

Abstract
FOXP3+ regulatory T cells (Tregs) are central to the maintenance of self-tolerance and immune homeostasis. The mechanisms of action and cellular targets for Treg mediated suppression remain controversial. The critical adhesion molecules utilized by Tregs for the interaction with their target cells have not been well characterized. We show that human CD4+FOXP3+CD25hi cells (hTregs) suppress the activation of mouse responders as efficiently as mouse Tregs. LFA-1 (CD11a/CD18) on the hTregs is critical for their suppressor function, since suppression can be reversed with blocking anti-hCD11a or -hCD18 mAb. Tregs from patients with LFA-1 deficiency fail to suppress human and mouse responders. Mouse CD4+ T cells deficient in ICAM-1 can be suppressed by hTregs, indicating that the hTregs target mouse DCs through the binding of human LFA-1 to mouse ICAM-1. Co-culture of mouse DCs with hTregs, but not hTregs from LFA-1 deficient patients, prevented the upregulation of CD80/CD86 on the DCs and their capacity to activate responder T cells. Lastly, IL-2 is not required for hTreg suppressor function under optimal stimulatory condition and IL-2 consumption plays no role in hTreg-mediated suppression. Taken together, one of the mechanisms of Treg-mediated suppression functions across species and mediates an LFA-1/ICAM-1 dependent interaction between Tregs and DCs.
Keywords: Human regulatory T cells, Dendritic Cells, Adhesion Molecules, Tolerance/Suppression/Anergy, Autoimmunity
Introduction
Regulatory T cells (Tregs/CD4+FOXP3+ T cells) have now been shown to play critical roles in all aspects of normal and pathologic immune responses (1, 2). They are central to the maintenance of self-tolerance and the prevention of autoimmunity. A detailed cellular and molecular understanding of their mechanism of action would provide a strong foundation for manipulating their function therapeutically. Numerous mechanisms have been proposed to explain the suppressive functions of Tregs, but none appears to be unifying (3, 4). Most studies have used an in vitro coculture assay developed by us (5) and others (6) as the major experimental tool. Proposed mechanisms have included secretion of suppressor cytokines (IL-10, TGF-b, IL-35) (7-9), CTLA-4/CD80-CD86 interactions (10-13), transfer of cAMP from suppressors to responders via gap junctions (14), generation of adenosine (15), IL-2 consumption (16, 17) and cell contact-mediated suppression by a yet uncharacterized membrane molecule. Although transwell experiments suggest that the suppressive activity of Tregs is cell-contact dependent, the secretion of short-range mediators has not been ruled out. A considerable controversy also exists regarding the cellular target for Treg-mediated suppression. Some studies have strongly supported a Treg-T responder cell interaction (18, 19), while others favor a Treg-APC interaction (20, 21). Lastly, the relationship between any of the in vitro properties of Tregs and their in vivo behavior also has been challenged (22). For example, while Tregs are non-responsive to TCR stimulation in vitro, they proliferate in a fashion indistinguishable from conventional CD4+ T cells upon TCR activation in vivo.
