


Abstract
Mammalian cells repair DNA double-strand breaks (DSBs) via efficient pathways of direct, nonhomologous DNA end joining (NHEJ) and homologous recombination (HR). Prior work has identified a complex of two polypeptides, PSF and p54(nrb), as a stimulatory factor in a reconstituted in vitro NHEJ system. PSF also stimulates early steps of HR in vitro. PSF and p54(nrb) are RNA recognition motif-containing proteins with well-established functions in RNA processing and transport, and their apparent involvement in DSB repair was unexpected. Here we investigate the requirement for p54(nrb) in DSB repair in vivo. Cells treated with siRNA to attenuate p54(nrb) expression exhibited a delay in DSB repair in a γ-H2AX focus assay. Stable knockdown cell lines derived by p54(nrb) miRNA transfection showed a significant increase in ionizing radiation-induced chromosomal aberrations. They also showed increased radiosensitivity in a clonogenic survival assay. Together, results indicate that p54(nrb) contributes to rapid and accurate repair of DSBs in vivo in human cells and that the PSF·p54(nrb) complex may thus be a potential target for radiosensitizer development.
INTRODUCTION
The RNA recognition motif (RRM) is one of the most common protein domains in eukaryotes (1). Early work identified the motif in a variety of RNA binding proteins (2,3). More recent work has broadened our concept of the function of RRM-containing proteins. The RRM domain itself serves as a platform for RNA−protein, DNA−protein and protein−protein interactions (1). Although most RRM-containing proteins function in RNA metabolism, others bind to specific chromosomal loci (4–6), regulate transcription (7), mediate the response to DNA damaging agents (8) or modify chromatin (9). Polypyrimidine tract-binding protein associated splicing factor (PSF) and 54 kDa nuclear RNA binding protein [p54(nrb)] are members of a subfamily of RRM proteins defined by tandem RRM domains flanked by an additional region of homology. PSF and p54(nrb), which form a stable complex in vivo, have previously been implicated in nuclear retention of A-to-I edited RNA (10), nuclear retention of other RNAs (11), pre-mRNA 3′-end formation (12) and transcriptional activation mediated by the androgen, thyroid hormone and retinoid X receptors and by the transducers of regulated CREB activity proteins (13–16).
In addition to its functions in RNA metabolism, early studies showed that PSF promotes annealing of single-stranded DNA with homologous double-stranded DNA (17,18). Recently, PSF was shown to cooperate in vitro with the homologous recombination (HR) protein, Rad51, to promote homologous DNA pairing and strand displacement (19). We have also shown that the purified PSF·p54(nrb) complex stimulates DNA non-homologous end joining (NHEJ) 10-fold in a reconstituted cell-free DNA double-strand break (DSB) repair assay (20). PSF·p54(nrb) binds the repair substrate in vitro, internal to the ends, and cooperates functionally with Ku to form a preligation complex (21,22). HR and NHEJ are the two major pathways of DSB repair in human cells, and the biochemical data suggest that PSF or the PSF·p54(nrb) complex might be involved in both.
Prior studies of the role of PSF·p54(nrb) in the DNA damage response have used reconstituted in vitro systems. Here, we report the results of a direct genetic investigation of whether p54(nrb) is involved in the DNA damage response in vivo in human cells. We show that attenuation of p54(nrb) expression produces a DSB repair-deficient, radiosensitive phenotype.
MATERIALS AND METHODS
Cells and culture conditions
HeLa cervical carcinoma cells were grown in Dulbecco’s Minimal Essential Medium supplemented with 10% FBS, 2 mM glutamine and antibiotics. IMR-90 normal diploid human lung fibroblasts were grown in modified Eagle’s Minimal Essential Medium supplemented with 10% FBS, 1.5 g/l sodium bicarbonate, 0.1 mM non-essential amino acids, 1.0 mM sodium pyruvate and antibiotics. Experiments were performed at population doubling level of 32 or lower. HCT 116 near-diploid colorectal carcinoma cells were grown in McCoy’s 5A medium supplemented with 10% FBS and antibiotics.
siRNA treatment and p54(nrb) expression detection
Human NONO mRNA (accession no. NM_007363) was evaluated using the Ambion ‘siRNA Target Finder’ web tool (http://www.ambion.com). We synthesized complementary 21-mer oligonucleotides containing 19 nucleotides of complementary RNA sequence flanked at the 3′ side by two thymidylate residues (sense strand, GGCUUGACUAUUGACCUGATT; antisense strand UCAGGUCAAUAGUCAAGCCTT). These were annealed and transfected into IMR-90 human diploid fibroblasts or HeLa cervical carcinoma cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Control cell populations were transfected in parallel using Silencer Negative Control #1 siRNA (Ambion, Austin, TX). The concentration of siRNA was 30 nM. Attenuation of mRNA levels was determined by real-time reverse transcriptase PCR using β-actin mRNA as an internal standard. Total RNA was isolated using Trizol Reagent (Invitrogen, Carlsbad, CA). DNase treatment was performed to remove residual DNA before reverse transcriptase (RT) PCR. RT-PCR was performed in 25 µl using the Qiagen OneStep RT-PCR kit (QIAGEN, Valencia, CA. Reactions contained 800 ng of input RNA. Primers for p54(nrb) were d(TTGTGGGAAATCTTCCTCCCGACA) and d(GGGTTTCCAAGCGGATAAAGCCAA); primers for β-actin were d(AGTCCTGTGGCATCCACGAAACTA) and d(ACTGTGTTGGCGTACAGGTCTTTG). Levels of p54(nrb) protein were determined by immunofluorescence and immunoblotting using anti-p54 mouse monoclonal antibody (1:2000, Clone 3, BD Biosciences Pharmingen, San Diego, CA) and Alexa Fluor 488- or enzyme-conjugated secondary antibodies. Other antibodies were: mouse anti-β–actin (1:5000, Sigma-Aldrich, St. Louis, MO), mouse anti-Ku80 antibody (1:200, clone 111, Lab Vision Corporation, Fremont, CA), mouse anti-Ku70 antibody (1:200, clone N3H10, Lab Vision Corporation, Fremont, CA) and mouse anti-XRCC4 antibody (1:250, BD Bioscience, San Jose, CA).
