4Ms in CpG islands would regulate the level of DNA methylation by folding into G4 DNA. Although G4 structure has been proved to facilitate DNA methylation in vitro, the situation in living cells and whether opposing functions exist needs further investigation (Figure 7E).
G4 DNA may regulate nucleosome organization (Figure 7F). Nucleosome density and organization is an indicator of the chromatin state controlling the accessibility of regulator proteins to their target DNA (77). Nucleosome occupancy is a general mechanism for gene regulation since the function of many regulatory elements relies on binding of cognate proteins. A recent study showed that the frequency of PG4Ms is negatively correlated with nucleosome occupancy near the TSS in yeast, suggesting that PG4M is an anti-nucleosomal motif, although the underlying mechanism is unclear (30). It has been recognized that DNA sequence could potentially influence the architecture of the nucleosome (78–81). It is highly possible that formation of G4 DNA in PG4Ms could affect nucleosome architecture of the host GREs and, in turn, control their regulatory function. Two potential mechanisms might be involved (Figure 7F): a) the formation of G4 DNA could impede DNA wrapping around the histone octamer to form the nucleosome; b) G4 DNA can recruit chromatin remodelers which then exclude the histones from DNA. The observation that PG4Ms are negatively correlated with nucleosome occupancy at the TPR is reminiscent of our recent finding that PG4Ms located in the immediate downstream region of the TSS positively correlated with the gene expression level (29). We proposed previously that PG4Ms stimulate gene transcription through a DNA structural-mediated mechanism. It is also possible that formation of G4 DNA can block rehybridization with the complementary strand, hence influencing nucleosome assembly in the TPR and making the chromatin remain in an open state for high-rate transcription.
Figure 7.

Potential models for G4 DNA-mediated gene regulation. Proposed models for G4 DNA-mediated gene regulation. As described in the text, formation of G4 DNA structure could contribute to gene regulation by influencing the function of PG4M hosted regulatory region. G4 structure could influence binding patterns of corresponding regulatory proteins in regulatory regions (A, B and C), modulate the role of regulatory elements by its unique biochemical and biophysical properties (D) and regulate the epigenetic properties of target sites such as DNA methylation status in CpG islands (E) and chromatin architecture of regulatory regions (F). G4 DNA-forming sequence is plotted in blue and corresponding regulatory proteins are plotted in red.
With all of our findings considered herein, we hypothesize that the G4 motifs are ‘colonized’ in regulatory elements as structure-based regulatory apparatus for gene regulation. The transition of DNA structure (spontaneously or with the help of G4-promoting or stabilizing proteins) may mediate the gene regulation process. While the regulatory roles of DNA sequence motifs/elements and epigenetic regulators have been extensively studied recently, the role of DNA secondary structures remains largely unknown in this context. Our findings highlight that G4 DNA structural motifs would provide regulatory signals for genome-wide gene regulation and suggest that DNA structure-mediated gene regulation might be a common strategy in the human genome. There are still outlying and unknown concerns, since the correlation coefficient between PG4Ms and genes only decreased from 0.589 to 0.488 when masking all PG4Ms in GREs. Our results, thus, indicate that a) PG4Ms are enriched in many other GREs that were not included in the seven representative types of GREs analyzed and b) PG4Ms may have other roles involving the gene body and gene proximal regions. Consistent with this idea, a recent study reported that PG4Ms are over-represented in the 3′-end of genes and suggested that G4 DNA would be involved in transcription termination (82). We are just beginning to understand the mystery of the role of G4 DNA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Major Basic Research Program of China (973 program) (2006CB102100) and the National High Technology Research and Development Program of China (863 Program) (2006AA10A120). Funding for open access charge: National Major Basic Research Program of China (2006CB102100).