While there are several studies addressing the target cells and the role of IL-2 and LFA-1 on mouse Tregs, the characterization of human Tregs is limited. A detailed understanding of the primary target cell and the adhesion molecules involved in Treg cell-contact-mediated suppression would provide a valuable opportunity to design therapeutic methods for manipulating Treg function. One of the major obstacles to defining the molecules involved in Treg-target cell interaction is that adhesion molecules, such as LFA-1/ICAM-1, are also necessary for the interaction of responder T cells with APCs. Therefore, blocking integrins such as LFA-1 in an in vitro suppression assay would affect the activation of the responder cells and the ability to measure suppression. Moreover, neutralization of IL-2 or blocking the IL-2 receptor in the conventional human suppression assay would not permit the analysis of the role of IL-2 in Treg-mediated supppression, since it would affect the activation of the responder cells. To circumvent these obstacles, we have developed a novel in vitro suppression assay consisting of human (h) Tregs as suppressors and mouse (m) CD4+CD25− T cells and APCs as responders. We demonstrate that suppression of T cell activation across the species is highly efficient in vitro, is cell contact-dependent, and is not mediated by IL-10 or TGF-b. Importantly, we define a critical role for LFA-1(CD11a-CD18)/ICAM-1(CD54) interactions in human Treg function. Use of responder CD4+ T cells from mice deficient in ICAM-1 expression demonstrated that human LFA-1 specifically interacts with ICAM-1 on the mouse DCs rather than on the responder T cells and that this interaction is sufficient for suppression of T cell activation. Since activated human T cells respond poorly to mouse IL-2, this assay system also allowed us to determine the requirements for IL-2 in the activation of Treg suppressor function and the contribution of IL-2 consumption to Treg-mediated suppression. These results provide new insights into the cell surface antigens, cellular targets, and cytokines involved in human Treg-mediated, cell contact-dependent suppression and offer a potential therapeutic approach to both augment and reverse Treg suppressor function.
Materials and Methods
Mouse cell purification
C57BL/6, BALB/c, and ICAM-1−/− mice were purchased from Jackson Laboratories (Bar Harbor, ME). Hemagglutinin (HA) TCR Tg mice were maintained at Taconic (Germantown, NY) under National Institute of Allergy and Infectious Diseases (NIAID) contract. Mice were used at 6-10 week of age and housed under specific pathogen-free conditions in the NIAID animal facility in accordance with institutional guidelines. The CD4+CD25−, CD4+CD25+, and CD8+CD25− populations were obtained from peripheral lymph nodes and labeled with CD4 FITC or CD8 FITC and CD25 PE (BD Biosciences) for FACS sorting. APCs were obtained from the spleen by depleting T cells with mouse CD90 microbeads (Miltenyi Biotec) over the AutoMACS using deplete-sensitive program and irradiated at 3,000 rads.
Human cell purification
Peripheral blood was obtained from healthy adult donors through the Department of Transfusion Medicine at the National Institutes of Health. LAD-1 patients were enrolled on protocol 93-I-0119 approved by the NIAID institutional review board. The entire clinical investigation was conducted and informed consent obtained from all patients in accordance with the Declaration of Helsinki. PBMCs were prepared over Ficoll-Paque Plus gradients (GE Healthcare). The CD4+ cells were enriched over the AutoMACS by positive selection with human CD4 microbead (Miltenyi Biotec). The cells were labeled with CD4 FITC, CD127 PE (both BD Biosciences) and CD25 PE-Alexa 700 (Invitrogen) then FACS-sorted with the FACSVantage DiVa or FACSAria flow cytometer for CD4+CD25−, CD4+CD25int (intermediate), and CD4+CD127−CD25hi (high). The purity of the Tregs was assessed by fixing and permeabilizing the cells with a Fixation/Permeabilization kit and staining for FOXP3 with anti-FOXP3 Alexa Fluor 647 mAb clone 236A/E7 (all from eBioscience). Human APCs were obtained by depleting T cells from PBMCs with CD3 microbead (Miltenyi Biotec) using the AutoMACS deplete-sensitive program and irradiating at 4,000 rads.
Antibodies
Anti-human CD25 mAb (daclizumab, Roche) was used at 25 μg/ml. For neutralization of IL-10 and TGFβ, 25 μg/ml anti-hIL10 (25209, R&D Systems), 25 μg/ml anti-mIL-10R (1B1.3a, BD Biosciences), 50 μg/ml anti-TGFβ (1D11, R&D Systems) or 5 μg/ml recombinant human (rh) LAP (R&D Systems) were used. For blocking CTLA-4, 25 μg/ml anti-human CTLA-4 was used (BNI3, BD Biosciences). For blocking CD11a, CD18 and ICAM, 10 μg/ml were used for anti-human CD11a (38 and MEM-83 from Genetex; efalizumab from Genentech), anti-human CD18 (TS1/18, Biolegend and MEM-48, Genetex), anti-human ICAM-1 (15.2, Genetex), anti-human ICAM-2 (CBR-IC2/2, Abcam), anti-human ICAM-3 (76205, R&D Systems) and anti-mouse ICAM-2 (3C4, BD Biosciences).