γ-H2AX focus formation assay
At 36 h post-transfection, cells received 0.5 Gy of 137Cs gamma radiation at a dose rate of 1 Gy min−1. They were allowed to recover for the indicated times, fixed with 4% paraformaldehyde, permeabilized and blocked by incubation in PBS containing 0.5% Triton X-100, 15% goat serum, 0.2% fish skin gelatin and 0.03% NaN3. Cells were then stained with mouse monoclonal anti-γ-H2AX antibody (1:500, clone JBW301, Millipore Corporation, Billerica, MA) and Alexa Fluor 488-conjugated secondary antibody and counterstained with 4′,6-diamidino-2-phenylindole (DAPI). A minimum of 120 nuclei per experimental group were scored by a blinded observer.
Chromosomal aberration assay
To create stable cell lines, d(TGCTGTATAACAGAACCGTATGTACGGTTTTGGCCACTGACTGACCGTACATAGTTCTGTTATA) and d(CCTGTATAACAGAACTATGTACGGTCAGTCAGTGGCCAAAACCGTACATACGGTTCTGTTATAC) were annealed and inserted into pcDNA6.2-GW/EmGFP-miR (Invitrogen). This vector and pcDNA6.2-GW/EmGFP-miR-neg control plasmid were transfected into HCT 116 cells, incubated overnight in McCoy’s 5A medium without antibiotics, and subcultured in complete McCoy’s 5A medium supplemented with 18 mg/ml Blasticidin (Invitrogen). Cells were cultured for 2 weeks with replacement of the Blasticidin-containing medium every 3–4 days. EmGFP-expressing colonies were captured on Whatman 3 MM filter paper (GE Healthcare) and expanded with subculturing in Blasticidin-containing medium. Proteins were extracted and analyzed by immunoblotting.
HCT 116-derived stable cell lines were subcultured in T-25 flasks and allowed to reach 60–80% confluency, irradiated using the 137Cs source, and incubated in fresh medium for 18 h. Colcemid was added to 0.1 μg/ml, and incubation was continued for 2 h at 37°C. Cells were trypsinized, collected and resuspended in 2 ml of pre-warmed 0.075 M KCl. The suspension was diluted to 45 ml with 0.075 M KCl and incubated for 45 min at 37°C. Five drops of freshly prepared fixative MeOH:HOAc [3:1 (v:v)] were added, and cells were collected by centrifugation. Supernatant was removed, leaving 0.5 ml, and 5 ml of fixative was gradually added. Cells were collected, washed twice in fixative and stored at 4°C until slides were prepared. The cell suspension was dropped onto slides and air-dried, then stained with Giemsa. One hundred metaphase spreads were scored by a cytogeneticist for abnormalities, including chromosome fragments, dicentric chromosomes and translocations. Significance was evaluated using Fisher’s exact test.
Clonogenic survival assay
Clonogenic survival assays were performed essentially as described (23). Cultures were trypsinized and 500–4000 cells were seeded in six-well plates, allowed to attach and irradiated using a 137Cs source at a dose rate of 0.85 Gy min−1. Cultures were incubated at 37°C in a humidified 5% CO2 atmosphere for 7 days with one change of medium. Staining was with 0.25% crystal violet and 3.7% formaldehyde in 80% methanol. Colonies of ≥50 cells were counted. Survival rates were determined as a function of radiation dose [S(D)] = colonies/cells plated, where D is expressed in units of Gray (Gy). Surviving Fractions [SF = S(D)/S(0)] were calculated, where S(0) is survival at 0 Gy (mock-irradiated). The radiation survival curves were fitted according to the linear-quadratic model: S(D)/S(0) = exp(αD + βD2), where α and β parameters are determined by weighted, stratified and linear regression (23). Differences between treatment groups were evaluated for significance based on an F test. The mean inactivation dose (MID, integral under survival curve from 0 to infinity) was calculated by numeric integration using the Gaussian-Laguerre quadrature (24). The sensitizing enhancement ratio (SER) was defined as MID(control miRNA)/MID[p54(nrb)RNA].
RESULTS
Attenuation of p54(nrb) expression with siRNA
To investigate the function of the PSF·p54(nrb) complex in vivo, we first determined the ability to attenuate expression of each subunit using siRNA. We chemically synthesized and annealed 21-nucleotide double-strand RNAs corresponding to the coding region of each gene and transfected them individually into IMR-90 human diploid fibroblast cells. Attenuation of PSF expression was variable and incomplete (data not shown) and was not investigated further. A previous report also described difficulty in attenuating PSF expression in cultured cells (12). Transfection with p54(nrb) siRNA, by contrast, consistently reduced mRNA expression by about 75%, as measured by real-time reverse transcriptase PCR with β-actin mRNA as an internal standard (Figure 1A). There was a similar reduction in p54(nrb) protein levels (Figure 1B). Transfection with p54(nrb) siRNA did not affect PSF, Ku70, Ku80 or XRCC4 protein levels, indicating specificity. Transfection with p54(nrb) siRNA attenuated p54(nrb) expression in at least 80% of cells in the IMR-90 population, with no apparent affect on morphology or viability (Figure 1C). Essentially identical RT-PCR and immunoblotting results were obtained using the same siRNAs to transfect HeLa cells (Supplementary Figure S1).
