

Abstract
In the last decade polycistronic vectors have become essential tools for both basic science and gene therapy applications. In order to co-express heterologous polypeptides, different systems have been developed from Internal Ribosome Entry Site (IRES) based vectors to the use of the 2A peptide. Unfortunately, these methods are not fully suitable for the efficient and reproducible modulation of the ratio between the proteins of interest. Here we describe a novel bicistronic vector type based on the use of alternative splicing. By modifying the consensus sequence that governs splicing, we demonstrate that the ratio between the synthesized proteins could easily vary from 1 : 10 to 10 : 1. We have established this system with luciferase genes and we extended its application to the production of recombinant monoclonal antibodies. We have shown that these vectors could be used in several typical cell lines with similar efficiencies. We also present an adaptation of these vectors to hybrid alternative splicing/IRES constructs that allow a ratio-controlled expression of proteins of interest in stably transfected cell lines.
INTRODUCTION
Many applications require co-expression of heterologous polypeptides from basic research to gene therapy experiments. In this purpose, numerous approaches have been developed from co-transfection with two independent constructs to single vectors where co-expression is achieved through the use of several promoters, Internal Ribosome Entry Sites (IRES) or Foot-and-Mouth Disease-Virus (FMDV)-derived 2A peptides (1). All these strategies have various drawbacks but one particular disadvantage is that they do not allow easy, reproducible and great modulation of the expression ratio between the proteins of interest. However, in several cases, this property might be useful.
One particular example is the production of recombinant antibodies, which are formed by association of two light chains (LCs) and two heavy chains (HCs). Studies demonstrated that intracellular HC : LC ratio is of major importance regarding antibodies production efficiency (2,3). The optimum ratio for efficient production depends on many factors including the cell type used for expression, and whether production is performed in a transient or stable context (4,5). Therefore, this ratio has to be adaptable to allow optimal antibody production in any case.
The system described in this article is based on alternative splicing to ensure regulated co-expression of two polypeptides. Alternative splicing is the mechanism by which different mature mRNAs can be generated from one pre-mRNA through the use of alternative splice sites (6). Splice sites define the border of an intron and consist of the almost invariant GU dinucleotide, called 5′ splice site (5′SS) and the 3′ splice site (3′SS) that comprises three sequence elements: the branch point, followed by a polypyrimidine tract, and the terminal AG sequence. Both 5′SS and 3′SS are comprised within larger, less conserved consensus regions. Choice between alternative splice sites is regulated in many ways including the inherent strength of the splice sites, i.e. how close they are from the consensus sequences (7) and the presence of cis-regulating elements in the surrounding sequences, for example Exonic Splicing Enhancers (ESE) or Silencers (ESS) (8).
In the present study we tried to evaluate if alternative splicing could be a valuable mechanism to co-express two proteins in a regulated manner through the mutation of splice sites. The system was tested for different applications including multimeric proteins expression, in various cell types and both in a transient or stable context.
MATERIAL AND METHODS
Plasmid construction
The vector’s backbone was obtained from pCRFL previously described (9) containing the chimeric intron of pRL-CMV (Promega). Luc R was polymerase chain reaction (PCR) amplified from pRL-CMV (Promega) using HA-LucR -F and -R primers (primer sequences are available in Supplementary Data and Table S1). Primers contained the following restriction sites: AvrII/BamHI (forward primer) and PacI/SacII/NotI (reverse primer). The reverse primer included the sequence of a second 3′SS consisting in the following elements: branch point, pyrimidine tract and acceptor splice site sequence. These elements were included between the PacI and NotI restriction sites. Luc F was amplified from pGL3 (Promega) using HA-LucF -F and -R primers, containing PacI/NheI and BglII/EcoRV sites. The forward primers, used to amplify both fragments, contained the sequence coding the hemaglutinin (HA) tag in fusion with the luciferase open reading frames. Both PCR fragments were respectively digested with AvrII/PacI and PacI/BglII and cloned into the XbaI/BglII digested pCRFL. Three putative alternative translation initiation codons CTG present in the vector 5′-UTR were mutated into TTG sequences using the Quikchange XL site directed mutagenesis kit (Stratagene) with oligonucleotides CTG-mut1 to CTG-mut3 -F and -R designed according to the manufacturer’s instructions. The resulting plasmid, further called pV1, was checked by sequence analysis. Plasmid expressing the LCs and HCs of the anti-N-VEGF antibody further called pA1, was obtained by replacing HA-LucR and HA-LucF coding cistrons of pV1 by the PCR amplified LCs and HCs respectively using BamHI/XbaI and NheI/EcoRV restriction sites. The sequences of the LCs and HCs were PCR amplified with the LC -F/-R and HC -F/-R primers from two plasmids previously constructed and containing the sequences of each chain isolated from the hybridoma cDNA.
EMCV IRES and EF-1α promoter were PCR amplified from pCREL (9) and pCREF1L (10) with primers (EMCV -F/-R and EF1α -F/-R) inserting SacII (5′-end) and NheI (3′-end) restriction sites. The resulting fragments were digested with SacII/NheI and used in a tri-molecular ligation with fragments resulting from XhoI/SacII and NheI/XhoI digestion of pA1 thus replacing the 3′SS between the cistrons by the IRES (pCI) or the EF-1α promoter (pCE).
For stable transfection experiments, a cassette containing the VEGF-IRES-A and the neomycin coding sequence was inserted downstream of the HC ORF. Both elements were PCR amplified from pCRA121mSpHA (11) and pCDNA3 (Invitrogen), with primers IRES-VEGF-F/overlap-R and overlap-F/Neo-R, then overlapped. Further cloning of this cassette was performed using the EcoRV/BglII digested fragment in a tri-molecular ligation with BglII/PvuI and PvuI/EcoRV digested fragments of different plasmids: pA1, mutants pA2 to pA9 (described below), pCI and pCE. Resulting plasmids were called pAx-s, pCI-s and pCE-s.
