


Abstract
The functional and structural-dynamical properties of the Lys681Ala mutation in the human topoisomerase IB linker domain have been investigated by catalytic assays and molecular dynamics simulation. The mutant is characterized by a comparable cleavage and a strongly reduced religation rate when compared to the wild type protein. The mutant also displays perturbed linker dynamics, as shown by analysis of the principal components of the motion, and a reduced electrostatic interaction with DNA. Inspection of the inter atomic distances in proximity of the active site shows that in the mutant the distance between the amino group of Lys532 side chain and the 5′ OH of the scissile phosphate is longer than the wild type enzyme, providing an atomic explanation for the reduced religation rate of the mutant. Taken together these results indicate the existence of a long range communication between the linker domain and the active site region and points out the crucial role of the linker in the modulation of the catalytic activity.
INTRODUCTION
DNA topoisomerase IB catalyzes the relaxation of supercoiled DNA through the transient cleavage of one strand of a DNA duplex and is of fundamental importance to processes such as replication, recombination and transcription (1–3). The enzyme is found in monomeric form in all eukaryotic systems (except for the unusual bi-subunit enzyme from kinetoplastid protozoan parasite Leishmania donovani), and its presence has been reported also in bacteria, and viruses (4–7).
The human topoisomerase IB enzyme (Top1) is composed of 765 amino acids and has four distinct domains: the NH2-terminal domain (1–214), the core domain (215–635), the linker domain (636–712) and the COOH-terminal domain (713–765) (8–10). The three-dimensional structure of reconstituted N-terminal truncated versions of human Top1 in complex with a 22 bp DNA molecule shows the enzyme organized in multiple domains which ‘clamp on’ the DNA molecule (9). Changes in DNA topology are achieved by introducing a transient break in the phosphodiester bond of one strand in the duplex DNA. Phosphodiester bond energy is preserved during catalysis through the formation of a transient covalent phosphotyrosine bond between the catalytic Tyr723 and the 3′-end of the broken DNA strand. DNA relaxation has been proposed to proceed via ‘controlled rotation’: the catalytic tyrosine holds one end of the DNA duplex, and the enzyme then accompanies the end downstream of the cleavage site to rotate around the remaining phosphodiester bond (10).
Human Top1 is of significant medical interest being the only target of the antitumor agent camptothecin (CPT). CPT reversibly binds to the covalent intermediate DNA-enzyme, stabilizing the cleavable complex and reducing the rate of religation. The stalled topoisomerase I collides with the progression of the replication fork producing lethal double strand DNA breaks and cell death (1,11).
A crucial role in the catalytic mechanism is played by the linker domain, that has been proposed to be involved in the relaxation reaction, acting as a ‘brake’ during the rotation of the DNA, downstream of the cleavage site (12,13). The linker has been shown to be in direct contact with the DNA since it is more resistant to proteolysis when the enzyme is non-covalently bound to duplex DNA (8) and it is one of the most flexible protein regions, as evidenced by multiple non-isomorphous crystal structures (12). Upon removal of the linker domain the enzyme retains its activities in vitro but passes from a processive to a distributive DNA relaxation (13), suggesting the loss of interdomain communication as also indicated by an all atom molecular dynamics (MD) simulation of the enzyme in the presence or absence of the linker (14). Actually, it has been shown that even the single mutation of Thr729 in Lys abolishes the protein communications between the C-terminal and the linker domain, altering the interactions between helix 17 in the core domain and helix 19 in the linker domain (15,16). Interdomain communications between the linker and other protein domains have been recently confirmed by means of a long time scale coarse grained molecular dynamics simulation (17), while non-equilibrium molecular dynamics simulations have identified distinct mechanisms for positive and supercoiled DNA relaxation, where the linker domain plays an essential role (18). Finally, it has been reported that the linker must retain a minimum length in order to have a fully functional enzyme (19).
Top1 sensitivity to CPT is modulated by several mutations (20–22), most of them are located in the proximity of the active site and make the enzyme insensitive to CPT lowering the enzyme drug binding. Interestingly, also mutations located far away from the active site and in particular in the linker have been found to modulate the enzyme sensitivity toward the drug, confirming the important role played by this domain. In particular mutation of residue 653, from Ala to Pro has been shown to render the enzyme resistant to CPT and to confer an enhanced linker flexibility correlated to an increased religation rate that does not permit CPT to stabilize the enzyme–DNA covalent complex (23). Mutations of the linker domain have been shown also to increase the CPT enzyme sensitivity confirming the occurrence of long range communication between the linker domain and the active site region (24). The linker can also feel perturbations introduced in proximity of the active site, since mutation of Thr718, close to the active site Tyr723, induces altered linker flexibility (25). Evidence of a correlation between these two regions comes also from the absence of the linker domain electron density map in the enzyme–DNA binary but not in the enzyme–DNA–CPT ternary complex (20). Additional evidence for a long range communication between these two domains comes from the double mutant Ala653Pro-Thr718Ala, involving two residues, Ala653 and Thr718, located on the linker domain and in proximity of the active site, respectively. The combination of the two mutations abolishes the lethal phenotype of the single mutant Thr718Ala, demonstrating how a mutation on the linker domain can influence the active site region (26).