Figure 1.

Attenuation of p54(nrb) expression using siRNA. (A) Attenuation of p54(nrb) mRNA in IMR-90 cells 36 h after transfection with control or p54(nrb) siRNA. Total RNA was quantified by real time RT-PCR using an actin internal control. Bars denote standard error based on 3 replicate transfections. Asterisks indicate significant differences between groups at *P < 0.05 and **P < 0.01 levels based on Student’s t-test. (B) Attenuation of p54(nrb) expression detected by immunoblotting. Cells were transfected with siRNA as in A and extracts were analyzed as described in ‘Materials and Methods’ section. (C) Attenuation of p54(nrb) expression detected by immunofluorescence. Fixed cells were stained for anti-p54(nrb) expression and counterstained with DAPI.
Attenuation of p54(nrb) expression delays DSB repair
Prior in vitro studies predict that attenuation of p54(nrb) expression should affect DSB repair. To investigate the phenotype associated with attenuation of p54(nrb), we induced DSBs by exposure to ionizing radiation and monitored their fate using a histone γ-H2AX focus assay. Phosphorylation of H2AX serine 139 creates the γ-isoform, producing foci that can be visualized by immunostaining using anti-γ-H2AX antibody. These foci correspond 1:1 with unrepaired DSBs (25,26). An advantage to the use of this single-cell assay for siRNA-treated cells is that a small fraction of non-transfected cells (if present) would not alter the modal number of foci per cell.
Following transfection with p54(nrb) or control siRNA, IMR-90 cells were treated with 0.5 Gy of 137Cs gamma radiation. Figure 2A shows the appearance of typical immunostained cells either without irradiation or with irradiation and a 0.5 h or 2 h recovery period. Figure 2B and C provide quantification based on scoring by a blinded observer. Transfected cell populations had a background of about 3 foci/cell in the absence of irradiation. This was the same for p54 siRNA and control siRNA. Transfected cells had a mode of about 14 foci/cell at 30 min after irradiation, roughly consistent with the expected number of DSBs at this radiation dose (27,28). Again, there was no difference between p54 siRNA and control siRNA. As the cells recovered, a marked difference between the two populations emerged. At the 2 h time point, control cells showed significant recovery, but p54(nrb)-attenuated cells did not. At the 4 h time point, the difference between the control and p54(nrb) groups persisted, although it had narrowed. At the 8 h time point, only a few repair resistant foci remained, with again no significant difference between groups. Results suggest that although attenuation of p54(nrb) has no effect on the initial level of DNA damage, it delays DSB repair.
Figure 2.

Delay in resolution of γ-H2AX foci in IMR-90 cells treated with p54(nrb) siRNA. IMR-90 cells were irradiated with 0.5 Gy at 36 h post-transfection, allowed to recover for indicated times, fixed and stained with anti-γ-H2AX. The antibody detects phosphorylation at H2AX Ser 139, a marker of unrepaired DNA double-strand breaks. (A) Representative nuclei treated as indicated. (B) Histogram showing number of foci in mock-irradiated cells. (C) Histograms showing number of foci in cells that were exposed to 0.5 Gy of ionizing radiation and allowed to recover for the indicated times at 37°C.
Attenuation of p54(nrb) expression increases the frequency of radiation-induced chromosomal aberrations
The finding that p54(nrb) siRNA treatment delayed, but did not abolish, DSB repair, suggests that the primary effect is on the rapid, initial phase of DSB repair, which is believed to be mediated by the NHEJ pathway. Prior studies have shown that most DSBs are repaired eventually, even in severely compromised genetic backgrounds [e.g. DNA ligase IV-null (29)]. However, this delayed repair, which has been attributed to ‘backup’ or ‘alternative’ pathways, may be less accurate and does not assure reproductive survival (30–33).
To investigate whether the effect on the rapid, initial phase of DSB repair was biologically significant, it was necessary to establish stable cell lines with reduced p54(nrb) expression. Because IMR-90 cells have a limited lifespan in culture, they were unsuitable for creating stable cell lines. Instead, we used HCT 116 a, p53-positive human colorectal cancer cell line. HCT 116 cells have a near-diploid karyotype and are suitable for both chromosomal aberration and survival assays. We transfected cells with a vector that produces miRNA by processing of an EmGFP-encoding mRNA. The vector also expresses a selectable drug-resistance marker. Multiple clonal cell lines were isolated from each miRNA-transfected population. All were positive for EmGFP expression, which indicates the presence of miRNA-bearing transcripts. The cell lines expressing p54(nrb) miRNA showed markedly reduced levels of p54(nrb) protein, relative to cell lines expressing control miRNA (Figure 3A).
Figure 3.