Construction of mutants for the 3′SS
Degenerated oligonucleotides mut-V1-F and mut V1-R were used to create mutants on three bases of the second 3′SS in the pV1 vector. Equimolar amounts of each oligonucleotide were mixed and hybridized, leading to a short double stranded DNA fragment with cohesive 5′- and 3′-ends corresponding to sequences for NotI and PacI restriction sites. Different dilutions of the hybridization product were cloned into the NotI/PacI digested pV1. More than 200 clones were analyzed and DNA sequences were determined (Millegen sequencing services) in order to isolate different mutants for the second acceptor splice site. Eight mutants, called pV2 to pV9, were chosen for further experiments.
Construction of the first 3′SS mutants was performed by inserting point mutations in pA1 using the Quikchange XL site directed mutagenesis kit (Stratagene) according to the manufacturer’s instructions with the primers couples listed from MUT-A2 -F/-R to MUT-A9 -F/-R. Resulting plasmids were called pA2 to pA9.
Cell culture and transfection
CHO cells (ATCC CCL61) were cultivated in EMEM medium (Gibco), HeLa (ATCC CCL2) and NIH-3T3 (ATCC CRL-1658) cells in DMEM containing 1 g/l glucose (Gibco) and HEK 293T cells (GenHunter, Q401) in DMEM containing 4.5 g/l glucose (Gibco). All media were supplemented with 10% FCS, 1% glutamine and antibiotics. All cell types were cultivated at 37°C in a humidified 5% CO2 atmosphere. For transient transfection experiments, cells were seeded either onto six-well dishes or 10 cm plates 24 h prior to transfection. CHO cells were transfected with Fugene-6 transfection reagent (Roche) according to the manufacturer’s instructions. HeLa, NIH-3T3 and HEK293T cells were transfected using the JetPEI transfection reagent (Polyplus Transfection) according to the manufacturer’s instructions. For stable transfection, CHO cells were transfected as described earlier with plasmids pAx-s, pCI-s and pCE-s and treated with 0.75 mg/ml G418 (Euromedex) 48 h after transfection. Surviving cells were pooled and amplified for 25 more days with continuous G418 selection.
Western blot analysis
Twenty four hours after transfection, cells were collected and sonicated in 50 µl of sodium dodecylsulfate (SDS) sample buffer. Protein concentration in the cell lysates was determined using the bicinchoninic acid method (Interchim). Samples were boiled at 95°C for 5 min after addition of reducing agents and 30 µg of total proteins were separated on a NuPAGE 4–12% Bis-Tris gel (Invitrogen). After electrophoretic transfer, the nitrocellulose membranes (Schleicher & Schüell) were blocked with 3% skimmed milk. Tagged luciferases were immunodetected using mouse monoclonal anti-HA (dilution 1 : 1000) (Babco) as a primary antibody and peroxidase-conjugated sheep anti-mouse (dilution 1 : 10000) (Amersham) as a secondary antibody and the ECL detection kit (Amersham). HCs and LCs of the anti-N-VEGF antibody were detected using a peroxidase-conjugated sheep anti-mouse IgG antibody (dilution 1 : 2500) (Promega). Normalization was done using a monoclonal anti β-actin antibody (dilution 1 : 10000) (Sigma-Aldrich). Densitometric analysis was performed using Scion Image 1.63 software (Scion Corporation).
Total and polysomal RNA isolation and RT–PCR analysis
Sucrose gradient fractionation was performed essentially as described earlier (12). Cell extracts were prepared by lysis at 4°C in extraction buffer (10 mM Tris–HCl, pH 8.0, 140 mM NaCl, 1.5 mM MgCl2, 0.5% Nonidet-P40 and 500 U/ml RNAsin), and nuclei were removed by centrifugation (12 000g, 10 s, 4°C). The supernatant was supplemented with 20 mM dithiothreitol, 150 µg/ml cycloheximide, 665 µg/ml heparin and 1 mM phenylmethylsulfonyl fluoride and centrifuged (12 000g, 5 min, 4°C) to eliminate mitochondria. The supernatant was layered onto a 5 ml linear sucrose gradient (15–40% sucrose [w/v] supplemented with 10 mM Tris–HCl, pH 7.5, 140 mM NaCl, 1.5 mM MgCl2, 10 mM dithiothreitol, 100 µg/ml cycloheximide and 0.5 mg/ml heparin) and centrifuged in a SW50.1Ti rotor (Beckman) for 1 h 40 min at 140 000g and at 4°C, without brake. Fractions of 300 µl were collected and digested with 100 µg proteinase K in 1% SDS and 10 mM EDTA (30 min, 37°C). RNAs were then recovered by phenol-chloroform-isoamyl alcohol extraction, followed by ethanol precipitation. Finally, the fractions containing the mRNA, were precipitated with 2 M LiCl on ice at 4°C overnight. After centrifugation (12 000g, 15 min at 4°C), pellets were washed with 70% ethanol pre-stored at –20°C, air dried and resuspended in appropriate volumes of RNAse-free water.