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Sen D, Gilbert W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature. 1988;334:364–366. doi: 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- 2.Han H, Hurley LH. G-quadruplex DNA: a potential target for anti-cancer drug design. Trends Pharmacol. Sci. 2000;21:136–142. doi: 10.1016/s0165-6147(00)01457-7. [DOI] [PubMed] [Google Scholar]
- 3.Burge S, Parkinson GN, Hazel P, Todd AK, Neidle S. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006;34:5402–5415. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phan AT, Kuryavyi V, Patel DJ. DNA architecture: from G to Z. Curr. Opin. Struct. Biol. 2006;16:288–298. doi: 10.1016/j.sbi.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomez D, Lemarteleur T, Lacroix L, Mailliet P, Mergny JL, Riou JF. Telomerase downregulation induced by the G-quadruplex ligand 12459 in A549 cells is mediated by hTERT RNA alternative splicing. Nucleic Acids Res. 2004;32:371–379. doi: 10.1093/nar/gkh181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maizels N. Dynamic roles for G4 DNA in the biology of eukaryotic cells. Nat. Struct. Mol. Biol. 2006;13:1055–1059. doi: 10.1038/nsmb1171. [DOI] [PubMed] [Google Scholar]
- 7.Dexheimer TS, Fry M, Hurley LH. In: Quadruplex Nucleic Acids. Neidle S, Balasubramanian S, editors. Cambridge: RSC Publishing; 2006. pp. 180–207. [Google Scholar]
- 8.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl Acad. Sci. USA. 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phan AT, Modi YS, Patel DJ. Propeller-type parallel-stranded G-quadruplexes in the human c-myc promoter. J. Am. Chem. Soc. 2004;126:8710–8716. doi: 10.1021/ja048805k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hurley LH, Von Hoff DD, Siddiqui-Jain A, Yang D. Drug targeting of the c-MYC promoter to repress gene expression via a G-quadruplex silencer element. Semin. Oncol. 2006;33:498–512. doi: 10.1053/j.seminoncol.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 11.Yang D, Hurley LH. Structure of the biologically relevant G-quadruplex in the c-MYC promoter. Nucleosides Nucleotides Nucleic Acids. 2006;25:951–968. doi: 10.1080/15257770600809913. [DOI] [PubMed] [Google Scholar]
- 12.Grand CL, Han H, Munoz RM, Weitman S, Von Hoff DD, Hurley LH, Bearss DJ. The cationic porphyrin TMPyP4 down-regulates c-MYC and human telomerase reverse transcriptase expression and inhibits tumor growth in vivo. Mol. Cancer Ther. 2002;1:565–573. [PubMed] [Google Scholar]
- 13.Cogoi S, Xodo LE. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006;34:2536–2549. doi: 10.1093/nar/gkl286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paramasivam M, Membrino A, Cogoi S, Fukuda H, Nakagama H, Xodo LE. Protein hnRNP A1 and its derivative Up1 unfold quadruplex DNA in the human KRAS promoter: implications for transcription. Nucleic Acids Res. 2009;37:2841–2853. doi: 10.1093/nar/gkp138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lew A, Rutter WJ, Kennedy GC. Unusual DNA structure of the diabetes susceptibility locus IDDM2 and its effect on transcription by the insulin promoter factor Pur-1/MAZ. Proc. Natl Acad. Sci. USA. 2000;97:12508–12512. doi: 10.1073/pnas.97.23.12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qin Y, Rezler EM, Gokhale V, Sun D, Hurley LH. Characterization of the G-quadruplexes in the duplex nuclease hypersensitive element of the PDGF-A promoter and modulation of PDGF-A promoter activity by TMPyP4. Nucleic Acids Res. 2007;35:7698–7713. doi: 10.1093/nar/gkm538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernando H, Reszka AP, Huppert J, Ladame S, Rankin S, Venkitaraman AR, Neidle S, Balasubramanian S. A conserved quadruplex motif located in a transcription activation site of the human c-kit oncogene. Biochemistry. 