Chromosomal aberration assays. (A) Near-diploid HCT 116 colorectal carcinoma cells were transfected with indicated miRNA plasmids to create stable cell lines. Cell clones were isolated, expanded and characterized for p54(nrb) expression by immunoblotting as described in ‘Material and Methods’ section. Replicate immunoblots were probed with anti-p54(nrb) and anti-GAPDH as an internal control. (B) Metaphase spreads were prepared and analyzed from irradiated p54(nrb)-deficient HCT 116 cell line as described in ‘Materials and Methods’ section. A representative image is shown. (C) Summary of chromosomal aberration data. Fifty metaphases were analyzed for each clonal cell line. The two clonal cell lines of each type (control and p54(nrb)-deficient) gave very similar results and the data were pooled for analysis. P-values were determined using a two-tailed Fisher’s exact test.
The stably transfected cell lines were exposed to 2 Gy of ionizing radiation, allowed to recover, and arrested at the first metaphase following irradiation. Figure 3B shows a typical chromosome spread for irradiated, p54(nrb) siRNA-transfected cells, and Figure 3C shows a quantification of chromosomal abnormalities in each treatment group. In non-irradiated cells, there was no significant difference in the frequency of abnormal metaphases between p54(nrb) and control miRNA-transfected lines (P = 0.43). By contrast, in the 2 Gy-irradiated populations, p54(nrb) miRNA-expressing cells had a significantly increased frequency of abnormal metaphases (P = 0.014). The most common abnormalities were acentric chromosomal fragments, suggesting the presence of a subset of breaks that are not repaired prior to chromosome replication. Double minute chromosomes were the next most common aberration, with chromatid-type breaks, deletions, translocations and a dicentric chromosome present in a few instances (Supplementary Table S1).
Attenuation of p54(nrb) decreases clonogenic survival following irradiation
To measure radiation sensitivity, we performed clonogenic survival assays in stable miRNA-expressing HCT 116 cell lines as described in ‘Materials and Methods’ section. Colony size was appreciably smaller for p54(nrb) miRNA-expressing cells (Figure 4A), and the plating efficiency in the absence of radiation was lower (16.3% versus 45%). The reduced colony size indicates slower growth of the clones under the conditions used. Slower growth and the lower plating efficiency cannot be attributed to changes in DSB repair, because they were seen even in the absence of exposure to ionizing radiation. There must be some other cause, such as a defect in RNA synthesis, consistent with previous reports that p54(nrb) is involved in various aspects of RNA biogenesis (see ‘Introduction’ section).
Figure 4.
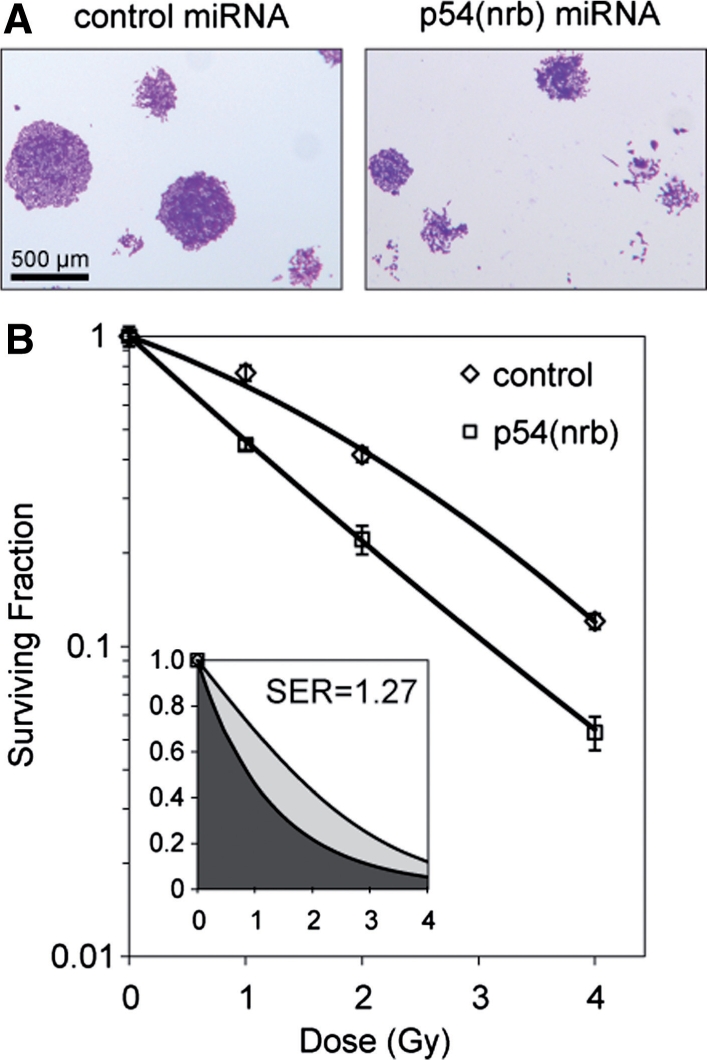
Clonogenic survival assay. (A) Clonogenic survival assays were performed as described in ‘Materials and Methods’ section. Note that p54(nrb) miRNA colonies were smaller. (B) Results of survival assays using clone number 2 for p54(nrb)-miRNA transfected cells. Assays were performed in triplicate with standard errors shown. Plating efficiency was 45% for non-irradiated control cells and 16.3% for non-irradiated p54(nrb) knockdown cells. Ratio of surviving fraction at 2 Gy was 1.97. Inset shows fitted linear-quadratic curves plotted using a linear axis. Mean inhibitory doses were determined based on integrals under the curves from 0 to ∞. MID for control cells was 2.35 Gy and for p54(nrb) cells was 2.98 Gy and the sensitization ratio was thus 1.27. Statistical analysis was performed as described in Materials and Methods section. Radiobiological parameters were as follows: control, α = −0.314 (95% CI from −0.218 to −0.410), β = −0.054 (95% CI from −0.028 to −0.080), R2 = 0.996; p54(nrb) α = −0.790 (95% CI from −0.652 to −0.928), β = 0.015 (95% CI from 0.057 to −0.027), R2 = 0.992. The null hypothesis that the survival data for the two different stable cell lines can best be described by the same fitted curve was rejected based on an F-test (P < 0.001).