Total RNA was extracted using the TriZol reagent protocol (Invitrogen). After DNAse treatment (DNA free, Ambion) and quantification, reverse transcription was done with 2 µg of total RNA using the First strand cDNA Synthesis kit (Invitrogen) and random hexamers. The resulting cDNA fragments were PCR amplified using three primers allowing simultaneous amplification of the three different mRNA species, i.e. spliced on the first 3′SS, spliced on the second 3′SS and unspliced (i.e. primers RT-CMV-F, RT-LucR-R and RT-LucF-R for the luciferases sequences or RT-CMV-F, RT-LC-R and RT-HC-R for the antibody’s sequences). PCR products were loaded on a 2% agarose gel and visualized using SYBR Safe DNA gel staining (Invitrogen). Control reactions were performed using β-actin control primers supplied in the Superscript III RT kit (Invitrogen). Cryptic splice sites were identified after cloning of the corresponding cDNA fragments purified from agarose into the pCR4-TOPO (Invitrogen), and sequencing with appropriate primers (T3 and T7). Alignment of the resulting sequences with the cDNA wild-type sequence allowed identification of aberrant splicing events. Corresponding cryptic splice sites were inactivated by inserting point mutations using the Quikchange site directed mutagenesis kit (Stratagene) according to the manufacturer’s instructions with primers couples listed from QKSC1 -F/-R to QKSC5 -F/-R.
Detection of secreted anti-N-VEGF antibody in the culture supernatants
Microtiter plates were coated overnight at 25°C with 1 µg/ml of recombinant N-VEGF diluted in phosphate buffered saline (PBS). After three washes with 250 µl of washing solution (PBS/0.05% Tween 20), plates were blocked with PBS containing 2% (w/v) skimmed milk for 2 h at 25°C and washed three times. Cell-free supernatants (different dilutions) were incubated for 2 h at 25°C. After extensive washing with PBS/T, the detection antibody (i.e. goat anti-mouse IgG, HRP conjugated; Promega) was diluted at 166 ng/ml in PBS/T and incubated for 2 h at 25°C. Further washes were followed by the addition of 100 µl of TMB substrate solution (BD OptEIA) and the reaction proceeded at 25°C in the dark for up to 30 min. The reaction was then stopped by the addition of 50 µl of 2N H2SO4 and the OD450 was measured immediately using a microplate reader (UVM340, Asys). Each experiment was repeated at least three times. Graphs represent the amount of secreted antibody for each mutant transfected relatively to the amount secreted with the pA1 vector for transient transfection experiments and relatively to the amount secreted with the pCE-s vector for stable transfection experiments. Errors bars represent standard deviation and asterisks depict statistically significant differences compared to pA1 or pCE-s (**P < 0.01 and *P < 0.05, ANOVA test).
RESULTS AND DISCUSSION
The aim of this study was to evaluate if alternative splicing could be a suitable mechanism to generate different ratios of expressed recombinant proteins from a bicistronic vector.
Evaluation of the efficiency of alternative splicing as a bicistronic mode of expression
In a first set of experiments, we wanted to test whether alternative splicing could lead to the co-expression of two proteins encoded by two cistrons in the same vector. For that purpose, we first elaborated a plasmid, called V1, comprising a complete intron in the 5′-UTR and an additional consensus acceptor splice site (3′SS) between the two cistrons (Figure 1A). The intron is constituted by consensus elements: a donor splice site (5′SS), a branch point, a pyrimidine tract and a 3′SS. The construction was carried out with the Renilla Luciferase (Luc R) and the Firefly Luciferase (Luc F) as reporter genes. Although expression of these proteins is usefully followed through their enzymatic activities, we wanted to evaluate their respective concentrations by western blotting. For this goal, we fused the HA tag to their amino-terminal ends. Consequently, we replaced their original start codon by a consensus AUG leading to a very similar initiation of translation (Figure 1A). Theoretically, transcription of the expression cassette can be followed by two distinct types of splicing events (resulting from the use of either the first or the second 3′SS) thus generating a first mature mRNA allowing Luc R expression (m1) and a second mRNA encoding Luc F (m2); whereas a third possibility could be generation of the unspliced mRNA (m-us) (Figure 1A).
Figure 1.

Development of a bicistronic vector based on alternative splicing. (A) Schematic representation of the bicistronic vector based on alternative splicing (pV1) and the corresponding matures mRNAs. The two cistrons consist of the HA-tagged Renilla Luciferase (Luc R) and Firefly Luciferase (Luc F). m1 corresponds to mRNA spliced on the first 3′SS leading to expression of Luc R, m2 corresponds to mRNA spliced on the second 3′SS leading to expression Luc F and m-us corresponds to unspliced mRNA. (B) An anti-HA antibody was used to perform western blot analysis on protein extracts from pV1 transiently transfected CHO cells. The upper band (61 kDa) corresponds to HA-LucF (second cistron) and the lower band (37 kDa) to HA-LucR (first cistron). Control (Ctrl) corresponds to mock-transfected cells.
A western blot analysis performed with lysates of CHO cells transiently transfected with the V1 plasmid revealed that the two luciferases were detected but HA-LucF (61 kDa) was much more expressed than HA-LucR (37 kDa) (Figure 1B). Our interpretation was that the second 3′SS was predominantly used by the splicing machinery compared with the first one. Repeated experiments also indicated that this ratio was very constant.
Modulation of the ratio between the two luciferases
In order to control the ratio between the two different mRNAs (and consequently the expression of the two luciferases), our strategy was to weaken efficiency of the second 3′SS by a directed mutagenesis approach. The −3, −5 and −11 positions of the consensus sequence (Figure 2A) were selected for mutagenesis for the following reasons: the −3 and −5 positions are the most conserved (apart the absolute AG) and the −11 is central in the polypyrimidine tract and could be changed into purines. All mutated constructs (53 mutants obtained out of the 64 possibilities) were tested for transient transfection in CHO cells followed by western blot analysis. Results revealed a large panel of Luc F/Luc R ratios between the different mutants. From these data, eight mutants that were representative of the whole range of expression ratio possibilities were chosen for further experiments. These mutants, whose second 3′SS sequences are provided in Figure 2A, were called V2 to V9. When used to transfect CHO cells, the different constructs generated important variations in the relative expression of the two luciferases. This was evaluated by measuring their enzymatic activities (Supplementary Figure S1). Since the specific activities of the two luciferases are not clearly established, we also determined their expression by western blotting (Figure 2B). Different ratios between HA-LucF and HA-LucR quantities can be observed from a large majority of HA-LucF to a large majority of HA-LucR and including intermediate ratios (e.g. ratio close to 1 : 1). In agreement with the proteins expression pattern, semi-quantitative-competitive reverse transcriptase (RT) PCR analyses of the different mRNA species of the same samples revealed an increased amount of RNA spliced on the first 3′SS (m1, Luc R) and a decrease in the amount of RNA spliced on the second 3′SS (m2, Luc F) from mutants V2 to V9 (Figure 2C).