2006;45:7854–7860. doi: 10.1021/bi0601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun D, Guo K, Rusche JJ, Hurley LH. Facilitation of a structural transition in the polypurine/polypyrimidine tract within the proximal promoter region of the human VEGF gene by the presence of potassium and G-quadruplex-interactive agents. Nucleic Acids Res. 2005;33:6070–6080. doi: 10.1093/nar/gki917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dexheimer TS, Sun D, Hurley LH. Deconvoluting the structural and drug-recognition complexity of the G-quadruplex-forming region upstream of the bcl-2 P1 promoter. J. Am. Chem. Soc. 2006;128:5404–5415. doi: 10.1021/ja0563861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dai J, Dexheimer TS, Chen D, Carver M, Ambrus A, Jones RA, Yang D. An intramolecular G-quadruplex structure with mixed parallel/antiparallel G-strands formed in the human BCL-2 promoter region in solution. J. Am. Chem. Soc. 2006;128:1096–1098. doi: 10.1021/ja055636a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Y, Sugiyama H. Formation of the G-quadruplex and i-motif structures in retinoblastoma susceptibility genes (Rb) Nucleic Acids Res. 2006;34:949–954. doi: 10.1093/nar/gkj485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qin Y, Hurley LH. Structures, folding patterns, and functions of intramolecular DNA G-quadruplexes found in eukaryotic promoter regions. Biochimie. 2008;90:1149–1171. doi: 10.1016/j.biochi.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Armond R, Wood S, Sun D, Hurley LH, Ebbinghaus SW. Evidence for the presence of a guanine quadruplex forming region within a polypurine tract of the hypoxia inducible factor 1alpha promoter. Biochemistry. 2005;44:16341–16350. doi: 10.1021/bi051618u. [DOI] [PubMed] [Google Scholar]
- 24.Yafe A, Shklover J, Weisman-Shomer P, Bengal E, Fry M. Differential binding of quadruplex structures of muscle-specific genes regulatory sequences by MyoD, MRF4 and myogenin. Nucleic Acids Res. 2008;36:3916–3925. doi: 10.1093/nar/gkn340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yafe A, Etzioni S, Weisman-Shomer P, Fry M. Formation and properties of hairpin and tetraplex structures of guanine-rich regulatory sequences of muscle-specific genes. Nucleic Acids Res. 2005;33:2887–2900. doi: 10.1093/nar/gki606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Todd AK, Johnston M, Neidle S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005;33:2901–2907. doi: 10.1093/nar/gki553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huppert JL, Balasubramanian S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005;33:2908–2916. doi: 10.1093/nar/gki609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eddy J, Maizels N. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006;34:3887–3896. doi: 10.1093/nar/gkl529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du Z, Zhao Y, Li N. Genome-wide analysis reveals regulatory role of G4 DNA in gene transcription. Genome Res. 2008;18:233–241. doi: 10.1101/gr.6905408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hershman SG, Chen Q, Lee JY, Kozak ML, Yue P, Wang LS, Johnson FB. Genomic distribution and functional analyses of potential G-quadruplex-forming sequences in Saccharomyces cerevisiae. Nucleic Acids Res. 2008;36:144–156. doi: 10.1093/nar/gkm986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verma A, Yadav VK, Basundra R, Kumar A, Chowdhury S. Evidence of genome-wide G4 DNA-mediated gene expression in human cancer cells. Nucleic Acids Res. 2009;37:4194–4204. doi: 10.1093/nar/gkn1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huppert JL, Balasubramanian S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007;35:406–413. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du Z, Kong P, Gao Y, Li N. Enrichment of G4 DNA motif in transcriptional regulatory region of chicken genome. Biochem. Biophys. Res. Commun. 2007;354:1067–1070. doi: 10.1016/j.bbrc.2007.01.093. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y, Du Z, Li N. Extensive selection for the enrichment of G4 DNA motifs in transcriptional regulatory regions of warm blooded animals. FEBS Lett. 2007;581:1951–1956. doi: 10.1016/j.febslet.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 35.Rawal P, Kummarasetti VB, Ravindran J, Kumar N, Halder K, Sharma R, Mukerji M, Das SK, Chowdhury S. Genome-wide prediction of G4 DNA as regulatory motifs: role in Escherichia coli global regulation. Genome Res. 2006;16:644–655. doi: 10.1101/gr.4508806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kikin O, Zappala Z, D'A;ntonio L, Bagga PS. GRSDB2 and GRS_UTRdb: databases of quadruplex forming G-rich sequences in pre-mRNAs and mRNAs. Nucleic Acids Res. 2008;36:D141–148. doi: 10.1093/nar/gkm982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verma A, Halder K, Halder R, Yadav VK, Rawal P, Thakur RK, Mohd F, Sharma A, Chowdhury S. Genome-wide computational and expression analyses reveal G-quadruplex DNA motifs as conserved cis-regulatory elements in human and related species. J. Med. Chem. 2008;51:5641–5649. doi: 10.1021/jm800448a. [DOI] [PubMed] [Google Scholar]