In vitro suppression assay
For optimal stimulation of fresh hTregs, 96 flat-bottom culture plates (NUNC) were coated overnight with 5 μg/ml anti-human CD3 (UCHT1) and 2.5 μg/ml anti-human CD28 (both eBioscience). For suboptimal stimulation of hTregs, 3 μg/ml anti-CD3 and 1 μg/ml anti-CD28 were plate bound. For suppression of mouse responders, CD4+CD25− or CD8+CD25− cells (5×104) were cultured with irradiated mouse T-depleted splenocytes as APCs (5×104) or mouse splenic DCs (5×103, 10:1 mouse CD4 to DC ratio) and 0.25 μg/ml soluble anti-mouse CD3 (145-2C11, eBiosciences) for 72 h in the presence of varying numbers of fresh hTregs or pre-activated hTregs. In this coculture, the fresh hTregs were activated with plate-bound anti-hCD3/CD28 as described above while the mouse responders were activated with APCs/DCs and soluble anti-mCD3. hTregs were pre-activated by stimulation for 48 h with plate-bound 5 μg/ml UCHT1, 2.5 μg/ml anti-CD28 and 100 U/ml rhu-IL-2 (Peprotech), washed and used in the suppression assay without restimulation. mTregs were pre-activated by stimulation for 48 h with plate-bound anti-mCD3 (2 μg/ml), anti-mCD28 (2 μg/ml) and 100 U/ml rhu-IL-2, washed and used in the suppression assay without restimulation. For suppression of human responders, 5×104 CD4+CD25− cells were cultured in 96 flat-bottom culture plates with irradiated human APCs (5×104) and 0.25 μg/ml soluble OKT3 for 72 h in the presence of varying numbers of fresh hTregs, fresh mTregs or pre-activated mTregs. Fresh mTregs in the cocultures were activated with plate-bound 2 μg/ml anti-mCD3 and 1 μg/ml anti-mCD28 (eBiosciences). Proliferation was measured in triplicates by the incorporation of tritiated thymidine over the last 6-8 h of the coculture. All cells were cultured in complete medium (RPMI 1640 supplemented with 10% heat-inactivated FCS (Atlanta Biologicals), 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, 10 mM HEPES, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate (all BioSource International) and 50 μM 2-mercaptoethanol (Sigma-Aldrich)).
Mouse splenic DCs assay
The splenic dendritic cells (DCs) were obtained from spleens by fragmenting and digesting for 30 min at 37°C in complete medium containing liberase blendzyme II and 2 μg/ml DNase (Roche). The DCs were isolated with CD11c microbead (Miltenyi) over the AutoMACS using posseld2 program. The purity was > 95% based on CD11c APC staining (BD Biosciences). 2×105 DCs were cultured with complete medium in 24-well culture plate (Corning) for 18 h alone or with 1×106 pre-activated hCD25hi or hCD25− cells. The level of CD80/CD86 on the DCs was detected by flow cytometry with the BD FACSCalibur after staining with CD80 FITC, CD86 PE and CD11c APC (BD Biosciences). All flow cytometric data were analyzed with FlowJo (Tree Star Inc). To test the function of the DCs after 18 h culture, the DCs were washed with complete medium containing 0.1 M EDTA to disrupt any DC-T cell complexes and positively selected with CD11c microbead over the AutoMACS. 5×103 DCs were cultured for 48 h with complete medium in 96 flat-bottom culture plates (Corning) plus 5×104 CD4+CD25− T cells from HA-TCR Tg mice and 5 μM HA110-119 peptides (National Institutes of Health Laboratory of Molecular Structure, Peptide Synthesis Laboratory). The cultures were pulsed with tritiated thymidine for the last 6-8 h. Flow cytometry was used to quantify the level of CD69 and CD25 on the mouse responders by staining the cultures with CD4 FITC, CD69 PE and CD25 APC (BD Biosciences).