In the present work we have continued our study of the role of linker domain, mutating the highly conserved residue Lys681, located on the tip of the linker, in Ala. Experimental characterization of the mutant indicates a reduced religation rate when compared to the wild type protein. MD simulation indicates that the linker is characterized by a decrease in flexibility correlated with a reduced linker-DNA electrostatic interaction. The simulation also points out an increased distance between Lys532 and the 5′ OH of the +1 base phosphate; this makes the proton transfer needed in the religation reaction more difficult and provides an atomistic hypothesis for the experimentally observed reduced religation rate.
MATERIALS AND METHODS
Yeast strains, plasmids and chemicals
Camptothecin (Sigma) was dissolved in dimethyl sulfoxide (Me2SO) to a final concentration of 4 mg/ml and stored at −20°C. Anti-FLAG M2 affinity gel, FLAG peptide and M2 monoclonal antibody were purchased from Sigma. Saccharomyces cerevisiae strain EKY3 (ura3-52, his3Δ200, leu2Δ1, trp1Δ63, top1::TRP1, MATα) was previously described (27). Plasmid YCpGAL1- wild type was described previously (28). Lys681Ala was generated by oligonucleotide-directed mutagenesis of the YCpGAL1-wild type in which the human topoisomerase I is expressed under the galactose inducible promoter in a single-copy plasmid. The epitope-tagged construct YCpGAL1-e-wild type contains the N-terminal sequence FLAG: DYKDDDY (indicated with ‘e’), recognized by the M2 monoclonal antibody. The epitope-tag was subcloned into YCpGAL1-Lys681Ala to produce the YCpGAL1-e-Lys681Ala construct.
Purification of DNA topoisomerase I
Epitope tagged EKY3 cells were transformed with YCpGAL1-e-wild type and YCpGAL1-e-Lys681Ala, grown on SC-uracil plus 2% dextrose and diluted 1 : 100 in SC-uracil plus 2% raffinose. At an optical density A595 of 1.0, the cells were induced with 2% galactose for 6 h. Cells were then harvested by centrifugation, washed with cold water and resuspended in 2 ml buffer/g cells using a buffer containing 50 mM Tris, pH 7.4, 1 mM EDTA, 1 mM ethylene glycol-bis(2-aminoethylether)-N,N,N',N'-tetraacetic acid, 10% (v/v) glycerol completed with protease inhibitors cocktail (Roche 1836153) and supplemented with 0.1 mg/ml sodium bisulfite and 0.8 mg/ml sodium fluoride. After addition of 0.5 volume of 425–600 µm diameter glass beads the cells were disrupted by vortexing for 30 s alternating with 30 s on ice. The lysate was centrifuged and KCl final concentration 0.15 M was added to the sample prior to loading onto 2 ml ANTI-FLAG M2 Affinity Gel column equilibrated as described in the technical bulletin (Sigma). The column was washed with 20 column volumes of TBS (50 mM Tris–HCl, 150 mM KCl, pH 7.4). Elution of FLAG-fusion topoisomerase I was performed by competition with five column volumes of a solution containing 100 µg/ml FLAG peptide in TBS. Fractions of 500 µl were collected and glycerol final concentration 40% was added; all preparations were stored at −20°C. The fractions were resolved by SDS-polyacrylamide gel electrophoresis; protein concentration and integrity were measured through immunoblot assay, using the epitope-specific monoclonal antibody M2. After normalization to the same amount of protein, the activity of the wild type and mutant DNA topoisomerase I, as assayed by relaxation of supercoiled DNA in 150 mM KCl, was found to be almost identical. In all experiments the same amount of wild type and mutated protein was used.
Kinetics of cleavage using oligonucleotide substrate
Oligonucleotide substrates CL14 (5′-GAAAAAAGACTTAG-3′) was radiolabeled with [γ32P] ATP at their 5′-end. The CP25 complementary strand (5′-TAAAAATTTTTCTAAGTCTTTTTTC-3′) was phosphorylated at its 5′-end with unlabeled ATP. CL14/CP25 was annealed as previously described (18). The suicide cleavage reactions were carried out by incubating 20 nM of the duplex with an excess of enzyme in 20 mM Tris pH 7.5, 0.1 mM Na2EDTA, 10 mM MgCl2, 50 µg/ml acetylated BSA and 150 mM KCl at 23°C in a final volume of 50 µl as described by Yang and Champoux (29). A 5 µl sample of the reaction mixture was removed before addition of the protein and used as the zero time point. At various time points 5 µl aliquots have been removed and the reaction stopped with 0.5% SDS. After ethanol precipitation samples were resuspended in 5 µl of 1 mg/ml trypsin and incubated at 37°C for 30 min. Samples have been analyzed by denaturing urea/polyacrylamide gel electrophoresis. The percentage of CL1 was determined by PhosphorImager and ImageQuant software and normalized on the total amount of radioactivity in each lane.
Kinetics of religation using oligonucleotide substrate
Twenty nanomolar CL14/CP25, was incubated with an excess of enzyme for 60 min at 23°C followed by 30 min at 37°C in 20 mM Tris pH 7.5, 0.1 mM Na2EDTA, 10 mM MgCl2, 50 µg/ml acetylated BSA, and 150 mM KCl. Religation reactions were initiated by adding a 200-fold molar excess of R11 oligonucleotide (5′-AGAAAAATTTT-3′) over the duplex CL14/CP25 in the presence or absence of 50 µM CPT. At 37°C a various time point 5 µl aliquots were removed and the reaction stopped with 0.5% SDS. After ethanol precipitation samples were resuspended in 5 µl of 1 mg/ml trypsin and incubated at 37°C for 30 min. Samples were analyzed by denaturing urea/polyacrylamide gel electrophoresis. The percentage of religation was determined by PhosphorImager and ImageQuant software and normalized on the total amount of radioactivity in each lane.