- 38.Mani P, Yadav VK, Das SK, Chowdhury S. Genome-wide analyses of recombination prone regions predict role of DNA structural motif in recombination. PLoS ONE. 2009;4:e4399. doi: 10.1371/journal.pone.0004399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu. Rev. Genomics Hum. Genet. 2006;7:29–59. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- 40.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bao L, Zhou M, Cui Y. CTCFBSDB: a CTCF-binding site database for characterization of vertebrate genomic insulators. Nucleic Acids Res. 2008;36:D83–87. doi: 10.1093/nar/gkm875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 45.Taylor J, Tyekucheva S, King DC, Hardison RC, Miller W, Chiaromonte F. ESPERR: learning strong and weak signals in genomic sequence alignments to identify functional elements. Genome Res. 2006;16:1596–1604. doi: 10.1101/gr.4537706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pennacchio LA, Loots GG, Nobrega MA, Ovcharenko I. Predicting tissue-specific enhancers in the human genome. Genome Res. 2007;17:201–211. doi: 10.1101/gr.5972507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karro JE, Yan Y, Zheng D, Zhang Z, Carriero N, Cayting P, Harrrison P, Gerstein M. Pseudogene.org: a comprehensive database and comparison platform for pseudogene annotation. Nucleic Acids Res. 2007;35:D55–60. doi: 10.1093/nar/gkl851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crawford GE, Holt IE, Whittle J, Webb BD, Tai D, Davis S, Margulies EH, Chen Y, Bernat JA, Ginsburg D, et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS) Genome Res. 2006;16:123–131. doi: 10.1101/gr.4074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blackwood EM, Kadonaga JT. Going the distance: a current view of enhancer action. Science. 1998;281:60–63. doi: 10.1126/science.281.5373.60. [DOI] [PubMed] [Google Scholar]
- 52.Fourel G, Magdinier F, Gilson E. Insulator dynamics and the setting of chromatin domains. Bioessays. 2004;26:523–532. doi: 10.1002/bies.20028. [DOI] [PubMed] [Google Scholar]
- 53.Geyer PK. The role of insulator elements in defining domains of gene expression. Curr. Opin. Genet. Dev. 1997;7:242–248. doi: 10.1016/s0959-437x(97)80134-7. [DOI] [PubMed] [Google Scholar]
- 54.Issa JP. CpG island methylator phenotype in cancer. Nat. Rev. Cancer. 2004;4:988–993. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 55.Galtier N. Gene conversion drives GC content evolution in mammalian histones. Trends Genet. 2003;19:65–68. doi: 10.1016/s0168-9525(02)00002-1. [DOI] [PubMed] [Google Scholar]
- 56.Eyre-Walker A, Hurst LD. The evolution of isochores. Nat. Rev. Genet. 2001;2:549–555. doi: 10.1038/35080577. [DOI] [PubMed] [Google Scholar]
- 57.Hurley LH. Secondary DNA structures as molecular targets for cancer therapeutics. Biochem. Soc. Trans. 2001;29:692–696. doi: 10.1042/0300-5127:0290692. [DOI] [PubMed] [Google Scholar]
- 58.Neidle S, Parkinson GN. Quadruplex DNA crystal structures and drug design. Biochimie. 2008;90:1184–1196. doi: 10.1016/j.biochi.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 59.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson JE, Smith JS, Kozak ML, Johnson FB. In vivo veritas: using yeast to probe the biological functions of G-quadruplexes. Biochimie. 2008;90:1250–1263. doi: 10.1016/j.biochi.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fry M. Tetraplex DNA and its interacting proteins. Front Biosci. 2007;12:4336–4351. doi: 10.2741/2391. [DOI] [PubMed] [Google Scholar]
- 62.Michelotti EF, Tomonaga T, Krutzsch H, Levens D. Cellular nucleic acid binding protein regulates the CT element of the human c-myc protooncogene. J Biol. Chem. 1995;270:9494–9499. doi: 10.1074/jbc.270.16.9494. [DOI] [PubMed] [Google Scholar]
- 63.Takimoto M, Tomonaga T, Matunis M, Avigan M, Krutzsch H, Dreyfuss G, Levens D. Specific binding of heterogeneous ribonucleoprotein particle protein K to the human c-myc promoter, in vitro. J. Biol. Chem. 1993;268:18249–18258. [PubMed] [Google Scholar]