Figure 2.

Mutations in the sequence of the second 3′SS allows efficient modulation of the expression ratio between the two luciferases. (A) Mutants of the second 3′SS listed from V1 to V9 and their corresponding sequences. V1 corresponds to the consensus splice site sequence. Mutated bases are bolded and the dinucleotide corresponding to the 3′ border of the splice site is underlined. The first dinucleotide of exon 2 is in italic. Positions −3, −5 and −11 that are chosen to be mutated are indicated under the V1 sequence. (B) Western blot analysis on protein extracts from CHO cells transiently transfected with the 3′SS mutants listed in Figure 2A. Ctrl corresponds to mock-transfected cells. In the lower panel, densitometric data from western blot X-ray films are used for graphical representation of the Luc F/Luc R ratio. (C) RT–PCR analysis on mRNAs extracted from CHO cells transiently transfected with the 3′SS mutants pV1 to pV9. Competitive PCR amplification was performed using a forward primer hybridizing upstream the intron 5′SS and two reverse primers hybridizing from bases 186 to 206 within the Luc R ORF and from bases 133 to 152 within the Luc F ORF, thus allowing simultaneous detection of the three mRNA species: m-us, m1 and m2. (D) Graphical representation of densitometric data calculating Luc F/Luc R expression ratios obtained with the different constructs in CHO, HeLa and NIH-3T3 cell lines.
Additionally, this RT–PCR analysis also showed that a significant part of the mRNAs were not spliced (‘m-us’ band). This mRNA could potentially be translated to generate HA-LucR. It was also interesting to point out the presence of additional PCR products distinct from the three described earlier. After cloning and sequencing of these bands, two cryptic 5′SS within the Luc R sequence were identified and it is worth noting that they were not predicted as putative splice sites in silico.
To assess if this alternative splicing mechanism was comparable among cells lines of different origins, the set of plasmids was tested, in parallel, in CHO (hamster), HeLa (human) and NIH-3T3 cells (mouse). Results clearly showed that all three cell types presented similar patterns of expression ratios between the two luciferases (Figure 2D). Taken together, these results tend to indicate that (i) the system developed here allows simultaneous expression of two proteins, (ii) easily permits the modulation of the ratio between the two proteins and (iii) could be used in various cell types with equal efficiency.
Expression of the two chains of an antibody
After validation of the vector’s functionality with two reporter genes, the construction was used to express a particular heteromultimeric protein consisting in an equimolar ratio of two polypeptides, i.e. an antibody. Previous studies have demonstrated that the ratio from LC to HC is crucial for correct antibodies folding and assembly (2). This ratio also influences the amount of functional secreted antibodies. The main problem is that the optimum ratio depends on the sequence of the antibodies, the cell type used for expression and whether production is performed in a stable or transient context (4,5,13). Consequently, for each antibody to be produced in a particular condition, the optimum ratio should be established. Furthermore, this ratio should be stable, especially when cell lines are created. For these reasons, the system developed in this study could be suitable for the expression of recombinant antibodies because of its adaptability. Indeed, a panel of mutants generating various expression ratios between the two chains can be easily created, thus allowing identification of the best construct for any given antibody, in any cell type chosen and in any condition.
To test the system, we chose a home-made monoclonal antibody directed against human N-VEGF, an intracellular protein that results from the cleavage of the L-VEGF precursor that also generates the secreted VEGF-A molecule (14) (Figure 3A). The cDNAs encoding the two chains of this antibody were cloned and sequenced. The LC and HC cDNAs were sub-cloned in the bicistronic vector comprising the consensus sequences for the different splice sites, respectively as the first and second cistron, leading to the pA1 construct (Figure 3B). A western blot analysis performed on a pA1 transfected CHO lysate showed that only free LC was detectable (data not shown). This indicated that, in this case, expression of the first cistron was higher than expression of the second cistron, contrary to what was observed with the pV1 construct. In agreement with knowledge in the field of alternative splicing mechanisms (8), this change in preferential 3′SS usage could be explained by the presence of putative regulatory sequences like ESE or ESS that generally lie within the coding regions. It may thus be explainable that replacing HA-LucR by the LC of an antibody may have such an effect on the splicing pattern.
Figure 3.

Use of the bicistronic vector for production of a murine recombinant antibody anti-N-VEGF. (A) Schematic representation of the precursor L-VEGF molecule which cleavage generates secreted VEGF-A and intracellular N-VEGF. (B) Schematic representation of the bicistronic vector constructed for antibody anti-N-VEGF expression (pA1). The LC was cloned as the first cistron and the HC as the second cistron. (C) Sequence of the anti-N-VEGF LC constant region and cryptic splice sites of the variable region. The signal peptide is showed in the black-lined box, followed by the variable region of the LC. The arrow delimits the variable region (upstream) and the constant region (downstream). Identified cryptic 5′ (GT) and 3′SS (AG) are underlined and mutations are represented in bold above the sequence.
In order to modulate the expression ratio between the LCs and HCs, we then chose to mutate the first acceptor site.