Results
Human Tregs suppress mouse T cell activation
To investigate whether hTregs could suppress the activation of mCD4+CD25− responder T cells, we FACS-sorted human CD4+CD25−, CD4+CD25int, and CD4+CD25hi cells. The mCD4+CD25− responders were stimulated with soluble anti-mouse CD3 and mouse APCs, while the hTregs were activated with plate-bound anti-human CD3/CD28. Human CD4+CD25hiFOXP3+ cells could suppress mouse responders, although the magnitude of suppression was somewhat less than that seen with mouse Tregs (Fig. 1A). No suppression was observed in the presence of human CD4+CD25− or CD4+CD25int cells ruling out that the suppression mediated by the hCD4+CD25hi cells was an artifact of the coculture of xenogeneic cells. In addition, only the human CD4+CD25hiFOXP3+ cells significantly suppressed the activation of mouse CD4+CD25− T cells based on their expression of CD69 and CD25, while the human CD4+CD25− and CD4+CD25int cells had no effect (Fig. 1B). We have shown previously that mTregs have enhanced suppressor activity in vitro following pre-activation with anti-CD3 and IL-2 and that pre-activated Tregs did not require reactivation via their TCRs to mediate suppression (23). Similarly, pre-activated hTregs were as suppressive as fresh mTregs for inhibiting the proliferation of mouse CD4+ (Fig. 1C) and CD8+ (Fig. 1D) T cells in the absence of restimulation by anti-hCD3/CD28 in the coculture. While the suppressive function of pre-activated hTregs was comparable to fresh mTregs, they were less suppressive when compared to pre-activated mTregs. The reason for this difference is unclear, but is likely due to mechanisms of suppression that might be operative only within species such as IL-2 consumption.
Figure 1.
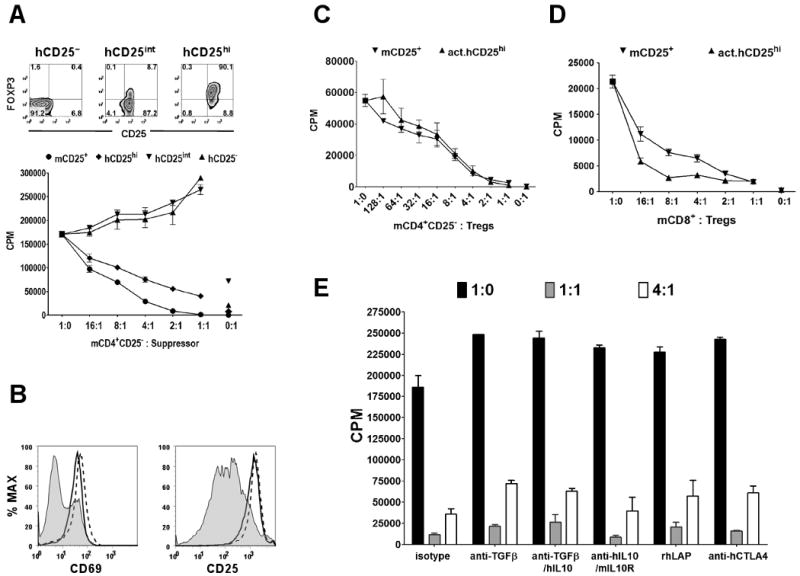
Human Treg suppression of mouse T cell activation and proliferation. A, In vitro suppression of CD4+CD25− T cells from BALB/c mice with FACS-sorted human CD4+CD127− CD25hi (hCD25hi), hCD25int, hCD25− or mouse mCD25+ T cells. In the assay, the mouse CD25− and CD25+ were activated with mouse APCs and soluble anti-mCD3, while the human cells were activated with plate-bound anti-hCD3 (5 μg/ml) and anti-hCD28 (2.5 μg/ml) in the cocultures. Top panel represents post-sort level of FOXP3 and CD25 in the three human T cell populations. B, Expression of CD69 and CD25 on mouse CD4+CD25− T cells cocultured for 3 days at 1:1 ratio with hCD25− (solid line histogram), hCD25int (dashed line histogram), and hCD25hi (shaded histogram). C. In vitro suppression of mouse, CD4+CD25− or D, CD8+CD25− T cells activated with mouse APCs and soluble anti-mCD3 alone or with fresh mouse Tregs (mCD25+) or pre-activated human Tregs (act.hCD25hi) without restimulation. E, In vitro suppression of mouse CD4+CD25− T cells with pre-activated human Tregs at 1:0, 1:1 and 4:1 ratios of mouse responder to hTreg in the presence of isotype control or neutralizing mAbs to TGFβ (anti-TGFβ), human IL-10 (anti-hIL10), mouse IL-10 receptor (mIL10R), and human CTLA-4 (anti-hCTLA4). Recombinant human latency associated peptide (rhLAP) also was used to neutralize TGFβ. Data are representative of three independent experiments.