Cleavage/religation equilibrium
Oligonucleotide CL25 (5′-GAAAAAAGACTTAGAAAAATTTTTA-3′) has been radiolabeled with [γ32P] ATP at its 5′-end. The CP25 complementary strand (5′-TAAAAATTTTTCTAAGTCTTTTTTC-3′) was phosphorylated at its 5′-end with unlabeled ATP. The two strands were annealed at a 2-fold molar excess of CP25 over CL25. A final concentration of 20 nM duplex CL25/CP25 was incubated with an excess of enzyme at 25°C in 20 mM Tris pH 7.5, 0.1 mM Na2EDTA, 10 mM MgCl2, 50 µg/ml acetylated BSA, and 50 or 150 mM KCl, in the presence or absence of 50 µM CPT. After 30 min, the reaction was stopped by adding 0.5% SDS and digested with trypsin after ethanol precipitation. Reaction products were resolved in 16% acrylamide-7 M urea gels.
MD
The initial configuration of Top1, in covalent complex with a 22 bp long linear double helix DNA substrate, has been modeled from the crystal structures 1K4T and 1K4S, (20). The starting positions for residues 201–631 and 708–765 are obtained from the 1K4S crystal structure and those for residues 632–707 from the 1K4T crystal structure (since the linker domain is not resolved in the former), following a mass-weighted fit of backbone atoms on 1K4S (RMSD between the two structures is 0.7 Å after the fit). Lysine 681 was mutated to alanine using the rotamer module present in the Chimera package (30). All the simulations were carried out with the GROMACS MD package version 3.3.3 (31) modeling the systems with the AMBER03 all atom force-field (using the porting of AMBER03 force field implemented by Sorin and Pande and available on the ffamber project web site) (32,33). The Top1-DNA complexes were placed in a rectangular box (93 × 110 × 130 Å3) filled with TIP3P water molecules (34), rendered electro neutrals by the addition of 23 or 24 Na+ counterions, for the wild type and for the Lys681Ala mutant respectively, using the genion program of the GROMACS package, that replaces solvent molecules by ions at the position with the most favorable electrostatic potential. The resulting total systems contained 9456 protein atoms, 1404 DNA atoms, 23 Na+ counterions and 40705 water molecules for the wild type, and 9444 protein atoms, 1404 DNA atoms 24 Na+ counterions and 40705 water molecules for the Lys681Ala mutant. Optimization and relaxation of solvent and ions was initially performed keeping the protein and DNA atoms constrained to their initial position with a force constant of 1000 kJ/(mol nm), for 2000 ps. The systems was then simulated for 15 ns in periodic boundary conditions, at a constant temperature of 300 K using the Berendsen's; method and at a constant pressure of one bar (35). The pressure was kept constant (1 bar) using the Rahman–Parrinello barostat (36) and with a 2 fs time step. Pressure and temperature coupling constants are 1 and 0.1 ps, respectively. The electrostatic interactions were taken into account by means of the Particle Mesh Ewald method (37), using a cut-off radius of 1.0 nm in real space, while a cut-off radius of 1.0 nm was chosen for the van der Waals interactions. The lengths of all bonds were kept constant with the LINCS algorithm (38). The neighbor list was updated every 10 steps. Direct hydrogen bonds, root mean square deviations (RMSD) and fluctuations (RMSF) and principal component analysis were carried out using the GROMACS MD package version 3.3.3. Figure 8 was created using the CHIMERA (30) and VMD (39) visualization packages, respectively; graphs were obtained with the Grace program (http://plasma-gate.weizmann.ac.il/Grace/). Secondary structure assignment was performed by means of the dssp program (40). Electrostatic interaction energy between the linker domain and the DNA or the other protein domains were calculated performing reruns of the trajectories with GROMACS, using a cut-off of 40 Å for the pairs interactions. Multiple sequence alignment was carried out with the TCOFFEE server (41).
Figure 8.
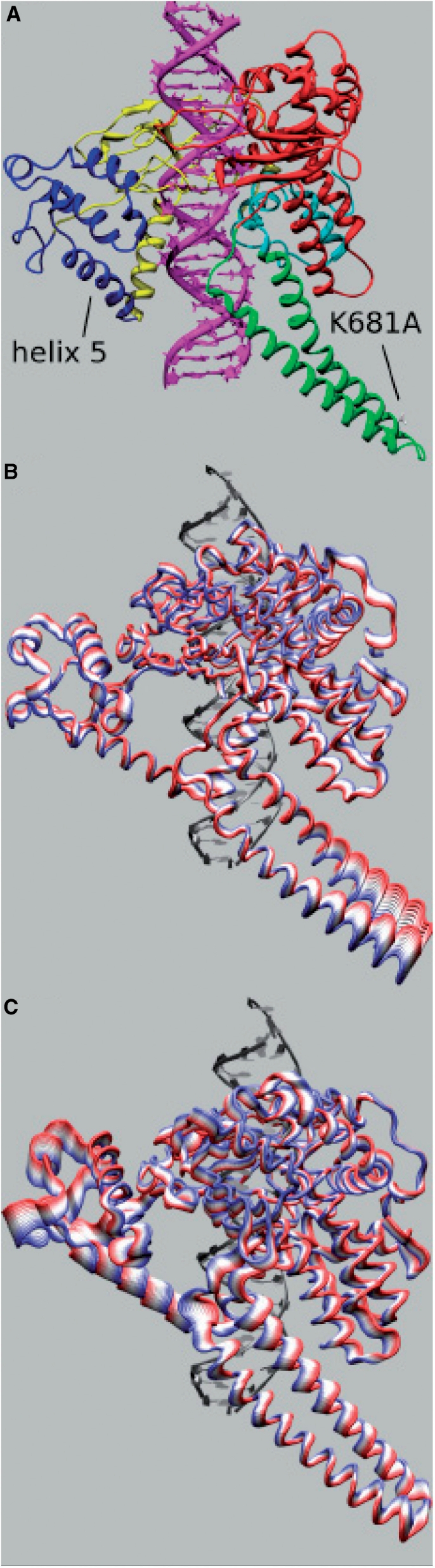
Three-dimensional structure of the Top1-DNA binary complex. The protein subdomain I, subdomain II, subdomain III are rappresented in blue, yellow and red respectively, the linker in green and C-terminal in cyan. The lateral chain of the mutated Lys681 residue is represented in ball and stick and evidenced by a line. Nose cone helix 5 is also evidenced by a line (A). Representation of ten projections, in different colors, of the MD motions along the second eigenvector for the wild-type protein (B) and Lys681Ala mutant (C).