The radiation survival data are quantified in Figure 3B. The survival curve for the control-transfected population is typical for mammalian cells, with a ‘shoulder’ in the 0–2 Gy region, reflecting repair of radiation-induced DNA damage. The data can be fitted almost exactly with a linear-quadratic model. By contrast, the p54(nrb) miRNA-transfected population shows a nearly log-linear response, with no evident shoulder. The difference between the two cell clones is highly significant (P < 0.001). Similar results were obtained with the other stable cell lines (data not shown). The effectiveness of a radiosensitizing treatment can be quantified based on the horizontal shift in the survival curve at an arbitrarily chosen level of survival (dose enhancement ratio), or more robustly, by taking the ratio of the integrals under the survival curves from zero to infinity (sensitization enhancement ratio) (24). The inset shows the survival data replotted to illustrate these integrals; the sensitization enhancement ratio thus determined is 1.27, indicative of a moderately strong effect.
We have also investigated the effect of p54(nrb) on clonogenic survival independently in siRNA-transfected IMR90 human diploid fibroblasts (Supplementary Figure 2S). There was an approximately 1.5-fold, statistically significant, decrease in survival at all radiation doses tested. In this experiment, the plating efficiencies were low, the shapes of the survival curves were atypical, and we were not able to fit the curves with a quantitative model. We believe that there was an interaction between toxicity of the transient transfection procedure and the stress of the clonogenic survival assay that limited the ability to perform a quantitative analysis. Despite this, results were qualitatively consistent with those obtained using the HCT 116 stable cell lines.
DISCUSSION
We have investigated the effect of p54(nrb) deficiency on repair and survival following induction of DNA double strand breaks. DSBs, which are produced by ionizing radiation and certain recombination enzymes, are more difficult to repair, more cytotoxic and more destabilizing to the genome than most other DNA damage (34). Three lines of evidence indicate that attenuation of p54(nrb) expression causes a deficiency in DSB repair in vivo: delay in resolution of γ-H2AX foci, an increase in chromosomal aberrations at the first metaphase following radiation exposure, and a decrease in clonogenic survival. These repair and survival phenotypes were seen in two different cell types and with two different approaches for gene silencing (siRNA and miRNA directed against distinct sequence regions). The genetic studies presented here complement previous findings that PSF·p54(nrb) stimulates DNA end joining in a reconstituted biochemical system (20,21). Taken together, the evidence strongly supports the involvement of p54(nrb) in the DNA damage and repair response. The available genetic tools have not, so far, allowed us to draw conclusions whether PSF is required for DNA damage and repair in intact cells.
To our knowledge, there are as yet no reports of germ-line p54(nrb) deficiency in humans or other mammalian species. There has been a report of a translocation of PSF and p54(nrb) to the TFE3 helix-loop-helix transcription factor gene in human papillary renal cell carcinoma lines (35). A hybrid transcript is expressed, but its role in oncogenic transformation has not been established. There have also been reports describing two different gene trap alleles affecting the gene encoding TLS/FUS, a different tandem RRM protein. In one case, deficiency resulted in male sterility, modest radiosensitivity in fibroblasts, and severe radiosensitivity in whole mice (36). In the other, deficiency resulted in defective B-cell development, spontaneous chromosomal instability and perinatal death (37). These reports support a more general hypothesis that multifunctional RRM proteins affect both gene regulation and genome maintenance.
The phenotypic characterization of p54(nrb) knockdown in human cells, presented here, does not provide direct insight into how this protein influences DSB repair. However, earlier studies showed that PSF·p54(nrb) binds DNA directly via DNA binding domain that overlaps the N-terminal region and RRM1 of the PSF subunit (38,39). In our previous studies, PSF·p54(nrb) and Ku bound linear DNA fragments independently, but cooperated to form a functional preligation complex (20). We suggest the same mechanism may apply in vivo. This is consistent with recent observations in our laboratory that PSF and p54(nrb) rapidly relocalize to sites of laser microbeam-induced DNA damage (Ha K. et al., manuscript in preparation). It will be of interest to further investigate the repair defects in p54-deficient cells, including whether both NHEJ and HR are affected and whether the cells are sensitized to DNA damaging agents other than ionizing radiation.
Another potential effect of p54(nrb) knockdown, which is not mutually exclusive, is an effect on the synthesis and processing of mRNAs for other repair proteins. Many studies have shown that p54(nrb) affects synthesis and processing of specific mRNAs, although to our knowledge there is no direct evidence for an effect on levels of mRNAs for DNA damage response genes. Indeed, we show here that siRNA transfection does not change the levels of the known DSB repair proteins, Ku70, Ku80 and XRCC4, all of which participate in the rapid early phase of DSB repair that is affected by p54(nrb) deficiency.