The majority of in vitro studies of mouse and human Tregs have shown that the Treg-mediated suppression was not due to IL-10 or TGFβ. Since human IL-10 and TGFβ can act on mouse cells, we attempted to reverse the suppressive effects of pre-activated hTregs by the addition of neutralizing anti-human IL-10 or anti-TGFβ mAbs. Neutralization of TGFβ and/or IL-10 and blocking the mouse IL-10 receptor did not abrogate suppression by hTregs (Fig. 1E). The suppressive effects of the hTregs on mouse responder cells were not even reversed at a concentration of anti-TGFβ mAb (50 μg/ml) that almost completely neutralized the suppressive effect of 2 ng/ml TGFβ1 on T cell proliferation (data not shown). Recombinant human latency associated peptide (rhLAP), a potent neutralizer of TGFβ (24), also did not have any effect. Although human CTLA-4 can bind mouse CD80/CD86 (25) and some studies have implicated CTLA-4 on the Tregs as a mediator of their suppressive function either by an unknown mechanism (26) or by induction of the indoleamine 2,3-dioxygenase (IDO)/tryptophan catabolism pathway (13), selective blocking of the CTLA-4 on the hTregs with anti-hCTLA-4 mAb did not abrogate suppression in the human-mouse assay (Fig. 1E).
LFA-1 on human Tregs is essential for cell-contact mediated suppression
Previous studies of Tregs have suggested that cell-contact between the Tregs and the responder T cells or APCs was necessary for Tregs to mediated suppression. The human-mouse suppression assay offers the unique opportunity to directly determine the contribution of cell surface antigens to cell contact-mediated suppression since we can use reagents directed to the hTregs without interfering with the activation of the mouse responders or the hTregs. One obvious candidate for a receptor/counter-receptor pair that would mediate the interaction of Tregs with their target cells would be LFA-1/ICAM-1 that mediates interactions between almost all immune cells (27, 28). One previous study (29) using CD18 deficient (−/−) mTregs concluded that LFA-1 was required for Treg contact-mediated suppression. However, this study could not distinguish whether LFA-1 was required for Treg interaction with their target cells or whether LFA-1 also was required for activation of the suppressive function of Tregs by the TCR stimulus in association with APCs. As the fresh hTregs in our model are activated with plate-bound anti-CD3/CD28, we can directly assess if the interaction between the hTregs and the mouse cells involves the LFA-1/ICAM-1 pathway. Since LFA-1 consists of CD11a and CD18 subunits, the addition of blocking anti-hCD11a or -hCD18 mAbs completely abrogated the capacity of freshly explanted human Tregs to suppress mouse responders (Fig. 2A). The addition of anti-hICAM-1, -hICAM-2 and -hICAM-3 mAbs did not reverse suppression, ruling out the possibility that the loss of suppression by blocking LFA-1 was due to the inhibition of LFA-1/ICAM-1 interactions between the hTregs. The suppressive capacity of pre-activated human Tregs in the absence of anti-hCD3/CD28 restimulation was also abrogated although not completely by the addition of anti-hCD11a therefore making it unlikely that LFA-1 engagement was required during the activation of the hTregs (Fig. 2B). Multiple mAbs to hCD11a and hCD18 that had been previously characterized as blocking human LFA-1/ICAM-1 interactions also reversed the suppressive effects of hTregs while mAb MEM-83 (anti-hCD11a), that has been reported to be non-blocking and to increase the high affinity conformation of LFA-1 (30), did not abrogate hTreg function (Fig. 2C). Further evidence in support of the binding of hLFA-1 to mICAM-1 was our finding that both fresh and pre-activated mouse Tregs failed to significantly suppress human CD4+ T cells (Fig. 2D), since it has been shown that hLFA-1 can bind mICAM-1 and mICAM-2, while the reverse does not occur (31, 32). Therefore, the lack of suppression by mTregs on human responders was due to the inability of mouse LFA-1 to interact with human ICAM-1.