RESULTS AND DISCUSSION
Lys681 is conserved in topoisomerases from different organisms
Gene sequences from topoisomerases IB from different organisms (Cenarchaeum symbiosum, Caenorhabditis elegans, Xenopus laevis, Cricetulus griseus, Mus musculus, Rattus norvegicus, Cercopithecus aethiops, Homo sapiens, Arabidopsis thaliana and Camptotheca acuminata) were multialigned. The alignment is shown only for the active site residues (Arg488, Lys532, Arg590, His632, Tyr723 for the human isoform) and the linker domain (Figure 1). The five active site residues are fully conserved in all the organisms, together with the flanking residues. In the linker domain only three residues are conserved in all the organisms: Lys638 (located in the loop connecting the core subdomain III with the linker domain), Ala668 and Lys681 (located in helices 18 and 19, respectively). This latter residue is likely to play a crucial role because of its position almost on the tip of the linker and because of its positive charge.
Figure 1.

Multiple sequence alignment of the topoisomerase IB sequences extracted from C. symbiosum, C. elegans, X. laevis, C. griseus, M. musculus, R. norvegicus, C. aethiops, H. sapiens, A. thaliana and C. acuminata, respectively. The alignment of the residues in proximity of the catalytic pentad (Arg488, Lys532, Arg590, His632, Tyr723 for the human isoform) and along the full linker domain is reported. Residues conserved in all sequences are highlighted by a grey column and evidenced by an asterisk. The residues belonging to the linker domain are underlined. The mutated residue Lys681 is evidenced by a black triangle.
Cleavage–religation equilibrium of the wild type and Lys681Ala mutant
The gel of the native and the Lys681Ala mutant Top1 corresponding to the cleavage–religation equilibrium experiment with the 25-mer full duplex oligonucleotide substrate CL25 (5′-GAAAAAGACTTAGAGAAAAATTTT-3′)/CP25 at medium (150 mM KCl) and low (50 mM KCl) ionic strength, in the absence or presence of 50 µM CPT is reported in Figure 2. At 150 mM KCl and in the absence of CPT (lanes 3 and 7) the intensity of the band due to the cleavable complex is identical for both enzymes indicating that in this condition the cleavage/religation equilibrium is shifted toward religation for both enzymes. Addition of CPT (lanes 4 and 8) strongly shifts the equilibrium toward cleavage as indicated by the increase of the intensity of the band diagnostic of the presence of the cleavable complex (11). At 50 mM KCl the equilibrium of the Lys681Ala is shifted toward cleavage when compared to the wild type enzyme (compare lane 5 with lane 1). Addition of CPT shifts the equilibrium toward cleavage for the wild type and to a lower extent for the mutated enzyme where the equilibrium is already shifted toward cleavage also in the absence of CPT (lanes 2 and 6).
Figure 2.

Cleavage/religation equilibrium of the full duplex DNA substrate. Gel electrophoresis of the products coming from the incubation of the wild type topoisomerase I with the [γ-32P] end-labelled duplex DNA, shown at the top of the figure in the absence and presence of CPT and at different KCl concentrations. The arrow at the DNA sequence indicates the CL1 preferred cleavage site. Lane 1–4 (wild type), lane 5–8 (Lys681Ala). The asterisk indicates the band corresponding to the CL1 site.
Since the cleavage–religation equilibrium constant (Keq), for a linear DNA substrate is determined by the ratio of the rate of cleavage (kcl) over the rate of religation (krl), the increase in the cleavable complex band (Figure 2, lane 5) indicates that Lys681Ala mutant when compared to the wild type (Figure 2, lane 1) either cleaves DNA more efficiently or is less efficient in the DNA religation step. To clarify this point the single steps of the enzyme catalytic cycle have been analyzed.
Kinetics of cleavage of the wild type and Lys681Ala mutant
The time course of the cleavage of the wild type and Lys681Ala mutant has been followed using a suicide cleavage substrate. In detail, a 5′-end radiolabeled oligonucleotide CL14 (5′-GAAAAAAGAC * TT↓AG-3′) has been annealed to the CP25 (5′-TAAAAATTTTTCTAAGTCTTTTTTC-3′) complementary strand, to produce a duplex with an 11-base 5′-single-strand extension. The enzyme preferentially cuts at the site indicated by the arrow and to a lower extent at the site indicated by the asterisk. In both cases the religation step is precluded because the AG-3′ or TTAG-3′ oligonucleotides are too short to be religated, leaving the enzyme covalently attached to the 12 or 10 oligonucleotide 3′-end.