Our clonogenic survival assay showed that attenuation of p54(nrb) expression largely eliminated the shoulder in the radiation survival curve. This has potential clinical significance, in that the shoulder region corresponds to the dose range used for single clinical radiation fractions (∼2 Gy). Inhibition of p54(nrb) in tumor cells would be predicted to significantly increase therapeutic gain in radiotherapy. In this respect, it is of interest that a small, natural, virally encoded RNA has been shown to inhibit the binding of the PSF·p54(nrb) to DNA (39). Although this prior study dealt with promoter binding activity, not with DNA repair effects, the finding that there is cross-talk between DNA and RNA-binding sites in the PSF·p54(nrb) complex suggests a possible approach for the development of radiosensitizers based on RNAs or RNA analogs.
A potential concern regarding the survival data is that, although the difference between wild-type and knockdown cells is highly significant (P < 0.001), the magnitude of this difference is relatively modest. It may be that the residual activity in knockdown cells provides sufficient repair function to explain the observed levels of survival. Alternatively, other proteins may compensate for the absence of p54(nrb). Human cells encode a closely related protein, PSP1α. It could be that PSP1α partially compensates for p54(nrb) function in deficient cells, a possibility that has not yet been tested.
In addition to the role in DSB repair defined in the present study, PSF and p54(nrb) participate, separately or together, in diverse aspects of RNA biogenesis. Although more than 25 published studies address these functions, there is no consensus on their exact role: PSF or p54(nrb) have been implicated in regulation of transcriptional initiation, 3′-end formation, splicing and nuclear-cytoplasmic transport. This apparent complexity may reflect interaction of PSF and p54(nrb) with diverse partner proteins. Androgen receptor (14,15,40), thyroid hormone receptor (13), retinoid X receptor (13), TORC protein (16) and RNA polymerase II (41) interactions contribute to effects of PSF·p54(nrb) on RNA synthesis initiation. XRN2 interaction contributes to effects on 3′-end formation (12). U5 and snRNP interactions contribute to effects on splicing (42–44), and matrin 3 (10) and PSP1 (11) interactions contribute to effects on nuclear-cytoplasmic transport.
These observations raise the question whether there are also specific partners involved in PSF·p54(nrb) repair function. Topoisomerase I interacts directly with PSF·p54(nrb) in vivo and in vitro (45,46) and has itself been shown to migrate to sites of laser-induced DNA damage (47). A more speculative candidate is PARP-1, which senses single-strand breaks and, under some conditions, DSBs. Like PSF·p54(nrb), PARP-1 is transiently recruited to sites of laser-induced DNA damage, suggesting a common mechanism (48). Further studies will be required to investigate these possibilities.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
US Public Health Service Grant, CA 98239, and by the NIH Roadmap for Biomedical Research. Funding for open access charge: US Public Health Service grant.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the Medical College of Georgia Imaging Core Facility and Office of Biostatistics and Bioinformatics for their services.
REFERENCES
- 1.Maris C, Dominguez C, Allain FH. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272:2118–2131. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 2.Birney E, Kumar S, Krainer AR. Analysis of the RNA-recognition motif and RS and RGG domains: conservation in metazoan pre-mRNA splicing factors. Nucleic Acids Res. 1993;21:5803–5816. doi: 10.1093/nar/21.25.5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kenan DJ, Query CC, Keene JD. RNA recognition: towards identifying determinants of specificity. Trends Biochem. Sci. 1991;16:214–220. doi: 10.1016/0968-0004(91)90088-d. [DOI] [PubMed] [Google Scholar]
- 4.Ding J, Hayashi MK, Zhang Y, Manche L, Krainer AR, Xu RM. Crystal structure of the two-RRM domain of hnRNP A1 (UP1) complexed with single-stranded telomeric DNA. Genes Dev. 1999;13:1102–1115. doi: 10.1101/gad.13.9.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moran-Jones K, Wayman L, Kennedy DD, Reddel RR, Sara S, Snee MJ, Smith R. hnRNP A2, a potential ssDNA/RNA molecular adapter at the telomere. Nucleic Acids Res. 2005;33:486–496. doi: 10.1093/nar/gki203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reim I, Stanewsky R, Saumweber H. The puff-specific RRM protein NonA is a single-stranded nucleic acid binding protein. Chromosoma. 1999;108:162–172. doi: 10.1007/s004120050365. [DOI] [PubMed] [Google Scholar]
- 7.Newberry EP, Latifi T, Towler DA. The RRM domain of MINT, a novel Msx2 binding protein, recognizes and regulates the rat osteocalcin promoter. Biochemistry. 1999;38:10678–10690. doi: 10.1021/bi990967j. [DOI] [PubMed] [Google Scholar]
- 8.Hamimes S, Bourgeon D, Stasiak AZ, Stasiak A, Van Dyck E. Nucleic acid-binding properties of the RRM-containing protein RDM1. Biochem. Biophys. Res. Commun. 2006;344:87–94. doi: 10.1016/j.bbrc.2006.03.154. [DOI] [PubMed] [Google Scholar]