Elimination of cryptic splice sites
Because we had to weaken the strength of the first 3′SS, a prerequisite was to identify putative cryptic 3′SS within the sequence of the LC encoding cistron. As seen with the study on luciferases, such sites can be used by the splicing machinery and thus generate aberrant splicing events that could potentially lead to the expression of truncated proteins (Supplementary Figure S2A). In order to identify these cryptic splice sites, an RT–PCR analysis was performed on mRNAs extracted from pA1 transfected CHO cells. As expected, this analysis revealed the presence of the m1 transcript (spliced on the first 3′SS and translated into LC) and the complete absence of the m2 transcript (spliced on the second 3′SS) (Supplementary Figure S2B). However, several other species of intermediate sizes were detected, most of them quite intense. This tends to indicate that many abnormal splicing events frequently occurred, generating an important pool of mis-spliced transcripts. After cloning and sequencing of corresponding cDNAs, several cryptic splice sites were identified. Most of them were 3′SS but we also identified cryptic 5′SS. One of them, a donor site located three bases upstream the LC STOP codon, was found to splice with the second constitutive acceptor site in almost 90% of the atypical spliced transcripts. Surprisingly, many of the identified sites were 3′SS despite the lack of a clearly defined pyrimidine tract. The sites were mutated with respect to the amino-acid sequence of the encoded polypeptide. As new cryptic splice sites may be activated if a mutation alters or removes a genuine nearby site, the experiment was repeated three times after each round of identification/mutation, and we clearly observed that aberrant splicing was considerably lowered after each step (data not shown).
Since we planed to mutate the first constitutive 3′SS to weaken its efficiency, it was of importance to test the presence of remaining cryptic splice sites under conditions of poor efficient intron 3′SS. In this goal, two mutants corresponding to variants V5 and V9 were generated. As expected, the analysis revealed activation of new cryptic splice sites which all were corrected. Finally, 10 3′SS and 5 5′SS were identified and mutated, as shown in Figure 3C.
Analysis of the mRNA profile of pA1 transfected CHO cells (that contained the LC mutated for all identified cryptic splice sites) revealed two major products corresponding to the expected spliced mRNAs and no extra bands. In these conditions, the m1 species was clearly more abundant than the m2 mRNA (Supplementary Figure S2C).
Modification of the ratio between the two chains of the antibody
In order to modify the ratio between the two different mRNAs (and consequently control the relative expression of the LCs and HCs) we mutated the sequence of the first acceptor splice site. The sequences chosen were those of the 8 mutants described earlier and mentioned as V2 to V9. These mutants (called A2 to A9) were used to transfect CHO and HEK293T cells, two cell types typically used for recombinant antibody expression because of easy adaptation to suspension and serum-free culture conditions, high transfection efficiency and high production rates (15). As described earlier with the luciferases system, mutants of the acceptor splice sites allowed efficient modulation and displayed a wide range of LC to HC mRNA ratios both in CHO (Figure 4A) and HEK cells (Figure 4C). As expected, these variations directly correlated with the amount of functional secreted antibody (determined by antigen-coated ELISA) in both cells types as shown in Figures 4B and 4D. Indeed, for CHO cells, mutants A6 and A8 showed significant increase in the level of secreted antibodies compared to A1 (Figure 4B), mutant A8 being the best producer with a 4-fold increase. In HEK cells, the same two mutants also led to the best productivity allowing a 2-fold increase in the level of secreted antibodies. These results are in accordance with western blot analysis of secreted antibodies (Supplementary Figure S3A). As controls, we demonstrated that LC alone could be efficiently secreted and form dimers, whereas HC alone could not be secreted as expected (16,17) (Supplementary Figure S3B). Obviously, we confirmed that single chains were not able to recognize the antigen in the ELISA test (data not shown).
Figure 4.

Effect of the different 3′SS mutants on expression efficiency in transiently transfected CHO or HEK293T cells lines. CHO (A and B) and HEK293T (C and D) cells were transfected with different mutants of the first 3′SS from pA2 to pA9. (A and C) upper panels: RT–PCR analysis on mRNAs. Competitive PCR amplification was performed using a forward primer hybridizing upstream the intron 5′SS and two reverse primers hybridizing from bases 419 to 458 of the LC ORF and from bases 303 to 324 of the HC ORF. (A and C) lower panels: western blot analysis on protein extracts from CHO (A) and HEK293T (C) cells transfected with the different 3′SS mutants. Anti β-actin was used to control protein loading. (B and D) Amount of functional antibodies secreted by CHO (B) and HEK293T (D) cells transfected with the different mutants pA2 to pA9 in comparison to pA1 containing the consensus sequences for all splice sites, determined by antigen specific ELISA. Data are presented as means ± SEM. Statistical significance was evaluated using one-way ANOVA test. *P < 0.05, **P < 0.01.
In conditions of transient transfection, it is not evident to determine an optimal ratio between the LC and HC encoding mRNAs since mutants A6 and A8 gave the best results and present reciprocal mRNA ratios. How inverse ratios could provide a comparable production of antibody is not really understood but similar results were provided by others (4). However, it is clear that extreme imbalanced ratios were deleterious for antibody production. Most of the mutants showed the same efficiency when compared between CHO and HEK293T. One major difference should be underlined: the unspliced form was more present in HEK293T than in CHO cells. This could be interpreted as a less efficient recognition of the 5′SS in HEK293T cells. This fact could be of general importance since most expression vectors possess the same kind of intron in their 5′-UTR. Thus, it could be of interest to optimize the efficiency of the 5′SS, especially for its use in these cells.