For a suicide substrate incubated with an excess of wild type or mutant enzyme, the cleaved DNA fragments have been resolved in a time course experiment in a denaturing polyacrylamide gel and are reported in Figure 3A. The amount of fragment, normalized to the plateau value of the wild type and Lys681Ala protein, plotted as a function of time in Figure 3B, shows that both enzymes have an almost identical cleavage rate (kcl), reaching the plateau in a comparable time.
Figure 3.

Suicide cleavage experiment for the wild type and Lys681Ala mutant. (A) Time course of the suicide cleavage reaction carried out with the substrate described on the top of the figure. CL1 and CL2 identify the cleaved complexes at the site indicated by the arrow and the asterisk respectively. Lane C, no protein added. (B) Percentage of the DNA substrate cleaved plotted at different time for the wild type (black line) and the Lys681Ala mutant (red line).
Kinetics of religation of the wild type and Lys681Ala mutant
The DNA religation step has been studied by testing the ability of both enzymes to religate the oligonucleotide R11 (5′-AGAAAAATTTT-3′) added to the cleaved suicide substrate incubated with an excess of enzyme in the presence or absence of CPT. Aliquots have been removed at different times, the reaction stopped by addition of SDS and the products analyzed by polyacrylamide gel electrophoresis (Figure 4A). The small percentage of religation product formed, plotted as a function of time in Figure 4B demonstrates that the Lys681Ala mutant shows a strong decrease in the religation rate when compared to the wild type protein. The religation rate decrease is so sharp that addition of CPT does not have relevant effects, at variance on what observed for the wild-type protein (Figure 4B).
Figure 4.

Religation experiment for the wild type and Lys681Ala mutant. (A) Time course of the religation experiment, carried out with the substrate described on the top of the figure, between the R11 substrate and the wild type or Lys681Ala covalent complexes, in the absence or presence of 50 µM CPT. The R11 oligonucleotide is selectively religated to the CL1. (B) Percentage of the religation plotted at different time for the wild type and Lys681Ala (black and red lines, respectively), in absence (full line) and presence (dashed line) of CPT.
Structural long range effects of the Lys681Ala mutation
The root mean square deviation (RMSD) of the native and mutated protein, calculated after a mass-weighted superposition on the starting structure, is shown in Figure 5A, as a function of simulation time. The trajectory of the wild type protein (full black line) shows a higher deviation from the starting structure as compared to the trajectory of the Lys681Ala mutant (full red line) (up to 4.5 and 3.0 Å for the wild type and Lys681Ala, respectively). However, upon elimination of the linker domain atoms contribution, the two systems show a comparable behavior and the amplitude of their deviations is similar. For both enzymes the RMSD reaches a plateau after 3000 ps, and therefore all the following analyses have been carried out on the last 12 000 ps (i.e. from 3 to 15 ns). The plot of Figure 5A indicates that the linker is the most deviating domain in both systems, but its contribution to the overall enzyme mobility is higher for the native protein, as compared to that of the Lys681Ala mutant.
Figure 5.

(A) RMSD from the starting structure plotted as a function of simulation time. The RMSD of the native protein and the Lys681Ala mutant are shown in black and red full lines, respectively. The RMSD calculated eliminating the linker domain are represented in black and red dotted lines for the wild type and Lys681Ala mutant, respectively. (B) Average per-residue RMSF represented as a function of the residue number for the wild type Top1 protein (full black line) and Lys681Ala mutant (dashed red line). Subdomain boundaries are shown as vertical black dot-dashed lines.
Figure 5B shows the per residue root mean square fluctuations (RMSF) of the two enzymes. Similar fluctuations are observed in the two trajectories, except in the linker domain and in the core subdomain I region (residues 201–214) where the native protein is more fluctuating than the Lys681Ala mutant. RMSF of the DNA bases in the two systems are similar in the scissile strand (Figure 6A) while the base −1, in the DNA intact strand, shows an increase of fluctuation (Figure 6B) due to the loss of the direct interaction between the DNA backbone and the lateral chain of Lys425, present in the wild type system (data not shown) (42).
Figure 6.

Average per-base RMSF as a function of the base number for the DNA in the wild type (full black line) and in the Lys681Ala mutant (dashed red line) Top1 DNA system. (A) scissile DNA strand. (B) Intact DNA strand.
The principal component analysis (PCA) has been applied to both the mutated and the native enzyme trajectories, in order to highlight protein concerted motions. The analysis is based on the diagonalization of the covariance matrix built from the atomic fluctuations, after the removal of the translational and rotational movements, and allows to identify the main 3N directions along which the majority of the protein motion is defined (43,44). PCA analysis has been carried out on the 565 Cα atoms of the protein. Up to 80% of total native enzyme motion is taken into account by the first 15 eigenvectors (among the 565 that are obtained from the covariance matrix diagonalization), while 20 eigenvectors are needed for the Lys681Ala mutant. The displacement of each Cα along the first eigenvector is shown in Figure 7A. The linker domain is the protein region with the maximum displacement and no differences along this direction are found between mutated and wild type proteins. Small differences are observed in the N-terminal and C-terminal regions of the core domain that show a slightly larger and a slightly smaller displacements respectively in the wild type than in the mutant enzyme.