- 9.Tresaugues L, Dehe PM, Guerois R, Rodriguez-Gil A, Varlet I, Salah P, Pamblanco M, Luciano P, Quevillon-Cheruel S, Sollier J, et al. Structural characterization of Set1 RNA recognition motifs and their role in histone H3 lysine 4 methylation. J. Mol. Biol. 2006;359:1170–1181. doi: 10.1016/j.jmb.2006.04.050. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Z, Carmichael GG. The fate of dsRNA in the nucleus: a p54(nrb)-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell. 2001;106:465–475. doi: 10.1016/s0092-8674(01)00466-4. [DOI] [PubMed] [Google Scholar]
- 11.Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, Bennett CF, Zhang MQ, Spector DL. Regulating gene expression through RNA nuclear retention. Cell. 2005;123:249–263. doi: 10.1016/j.cell.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 12.Kaneko S, Rozenblatt-Rosen O, Meyerson M, Manley JL. The multifunctional protein p54nrb/PSF recruits the exonuclease XRN2 to facilitate pre-mRNA 3′ processing and transcription termination. Genes Dev. 2007;21:1779–1789. doi: 10.1101/gad.1565207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mathur M, Tucker PW, Samuels HH. PSF is a novel corepressor that mediates its effect through Sin3A and the DNA binding domain of nuclear hormone receptors. Mol. Cell Biol. 2001;21:2298–2311. doi: 10.1128/MCB.21.7.2298-2311.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong X, Sweet J, Challis JR, Brown T, Lye SJ. Transcriptional activity of androgen receptor is modulated by two RNA splicing factors, PSF and p54nrb. Mol. Cell Biol. 2007;27:4863–4875. doi: 10.1128/MCB.02144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishitani K, Yoshida T, Kitagawa H, Ohta H, Nozawa S, Kato S. p54nrb acts as a transcriptional coactivator for activation function 1 of the human androgen receptor. Biochem. Biophys. Res. Commun. 2003;306:660–665. doi: 10.1016/s0006-291x(03)01021-0. [DOI] [PubMed] [Google Scholar]
- 16.Amelio AL, Miraglia LJ, Conkright JJ, Mercer BA, Batalov S, Cavett V, Orth AP, Busby J, Hogenesch JB, Conkright MD. A coactivator trap identifies NONO (p54nrb) as a component of the cAMP-signaling pathway. Proc. Natl Acad. Sci. USA. 2007;104:20314–20319. doi: 10.1073/pnas.0707999105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akhmedov AT, Lopez BS. Human 100-kDa homologous DNA-pairing protein is the splicing factor PSF and promotes DNA strand invasion. Nucleic Acids Res. 2000;28:3022–3030. doi: 10.1093/nar/28.16.3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akhmedov AT, Bertrand P, Corteggiani E, Lopez BS. Characterization of two nuclear mammalian homologous DNA-pairing activities that do not require associated exonuclease activity. Proc. Natl Acad. Sci. USA. 1995;92:1729–1733. doi: 10.1073/pnas.92.5.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morozumi Y, Takizawa Y, Takaku M, Kurumizaka H. Human PSF binds to RAD51 and modulates its homologous-pairing and strand-exchange activities. Nucleic Acids Res. 2009;37:4296–4307. doi: 10.1093/nar/gkp298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bladen CL, Udayakumar D, Takeda Y, Dynan WS. Identification of the polypyrimidine tract binding protein-associated splicing factor·p54(nrb) complex as a candidate DNA double-strand break rejoining factor. J. Biol. Chem. 2005;280:5205–5210. doi: 10.1074/jbc.M412758200. [DOI] [PubMed] [Google Scholar]
- 21.Udayakumar D, Bladen CL, Hudson FZ, Dynan WS. Distinct pathways of nonhomologous end joining that are differentially regulated by DNA-dependent protein kinase mediated phosphorylation. J. Biol. Chem. 2003;278:41631–41635. doi: 10.1074/jbc.M306470200. [DOI] [PubMed] [Google Scholar]
- 22.Udayakumar D. Augusta (GA): Medical College of Georgia; 2005. Identification and characterization of two new players in double-strand break repair – PSF and p54(nrb). Ph.D. Thesis. [Google Scholar]
- 23.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 24.Fertil B, Dertinger H, Courdi A, Malaise EP. Mean inactivation dose: a useful concept for intercomparison of human cell survival curves. Radiat. Res. 1984;99:73–84. [PubMed] [Google Scholar]
- 25.Rothkamm K, Löbrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc. Natl Acad. Sci. USA. 2003;100:5057–5062. doi: 10.1073/pnas.0830918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iliakis GE, Cicilioni O, Metzger L. Measurement of DNA double-strand breaks in CHO cells at various stages of the cell cycle using pulsed field gel electrophoresis: calibration by means of 125I decay. Int. J. Radiat. Biol. 1991;59:343–357. doi: 10.1080/09553009114550321. [DOI] [PubMed] [Google Scholar]