Study of mRNA species engaged in polysomes
Since a significant part of the mRNAs was unspliced, especially in HEK293T transfected cells, we tried to evaluate the translation efficiency of this species that have a larger 5′-UTR than spliced mRNAs. This study was performed by polysome-associated RNAs assay (Figure 5A). For this purpose, HEK293T cells were transfected with the A6 mutant and polysome fractionation was done. Different fractions from the sucrose gradient were collected and associated mRNAs corresponding to species from free mRNAs to mRNAs associated with poly-ribosomes were isolated (Figure 5B). Fractions were pooled into four distinct groups and analyzed by RT–PCR to detect the different species. As shown in Figure 5C, mRNA spliced on the first 3′SS, mRNA spliced on the second 3′SS but also unspliced mRNA are present in all the fractions. This indicates that the three species can be efficiently translated (fractions 3 and 4), including unspliced mRNA that generates the LC. It could be noticed that the ratio between unspliced/spliced mRNAs is lower in translated mRNAs (mostly fractions 3 and 4) than in untranslated mRNAs (fraction 1) or total RNA (Figure 5C, lane T). This reinforces a previous conclusion about the need to improve efficiency of the intron 5′SS recognition since, even if the unspliced form can be partially translated, it represents a loss in the efficiency of the total mRNA to produce full antibodies. However, an important part of transcribed mRNAs remained in fractions 1 and 2 suggesting that translational machinery may be saturated in these cell culture conditions and that increasing the cells translational capacity could be a way to improve their productivity.
Figure 5.
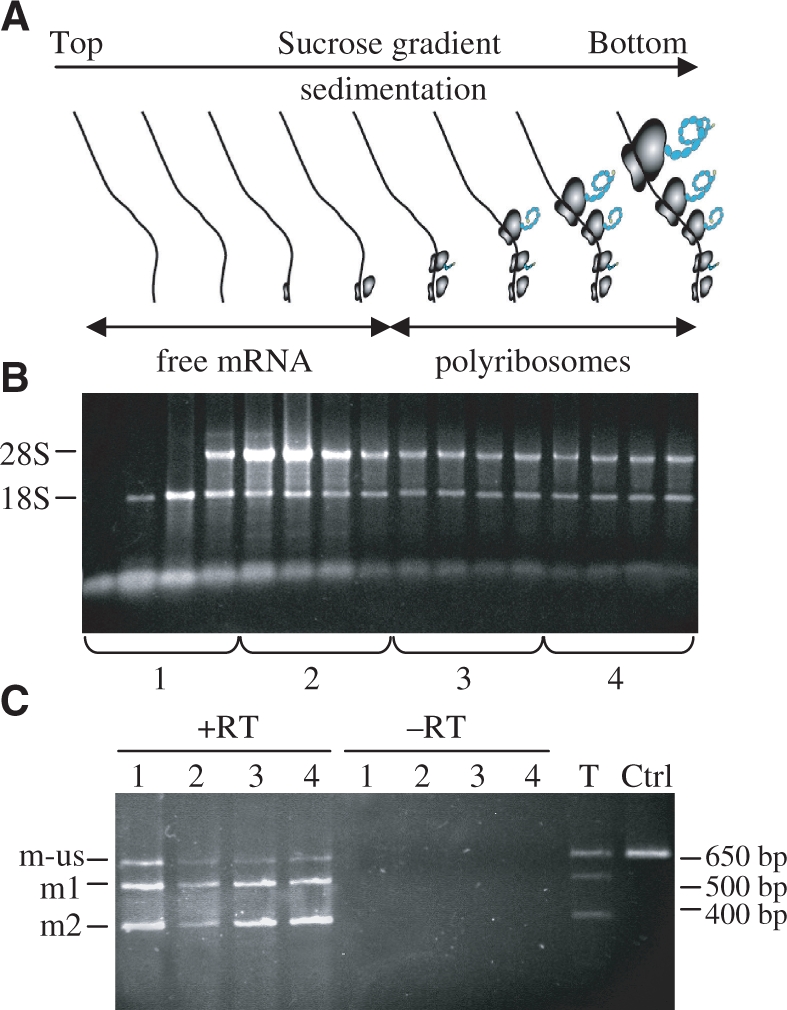
Study of mRNA species engaged in polysomes. (A) Schematic representation of polysomal and free mRNAs distribution after sucrose density gradient RNA fractionation of pA6 transfected CHO cells. (B) RNAs purified from each fraction, collected from top to bottom of the sucrose density gradient, were visualised on agarose gel by SyBRgreen staining. The fractions were pooled in four samples representing the different stages of mRNAs commitment in translation. (C) RT–PCR analysis was performed on mRNAs extracted from the four samples obtained after polysomes fractionation. To exclude any DNA contamination, PCR reactions were performed on the four RNA fractions lacking the Reverse Transcriptase step (−RT). The control T corresponds to the RT–PCR process with total RNA, i.e. before polysomes fractionation, and Ctrl to PCR amplification with pA1 as the matrix.
Comparison of the vector based on alternative splicing to other constructs in stably transfected cells
As many processes for recombinant antibody production involve the use of stable cell lines, we finally wanted to investigate if the vectors based on alternative splicing could be successfully utilized in such conditions. Expression cassettes including VEGF IRES-A and the gene coding for neomycin were cloned downstream of the HC in pA1 to pA9 constructs (Figure 6A) to allow selection of the stable transfectants under G418 treatment. Two additional constructs were generated in order to compare, in this context, the vectors based on alternative splicing to other types of vectors, classically used for recombinant antibody production, i.e. a vector with an IRES and a vector with two different promoters (Figure 6A). For the vector containing the IRES between the two cistrons, we chose the EMCV IRES, which is one of the most efficient IRES in a great number of cell types and therefore the most commonly used for expression of recombinant antibodies (18,19). For the vector in which transcription of the HC is under a separate promoter, we chose the EF1α promoter which was successfully used in vectors for IgG mammalian expression (20).
Figure 6.