Figure 7.

Displacements of each Cα atom along the first two eigenvectors are represented in black continuous lines and dashed red lines for the wild type and Lys681Ala mutant, respectively. (A) Displacements along the first eigenvector. (B) Displacements along the second eigenvector.
Figure 7B shows the Cα displacements along the second eigenvector. The main difference is found for the linker domain that shows a much higher displacement in the native enzyme than the mutated protein, where this region has a displacement of the same magnitude as the other domains. Some differences are also observed in the C-terminal domain that is more mobile in the wild type, while the core subdomains show overall comparable displacements, with the exception of helix 5 region (residues 303–319) in core subdomain II, one of the so called ‘nose cone’ helices, that is more mobile in the mutated enzyme. The loss of linker domain mobility can be appreciated looking at Figures 8B (native enzyme) and 8C (Lys681Ala mutant), where 10 projections of the protein motions along the second eigenvector are shown with different colors. The principal component analysis indicates that the regions more altered in their dynamics, as a result of the single mutation, are those contacting the DNA scissile strand, which therefore play a role during the DNA relaxation process. In particular, even though helix 5 has comparable average per residue RMSF in the two systems (Figure 5B), its fluctuations in the mutant are lower along the first eigenvector and higher along the second eigenvector, as can be well appreciated looking at motion projections along the second eigenvector (Figure 8).
Electrostatic long range perturbation
Even though it is well known that several single residue mutations, such as Ala653Pro, Thr718Ala, Glu418Lys, and Thr729Ala, have long range effects on the complex topoisomerase machinery (15,23,25,45), this is the first example in which the causes of the interdomain communication are not easily attributable to a direct effect, such as the alteration of direct or water mediated hydrogen bonds. Moreover secondary structure assignment shows that mutation of residue 681 from lysine to alanine does not cause alterations in the linker domain secondary structure (Figure 10 in the Supplementary Data) and the main chain conformation of the mutated residue does not significantly change in relation with the neighbor residues, when compared to the wild type protein. We have therefore investigated the alteration in the protein electrostatic field, caused by the elimination, in the mutant, of the positively charged residue. The average electrostatic interaction energy between the linker and the DNA, calculated for all the trajectories is about −10 200 kJ/mol in the wild type enzyme, while it decreases to −8600 kJ/mol in the Lys681Ala mutant (Figure 11 in the Supplementary Data). The elimination of the single highly conserved positive charge of Lys681, therefore, produce a reduction of 16% of the linker-DNA interaction energy. The electrostatic interaction between the linker and any other protein domains, on the contrary, does not show any significant difference between the native and the mutated enzyme.
Perturbation of active site architecture
The structure of the catalytic pentad is very stable since it maintains the same definite geometrical arrangement over the trajectory in both simulations. This result can be appreciated overlapping the active site Cα atoms of the last snapshots of the two simulations. The Cα atoms RMSD of these two configurations is 0.50 Å and the largest deviation from the crystallographic structure from any configuration sampled during the trajectories is 0.97 and 0.91 Å for the native and mutated simulation respectively.
An interesting difference is observed when comparing the distance between the O5′ oxygen atom on the guanosine in position +1 on the cleaved strand and the highly conserved Lys532 Nε atom (Figure 1). This distance is reported as a function of simulation time for the native and mutant enzyme in Figure 9. After the thermalization and equilibration phases, the two systems are in quite different conformations, the amino group of the lysine residue in the mutant being closer to the nucleotide by about 2.5 Å when compared to the wild type enzyme. During the trajectory the two enzymes behave in a completely different way: after 5 ns Lys532 in the native enzyme starts to get closer to the O5′ guanosine (reaching an average distance of 6 Å), while the opposite occurs in Lys681Ala where the same residue moves away from the O5′ guanosine group, (reaching an average distance of 8.5 Å). These last distance values are reached after 7.5 of simulation time and fully maintained afterwards. Lys532 has been proposed to act as a general acid catalyst in the cleavage phase, donating a proton to the leaving strand nucleotide, and as a general base catalyst in the religation step, accepting a proton from the same nucleotide (46–48). This residue has therefore a crucial role in the catalytic reaction. The longer average distance between the O5′ atom of the DNA guanosine +1 and Lys532 lateral chain, observed in the mutant system, constitutes a relevant obstacle to the proton transfer in the religation phase and provides a molecular explanation for the altered religation rate, experimentally observed for the Lys681Ala mutant.
Figure 9.

Distance between the O5′ atom of the nucleotide in position +1 on the cleaved strand and the Nε atom of the Lys532 residue calculated as a function of time. Wild type and Lys681Ala – DNA distances are shown as full black and red lines, respectively.
CONCLUSIONS
The linker domain has a crucial role in the control of the catalytic cycle of human topoisomerase I. Such a finding was initially proposed by Champoux and co-workers ten years ago, when they demonstrated that reconstitution of an enzyme, made by the core domain plus a COOH-terminal fragment containing the complete linker region, renders the linker non-functional and modulates the religation rate (13). Since then several papers appeared showing through experimental and computation approaches the interdependence between the linker and the active site region (14,23,26).