- 28.Ruiz de Almodovar JM, Steel GG, Whitaker SJ, McMillan TJ. A comparison of methods for calculating DNA double-strand break induction frequency in mammalian cells by pulsed-field gel electrophoresis. Int. J. Radiat. Biol. 1994;65:641–649. doi: 10.1080/09553009414550751. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Zeng ZC, Perrault AR, Cheng X, Qin W, Iliakis G. Genetic evidence for the involvement of DNA ligase IV in the DNA-PK-dependent pathway of non-homologous end joining in mammalian cells. Nucleic Acids Res. 2001;29:1653–1660. doi: 10.1093/nar/29.8.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Errami A, Finnie NJ, Morolli B, Jackson SP, Lohman PH, Zdzienicka MZ. Molecular and biochemical characterization of new X-ray-sensitive hamster cell mutants defective in Ku80. Nucleic Acids Res. 1998;26:4332–4338. doi: 10.1093/nar/26.19.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaganay-Juery S, Muller C, Marangoni E, Abdulkarim B, Deutsch E, Lambin P, Calsou P, Eschwege F, Salles B, Joiner M. Decreased DNA-PK activity in human cancer cells exhibiting hypersensitivity to low-dose irradiation. Br. J. Cancer. 2000;83:514–518. doi: 10.1054/bjoc.2000.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Girard PM, Kysela B, Harer CJ, Doherty AJ, Jeggo PA. Analysis of DNA ligase IV mutations found in LIG4 syndrome patients: the impact of two linked polymorphisms. Hum. Mol. Genet. 2004;13:2369–2376. doi: 10.1093/hmg/ddh274. [DOI] [PubMed] [Google Scholar]
- 33.Chen F, Peterson SR, Story MD, Chen DJ. Disruption of DNA-PK in Ku80 mutant xrs-6 and the implications in DNA double-strand break repair. Mutat. Res. 1996;362:9–19. doi: 10.1016/0921-8777(95)00026-7. [DOI] [PubMed] [Google Scholar]
- 34.Nickoloff JA, Hoekstra MF. DNA Damage and Repair. Totowa, N.J: Humana Press; 1998. [Google Scholar]
- 35.Clark J, Lu YJ, Sidhar SK, Parker C, Gill S, Smedley D, Hamoudi R, Linehan WM, Shipley J, Cooper CS. Fusion of splicing factor genes PSF and NonO (p54nrb) to the TFE3 gene in papillary renal cell carcinoma. Oncogene. 1997;15:2233–2239. doi: 10.1038/sj.onc.1201394. [DOI] [PubMed] [Google Scholar]
- 36.Kuroda M, Sok J, Webb L, Baechtold H, Urano F, Yin Y, Chung P, de Rooij DG, Akhmedov A, Ashley T, et al. Male sterility and enhanced radiation sensitivity in TLS(-/-) mice. EMBO J. 2000;19:453–462. doi: 10.1093/emboj/19.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hicks GG, Singh N, Nashabi A, Mai S, Bozek G, Klewes L, Arapovic D, White EK, Koury MJ, Oltz EM, et al. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat. Genet. 2000;24:175–179. doi: 10.1038/72842. [DOI] [PubMed] [Google Scholar]
- 38.Urban RJ, Bodenburg YH, Wood TG. NH2 terminus of PTB-associated splicing factor binds to the porcine P450scc IGF-I response element. Am. J. Physiol. Endocrinol. Metab. 2002;283:E423–E427. doi: 10.1152/ajpendo.00057.2002. [DOI] [PubMed] [Google Scholar]
- 39.Song X, Sun Y, Garen A. Roles of PSF protein and VL30 RNA in reversible gene regulation. Proc. Natl Acad. Sci. USA. 2005;102:12189–12193. doi: 10.1073/pnas.0505179102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuwahara S, Ikei A, Taguchi Y, Tabuchi Y, Fujimoto N, Obinata M, Uesugi S, Kurihara Y. PSPC1, NONO, and SFPQ are expressed in mouse Sertoli cells and may function as coregulators of androgen receptor-mediated transcription. Biol. Reprod. 2006;75:352–359. doi: 10.1095/biolreprod.106.051136. [DOI] [PubMed] [Google Scholar]
- 41.Emili A, Shales M, McCracken S, Xie W, Tucker PW, Kobayashi R, Blencowe BJ, Ingles CJ. Splicing and transcription-associated proteins PSF and p54nrb/nonO bind to the RNA polymerase II CTD. RNA. 2002;8:1102–1111. doi: 10.1017/s1355838202025037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng R, Dye BT, Perez I, Barnard DC, Thompson AB, Patton JG. PSF and p54nrb bind a conserved stem in U5 snRNA. RNA. 2002;8:1334–1347. doi: 10.1017/s1355838202022070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gozani O, Patton JG, Reed R. A novel set of spliceosome-associated proteins and the essential splicing factor PSF bind stably to pre-mRNA prior to catalytic step II of the splicing reaction. EMBO J. 1994;13:3356–3367. doi: 10.1002/j.1460-2075.1994.tb06638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kameoka S, Duque P, Konarska MM. p54(nrb) associates with the 5′ splice site within large transcription/splicing complexes. EMBO J. 2004;23:1782–1791. doi: 10.1038/sj.emboj.7600187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Straub T, Grue P, Uhse A, Lisby M, Knudsen BR, Tange TO, Westergaard O, Boege F. The RNA-splicing factor PSF/p54 controls DNA-topoisomerase I activity by a direct interaction. J. Biol. Chem. 1998;273:26261–26264. doi: 10.1074/jbc.273.41.26261. [DOI] [PubMed] [Google Scholar]
- 46.Trzcinska-Daneluti AM, Gorecki A, Czubaty A, Kowalska-Loth B, Girstun A, Murawska M, Lesyng B, Staron K. RRM proteins interacting with the cap region of topoisomerase I. J. Mol. Biol. 2007;369:1098–1112. doi: 10.1016/j.jmb.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 47.Mielke C, Kalfalah FM, Christensen MO, Boege F. Rapid and prolonged stalling of human DNA topoisomerase I in UVA-irradiated genomic areas. DNA Repair (Amst) 2007;6:1757–1763. doi: 10.1016/j.dnarep.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 48.Mortusewicz O, Ame JC, Schreiber V, Leonhardt H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007;35:7665–7675. doi: 10.1093/nar/gkm933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.