Efficiency of alternative-splicing based vectors in CHO stable transfectants and comparison to other constructs. (A) Schematic representation of the different vectors used to generate stable pools of CHO transfectants: bicistronic vectors based on alternative splicing (pAx-s), IRES-containing bicistronic vector (pCI-s) and double-promoter vector (pCE-s). (B) RT–PCR analysis of mRNAs extracted from CHO cells stably transfected with the different 3′SS mutants pA1-s to pA9-s. (C) Amount of functional antibodies secreted by pools of CHO cells stably transfected with mutants pA1-s to pA9-s in comparison to the IRES-containing bicistronic vector (pCI-s) and the bigenic vector (pCE-s). Data are presented as means ± SEM. Statistical significance was evaluated using one-way ANOVA test. *P < 0.05, **P < 0.01.
RT–PCR analysis performed on mRNAs extracted from stable pools of cells transfected with vectors pA1-s to pA9-s showed a clear excess in the LC mRNA from mutants A1-s to A5-s, an excess of HC mRNA for mutants A7-s and A9-s and a more balanced ratio for mutants A6-s and A8-s (Figure 6B).
In order to test the stability of the splicing efficiency throughout the cell divisions, a RT–PCR experiment was performed on mRNAs extracted from cells at passages 2, 5, 10 and 20. The profiles obtained for three representative mutants (A4, A7 and A8) clearly indicate that the ratio between the different mRNA species was stable throughout passages (Supplementary Figure S4).
As shown in Figure 6C, mutants leading to the best productivity were mutants A7-s and A9-s, indicating that, in a stable context, an excess for HC mRNA tended to be more favourable for antibody productivity. Previous results have also reported that an excess of HC mRNA or polypeptide can lead to a more efficient IgG production in HEK or CHO cells both in transient (4,5) or stable transfection (3). The fact that the best producers were different in a transient versus a stable context might be interpreted with potential differences in the splicing efficiency in the two contexts, thus leading to slight differences in the m1/m2 ratio. Another explanation could be, as it has been described by Schlatter et al. (4) that the optimal ratio between the two mRNAs is different depending on whether production is performed in transiently or stably transfected cells.
Figure 6C also showed that, compared to the two ‘reference’ constructs, pCE-s and pCI-s, some mutants of the vector based on alternative splicing, mutants A7-s to A9-s, allow secretion of a greater amount of functional antibodies. This clearly indicates that vectors based on alternative splicing can be an interesting alternative to conventionally used vectors. Moreover, only a restricted number of 3′SS mutants have been tested here. One can imagine that other mutations might allow to control more tightly the ratio between the two chains and thus permit even greater antibody productivity.
In accordance to studies performed with other antibodies sequences in different expression systems, our results emphasize the fact that the optimum ratio for maximal antibody productivity is poorly predictable. The system developed in this study overcomes this problem by allowing rapid screening of several mutants, generating different possibilities of ratios between the two chains. In addition, our results indicate that if an excess of HC encoding mRNA is favourable for antibody secretion when expressed after permanent transfection, it seems that no clear rule can be established for transient transfection. In this last case, an empirical approach consisting of testing several possibilities remains appropriate. However, finding the best combination for each antibody to be expressed in a chosen cell type could be easily achieved. Moreover, this method, involving the use of only one plasmid, seems more reproducible than co-transfection of two plasmids in various ratios (4) because this technique could easily be biased by (i) various transfection efficiencies in transient experiments, (ii) extrapolation for stable transfections and (iii) chromosomal insertion sites of the two plasmids because these sites have a major effect on expression efficiency (21). Using a single vector with two independent genes generates invariable expression ratio between the two proteins but also (i) two identical promoters may lead to recombination events in the case of stable expression, (ii) different promoters could be perturbed by transcriptionnal interference or suppression events (1). In addition, plasmids with the EMCV IRES have also been successfully used to generate different mutants expressing various ratios of HC to LC (5). However, Mizuguchi et al. (22) showed that the translation efficiency of the IRES can only reach maximum 60% of the cap-dependent translation efficiency of the first cistron. On the opposite, our system clearly allows a larger panel of ratios from 10 : 1 to 1 : 10, thus permitting extended possibilities compared to IRES-driven systems but also to 2A peptide-containing systems that only provide a unique ratio (23).
CONCLUSION
During the establishment of the Alternative Splicing-based Bicistronic (ASB) vector we have provided information concerning the presence of cryptic splice sites that could be usefully integrated for classical vectors that contain an intron in their 5′-UTR. In the particular example of antibody production, this work would facilitate further production of other murine antibodies, since the majority of the identified cryptic splice sites (11/15 identified sites) are located in the sequence that encodes the constant region of the LC. Indeed, simple cloning of different variable regions in frame with the previously mutated constant region can be done allowing an easier and faster research for additional cryptic splice sites and further optimize antibodies production efficiency.
We provide here a novel system to elaborate bicistronic vectors that present several advantages in addition to its main interest which is the possibility to control the ratio of two expressed proteins: (i) the expression and the ratio are stable and very similar for several cell lines for both transient and stable transfection (ii) the vector is small and easy to manipulate, (iii) the vector could be easily adapted to study ESE or ESS.