- 64.Etzioni S, Yafe A, Khateb S, Weisman-Shomer P, Bengal E, Fry M. Homodimeric MyoD preferentially binds tetraplex structures of regulatory sequences of muscle-specific genes. J. Biol. Chem. 2005;280:26805–26812. doi: 10.1074/jbc.M500820200. [DOI] [PubMed] [Google Scholar]
- 65.Todd AK, Neidle S. The relationship of potential G-quadruplex sequences in cis-upstream regions of the human genome to SP1-binding elements. Nucleic Acids Res. 2008;36:2700–2704. doi: 10.1093/nar/gkn078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eddy J, Maizels N. Conserved elements with potential to form polymorphic G-quadruplex structures in the first intron of human genes. Nucleic Acids Res. 2008;36:1321–1333. doi: 10.1093/nar/gkm1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Travers A, Muskhelishvili G. DNA supercoiling – a global transcriptional regulator for enterobacterial growth? Nat. Rev. Microbiol. 2005;3:157–169. doi: 10.1038/nrmicro1088. [DOI] [PubMed] [Google Scholar]
- 68.Peter BJ, Arsuaga J, Breier AM, Khodursky AB, Brown PO, Cozzarelli NR. Genomic transcriptional response to loss of chromosomal supercoiling in Escherichia coli. Genome Biol. 2004;5:R87. doi: 10.1186/gb-2004-5-11-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheung I, Schertzer M, Rose A, Lansdorp PM. Disruption of dog-1 in Caenorhabditis elegans triggers deletions upstream of guanine-rich DNA. Nat. Genet. 2002;31:405–409. doi: 10.1038/ng928. [DOI] [PubMed] [Google Scholar]
- 70.Jinks-Robertson S. The genome's; best friend. Nat. Genet. 2002;31:331–332. doi: 10.1038/ng936. [DOI] [PubMed] [Google Scholar]
- 71.Duquette ML, Huber MD, Maizels N. G-rich proto-oncogenes are targeted for genomic instability in B-cell lymphomas. Cancer Res. 2007;67:2586–2594. doi: 10.1158/0008-5472.CAN-06-2419. [DOI] [PubMed] [Google Scholar]
- 72.Duquette ML, Handa P, Vincent JA, Taylor AF, Maizels N. Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev. 2004;18:1618–1629. doi: 10.1101/gad.1200804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33(Suppl.):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 74.Fazzari MJ, Greally JM. Epigenomics: beyond CpG islands. Nat. Rev. Genet. 2004;5:446–455. doi: 10.1038/nrg1349. [DOI] [PubMed] [Google Scholar]
- 75.Smith SS, Kan JL, Baker DJ, Kaplan BE, Dembek P. Recognition of unusual DNA structures by human DNA (cytosine-5)methyltransferase. J. Mol. Biol. 1991;217:39–51. doi: 10.1016/0022-2836(91)90609-a. [DOI] [PubMed] [Google Scholar]
- 76.Smith SS, Laayoun A, Lingeman RG, Baker DJ, Riley J. Hypermethylation of telomere-like foldbacks at codon 12 of the human c-Ha-ras gene and the trinucleotide repeat of the FMR-1 gene of fragile X. J. Mol. Biol. 1994;243:143–151. doi: 10.1006/jmbi.1994.1640. [DOI] [PubMed] [Google Scholar]
- 77.Henikoff S. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat. Rev. Genet. 2008;9:15–26. doi: 10.1038/nrg2206. [DOI] [PubMed] [Google Scholar]
- 78.Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, Leproust EM, Hughes TR, Lieb JD, Widom J, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, Tomsho LP, Qi J, Glaser RL, Schuster SC, et al. Nucleosome organization in the Drosophila genome. Nature. 2008;453:358–362. doi: 10.1038/nature06929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen K, Meng Q, Ma L, Liu Q, Tang P, Chiu C, Hu S, Yu J. A novel DNA sequence periodicity decodes nucleosome positioning. Nucleic Acids Res. 2008;36:6228–6236. doi: 10.1093/nar/gkn626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Levitsky VG, Podkolodnaya OA, Kolchanov NA, Podkolodny NL. Nucleosome formation potential of eukaryotic DNA: calculation and promoters analysis. Bioinformatics. 2001;17:998–1010. doi: 10.1093/bioinformatics/17.11.998. [DOI] [PubMed] [Google Scholar]
- 82.Huppert JL, Bugaut A, Kumari S, Balasubramanian S. G-quadruplexes: the beginning and end of UTRs. Nucleic Acids Res. 2008;36:6260–6268. doi: 10.1093/nar/gkn511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ji X, Li W, Song J, Wei L, Liu XS. CEAS: cis-regulatory element annotation system. Nucleic Acids Res. 2006;34:W551–W554. doi: 10.1093/nar/gkl322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.