In this work we show how mutation of a residue Lys681, located on the tip of the linker domain and that apparently should not have any role in the functional properties of the enzyme, dramatically reduces the religation rate. The mutant is characterized by an altered dynamics of the linker and a varied linker-DNA interaction energy, correlated with a lengthening of the +1 base O5′-Lys532 distance and thus providing an atomic explanation for the reduced religation rate. These results demonstrate the crucial role played by this domain and the occurrence of a long range communication between the linker and the active site region.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Grants from AIRC (Associazione Italiana Ricerca Cancro). Funding for open access charge: Associazione Italiana Ricerca Cancro.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank S. Z. Pedersen for critical reading. The experimental contribution of A. Galbiati in the laboratory is also acknowledged. They acknowledge CASPUR Supercomputing Consortium for computational resources.
REFERENCES
- 1.Chen AY, Liu LF. DNA topoisomerases: essential enzymes and lethal targets. Ann. Rev. Pharmacol. Toxicol. 1994;34:191–218. doi: 10.1146/annurev.pa.34.040194.001203. [DOI] [PubMed] [Google Scholar]
- 2.Nitiss JL. Investigating the biological functions of DNA topoisomerases in eukaryotic cells. Biochim. Biophys. Acta. 1998;1400:63–82. doi: 10.1016/s0167-4781(98)00128-6. [DOI] [PubMed] [Google Scholar]
- 3.Wang JC. DNA topoisomerases. Ann. Rev. Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- 4.Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Ann. Rev. Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 5.Das B, Sen N, Ganguly A, Majumder HK. Reconstitution and functional characterization of the unusual bi-subunit type I DNA topoisomerase from Leishmania donovani. FEBS Lett. 565:81–88. doi: 10.1016/j.febslet.2004.03.078. [DOI] [PubMed] [Google Scholar]
- 6.Krogh B, Shuman S. A poxvirus-like type IB topoisomerase family in bacteria. Proc. Natl. Acad. Sci. 2002;99:1853–1858. doi: 10.1073/pnas.032613199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shuman S, Moss B. Identification of a vaccinia virus gene encoding a type I DNA topoisomerase. Proc. Natl Acad. Sci. 1987;84:7478–7482. doi: 10.1073/pnas.84.21.7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stewart L, Ireton GC, Parker LH, Madden KR, Champoux JJ. Biochemical and biophysical analysis of recombinant forms of human DNA topoisomerase I. J. Biol. Chem. 1996;271:7593–7601. doi: 10.1074/jbc.271.13.7593. [DOI] [PubMed] [Google Scholar]
- 9.Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WGJ. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science. 1998;279:1504–1513. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- 10.Stewart L, Redinbo MR, Qiu X, Hol WGJ, Champoux JJ. A model for the mechanism of human topoisomerase I. Science. 1998;279:1534–1541. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- 11.Pommier Y, Pourquier P, Fan Y, Strumberg D. Mechanism of action of eukaryotic DNA topoisomerase I and drugs targeted to the enzyme. Biochim. Biophys. Acta. 1998;1400:83–106. doi: 10.1016/s0167-4781(98)00129-8. [DOI] [PubMed] [Google Scholar]
- 12.Redinbo MR, Stewart L, Champoux JJ, Hol WG. Structural flexibility in human topoisomerase I revealed in multiple non-isomorphous crystal structures. J. Mol. Biol. 1999;292:685–696. doi: 10.1006/jmbi.1999.3065. [DOI] [PubMed] [Google Scholar]
- 13.Stewart L, Ireton GC, Champoux JJ. A functional linker in human topoisomerase I Is required for maximum sensitivity to camptothecin in a DNA relaxation assay. J. Biol. Chem. 1999;274:32950–32960. doi: 10.1074/jbc.274.46.32950. [DOI] [PubMed] [Google Scholar]
- 14.Chillemi G, Redinbo M, Bruselles A, Desideri A. Role of the linker domain and the 203–214 N-terminal residues in the human topoisomerase I DNA complex dynamics. Biophys. J. 2004;87:4087–4097. doi: 10.1529/biophysj.104.044925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chillemi G, D’Annessa I, Fiorani P, Losasso C, Benedetti P, Desideri A. Thr729 in human topoisomerase I modulates anti-cancer drug resistance by altering protein domain communications as suggested by molecular dynamics simulations. Nucleic Acids Res. 2008;36:5645–5651. doi: 10.1093/nar/gkn558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Losasso C, Cretaio E, Fiorani P, D’Annessa I, Chillemi G, Benedetti P. A single mutation in the 729 residue modulates human DNA topoisomerase IB DNA binding and drug resistance. Nucleic Acids Res. 2008;36:5635–5644. doi: 10.1093/nar/gkn557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szklarczyk O, Staron K, Cieplak M. Native state dynamics and mechanical properties of human topoisomerase I within a structure-based coarse-grained model. Proteins. 2009;77:420–431. doi: 10.1002/prot.22450. [DOI] [PubMed] [Google Scholar]
- 18.Sari L, Andricioaei I. Rotation of DNA around intact strand in human topoisomerase I implies distinct mechanisms for positive and negative supercoil relaxation. Nucleic Acids Res. 2005;33:6621–6634. doi: 10.1093/nar/gki935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frøhlich R, Juul S, Nielsen MB, Vinther M, Veigaard C, Hede MS, Andersen FF. Identification of a minimal functional linker in human topoisomerase I by domain swapping with Cre recombinase. Biochemistry. 2008;47:7127–7136. doi: 10.1021/bi800031k. [DOI] [PubMed] [Google Scholar]