Further modifications could be performed in order to optimize the production yield. Finally this system might be easily adapted for expression of several heteromeric proteins for which the ratio between two subunits has to be tuned or for expression of two distinct proteins with synergistic effects at variable ratios, e.g. in gene therapy experiments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
‘Région Midi-Pyrénées’ grants; fellowships from Millegen SA and the ‘Association Nationale de la Recherche Technologique’ (to S.F.). Funding for open access charge: INSERM annual grant.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.de Felipe P. Polycistronic viral vectors. Curr. Gene Ther. 2002;2:355–378. doi: 10.2174/1566523023347742. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalez R, Andrews BA, Asenjo JA. Kinetic model of BiP and PDI-mediated protein folding and assembly. J. Theor. Biol. 2002;214:529–537. doi: 10.1006/jtbi.2001.2478. [DOI] [PubMed] [Google Scholar]
- 3.Jiang Z, Huang Y, Sharfstein ST. Regulation of recombinant monoclonal antibody production in Chinese Hamster Ovary cells: a comparative study of gene copy number, mRNA level, and protein expression. Biotechnol. Prog. 2006;22:313–318. doi: 10.1021/bp0501524. [DOI] [PubMed] [Google Scholar]
- 4.Schlatter S, Stansfield SH, Dinnis DM, Racher AJ, Birch JR, James DC. On the optimal ratio of heavy to light chain genes for efficient recombinant antibody production by CHO cells. Biotechnol. Prog. 2005;21:122–133. doi: 10.1021/bp049780w. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Zhang C, Jostock T, Dübel S. Analysis of IgG heavy chain to light chain ratio with mutant Encephalomyocarditis virus internal ribosome entry site. Protein Eng. Des. Sel. 2007;20:491–496. doi: 10.1093/protein/gzm038. [DOI] [PubMed] [Google Scholar]
- 6.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 7.Garg K, Green P. Differing patterns of selection in alternative and constitutive splice sites. Genome Res. 2007;17:1015–1022. doi: 10.1101/gr.6347907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bornes S, Prado-Lourenco L, Bastide A, Zanibellato C, Iacovoni JS, Lacazette E, Prats AC, Touriol C, Prats H. Translational induction of VEGF internal ribosome entry site elements during the early response to ischemic stress. Circ. Res. 2007;100:305–308. doi: 10.1161/01.RES.0000258873.08041.c9. [DOI] [PubMed] [Google Scholar]
- 10.Martineau Y, Le Bec C, Monbrun L, Allo V, Chiu IM, Danos O, Moine H, Prats H, Prats AC. Internal ribosome entry site structural motifs conserved among mammalian fibroblast growth factor 1 alternatively spliced mRNAs. Mol. Cell Biol. 2004;24:7622–7635. doi: 10.1128/MCB.24.17.7622-7635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bornes S, Boulard M, Hieblot C, Zanibellato C, Iacovoni JS, Prats H, Touriol C. Control of the vascular endothelial growth factor internal ribosome entry site (IRES) activity and translation initiation by alternatively spliced coding sequences. J. Biol. Chem. 2004;279:18717–18726. doi: 10.1074/jbc.M308410200. [DOI] [PubMed] [Google Scholar]
- 12.Bastide A, Karaa Z, Bornes S, Hieblot C, Lacazette E, Prats H, Touriol C. An upstream open reading frame within an IRES controls expression of a specific VEGF-A isoform. Nucleic Acids Res. 2008;36:2434–2445. doi: 10.1093/nar/gkn093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hotta A, Kamihira M, Itoh K, Morshed M, Kawabe Y, Ono K, Matsumoto H, Nishijima K, Iijima S. Production of anti-CD2 chimeric antibody by recombinant animal cells. J. Biosci. Bioeng. 2004;98:298–303. doi: 10.1016/S1389-1723(04)00285-3. [DOI] [PubMed] [Google Scholar]
- 14.Huez I, Bornes S, Bresson D, Créancier L, Prats H. New vascular endothelial growth factor isoform generated by internal ribosome entry site-driven CUG translation initiation. Mol. Endocrinol. 2001;15:2197–2210. doi: 10.1210/mend.15.12.0738. [DOI] [PubMed] [Google Scholar]
- 15.Baldi L, Hacker DL, Adam M, Wurm FM. Recombinant protein production by large-scale transient gene expression in mammalian cells: state of the art and future perspectives. Biotechnol. Lett. 2007;29:677–684. doi: 10.1007/s10529-006-9297-y. [DOI] [PubMed] [Google Scholar]
- 16.Leitzgen K, Knittler MR, Haas IG. Assembly of immunoglobulin light chains as a prerequisite for secretion. A model for oligomerization-dependent subunit folding. J. Biol. Chem. 1997;272:3117–3123. doi: 10.1074/jbc.272.5.3117. [DOI] [PubMed] [Google Scholar]
- 17.Lee YK, Brewer JW, Hellman R, Hendershot LM. BiP and immunoglobulin light chain cooperate to control the folding of heavy chain to ensure the fidelity of immunoglobulin assembly. Mol. Biol. Cell. 1999;10:2209–2219. doi: 10.1091/mbc.10.7.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolb AF, Siddell SG. Expression of a recombinant monoclonal antibody from a bicistronic mRNA. Hybridoma. 1997;16:421–426. doi: 10.1089/hyb.1997.16.421. [DOI] [PubMed] [Google Scholar]
- 19.Jostock T, Vanhove M, Brepoels E, Van Gool R, Daukandt M, Wehnert A, Van Hegelsom R, Dransfield D, Sexton D, Devlin M, et al. Rapid generation of functional human IgG antibodies derived from Fab-on-phage display libraries. J. Immunol. Methods. 2004;289:65–80. doi: 10.1016/j.jim.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Menzel C, Meier D, Zhang C, Dübel S, Jostock T. A comparative study of different vector designs for the mammalian expression of recombinant IgG antibodies. J. Immunol. Methods. 2007;318:113–124. doi: 10.1016/j.jim.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 21.Wurm FM. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat. Biotechnol. 2004;22:1393–1398. doi: 10.1038/nbt1026. [DOI] [PubMed] [Google Scholar]
- 22.Mizuguchi H, Xu Z, Ishii-Watabe A, Uchida E, Hayakawa T. IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol. Ther. 2000;1:376–382. doi: 10.1006/mthe.2000.0050. [DOI] [PubMed] [Google Scholar]
- 23.Fang J, Qian JJ, Yi S, Harding TC, Tu GH, VanRoey M, Jooss K. Stable antibody expression at therapeutic levels using the 2A peptide. Nat. Biotechnol. 2005;23:584–590. doi: 10.1038/nbt1087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.