- 20.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl Acad. Sci. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pommier Y, Pourquier P, Yoshimasa U, Wu J, Laco GS. Topoisomerase I inhibitors: selectivity and cellular resistance. Drug Resist. Updates. 1999;2:307–318. doi: 10.1054/drup.1999.0102. [DOI] [PubMed] [Google Scholar]
- 22.Chrencik JE, Staker BL, Burgin AB, Pourquier P, Pommier Y, Stewart L, Redinbo MR. Mechanisms of camptothecin resistance by human topoisomerase I mutations. J. Mol. Biol. 2004;339:773–784. doi: 10.1016/j.jmb.2004.03.077. [DOI] [PubMed] [Google Scholar]
- 23.Fiorani P, Bruselles A, Falconi M, Chillemi G, Desideri A, Benedetti P. Single mutation in the linker domain confers flexibility and camptothecin resistance to human topoisomerase I. J. Biol. Chem. 2003;278:43268–43275. doi: 10.1074/jbc.M303899200. [DOI] [PubMed] [Google Scholar]
- 24.Scaldaferro S, Tinelli S, Borgnetto ME, Azzini A, Capranico G. Directed evolution to increase camptothecin sensitivity of human DNA topoisomerase I. Chem. Biol. 2001;8:871–881. doi: 10.1016/s1074-5521(01)00059-x. [DOI] [PubMed] [Google Scholar]
- 25.Chillemi G, Fiorani P, Castelli S, Bruselles A, Benedetti P, Desideri A. Effect on DNA relaxation of the single Thr718Ala mutation in human topoisomerase I: a functional and molecular dynamics study. Nucleic Acids Res. 2005;33:3339–3350. doi: 10.1093/nar/gki642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Losasso C, Cretaio E, Palle K, Pattarello L, Bjornsti MA, Benedetti P. Alterations in linker flexibility suppress DNA topoisomerase I mutant-induced cell lethality. J. Biol. Chem. 2007;282:9855–9864. doi: 10.1074/jbc.M608200200. [DOI] [PubMed] [Google Scholar]
- 27.Bjornsti MA, Benedetti P, Viglianti GA, Wang JC. Expression of human DNA topoisomerase I in yeast cells lacking yeast DNA topoisomerase I: restoration of sensitivity of the cells to the antitumor drug camptothecin. Cancer Res. 1989;49:6318–6323. [PubMed] [Google Scholar]
- 28.Kauh EA, Bjornsti MA. CT1 mutants suppress the camptothecin sensitivity of yeast cells expressing wild-type DNA topoisomerase I. Proc. Natl Acad. Sci. 1995;92:6299–6303. doi: 10.1073/pnas.92.14.6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Z, Champoux JJ. Reconstitution of enzymatic activity by the association of the cap and catalytic domains of human topoisomerase I. J. Biol. Chem. 2002;277:30815–30823. doi: 10.1074/jbc.M205302200. [DOI] [PubMed] [Google Scholar]
- 30.Pettersen E, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera – a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 31.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 32.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 33.Sorin E, Pande VS. Empirical force-field assessment: the interplay between backbone torsions and noncovalent term scaling. J. Comput. Chem. 2005;26:682–690. doi: 10.1002/jcc.20208. [DOI] [PubMed] [Google Scholar]
- 34.Jorgensen WL, Chandrasekhar J, Madura JD, Impery RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 35.Berendsen HJC, Postma JPM, van Gusteren WF, Di Nola A, Haak JR. Molecular dynamics with coupling to an external bath. J. Comput. Phys. 1984;81:3684–3690. [Google Scholar]
- 36.Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- 37.Darden T, York D, Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 38.Hess B, Bekker H, Berendsen HJC, Fraaije J. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 39.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;33:27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 40.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 41.Notredame C, Higgins D, Heringa J. T-Coffee: A novel method for multiple sequence alignments. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 42.Chillemi G, Castrignano T, Desideri A. Structure and hydration of the DNA-human topoisomerase I covalent complex. Biophys J. 2001;81:490–500. doi: 10.1016/S0006-3495(01)75716-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia AE. Large amplitude nonlinear motions in proteins. Phys. Rev. Lett. 1992;68:2696–2699. doi: 10.1103/PhysRevLett.68.2696. [DOI] [PubMed] [Google Scholar]
- 44.Chillemi G, Fiorani P, Benedetti P, Desideri A. Protein concerted motions in the DNA-human topoisomerase I complex. Nucleic Acids Res. 2003;31:1525–1535. doi: 10.1093/nar/gkg242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fiorani P, Chillemi G, Losasso C, Castelli S, Desideri A. The different cleavage DNA sequence specificity explains the camptothecin resistance of the human topoisomerase I Glu418Lys mutant. Nucleic Acids Res. 2006;34:5093–5100. doi: 10.1093/nar/gkl670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Interthal H, Quigley P, Hol WG, Champoux JJ. The role of lysine 532 in the catalytic mechanism of human topoisomerase I. J. Biol. Chem. 2004;279:2984–2992. doi: 10.1074/jbc.M309959200. [DOI] [PubMed] [Google Scholar]
- 47.Krogh BO, Shuman S. Catalytic mechanism of DNA topoisomerase IB. Mol. Cell. 2000;5:1035–1041. doi: 10.1016/s1097-2765(00)80268-3. [DOI] [PubMed] [Google Scholar]
- 48.Krogh BO, Shuman S. Proton relay mechanism of general acid catalysis by DNA topoisomerase IB. J. Biol. Chem. 2002;277:5711–5714. doi: 10.1074/jbc.C100681200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.