Figure 2.
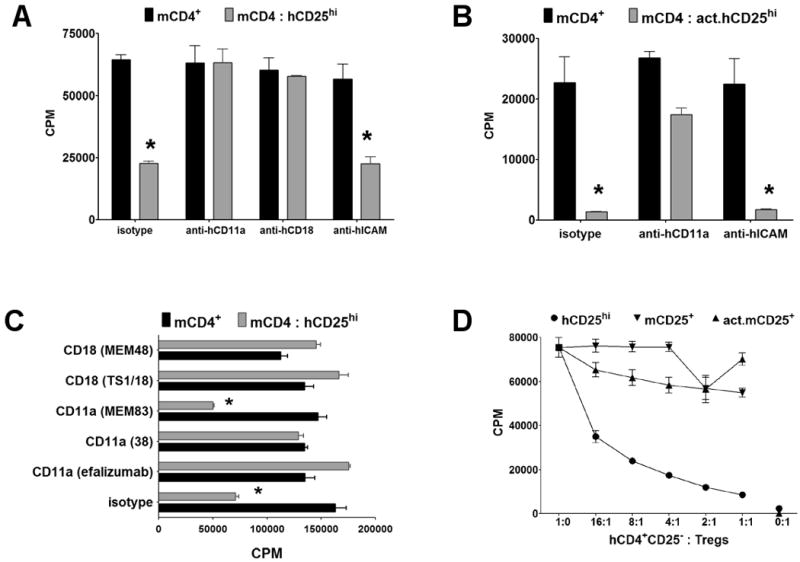
LFA-1 on human Tregs is essential for cell-contact mediated suppression. A, Proliferation of mouse CD4+CD25− T cells alone (black bar), or with a 1:1 ratio of fresh hTregs (hCD25hi, gray bar) or B, pre-activated hTregs (act.hCD25hi) in the presence of isotype control or anti-hCD11a (efalizumab), -hCD18 (TS1/18), or -hICAM-1/2/3 blocking mAbs. C, Proliferation of mouse CD4+CD25− T cells alone (black bar), or with a 1:1 ratio of fresh hTregs (hCD25hi, gray bar) in the presence of isotype control or different clones of anti-hCD11a or -hCD18 mAbs. For the above assays, the mouse responders were stimulated with mouse APCs and soluble anti-mCD3 while the fresh hTregs were optimally activated in the cocultures with plate-bound anti-hCD3/CD28. The pre-activated hTregs were not restimulated. D, Suppression of human CD4+CD25− T cells with FACS-sorted hTregs (hCD25hi), mTregs (mCD25+), or pre-activated mTregs (act.mCD25+). In this assay, the human responders and Tregs were stimulated with human APCs and soluble anti-hCD3, while the fresh mTregs were optimally activated in the cocultures with plate-bound anti-mCD3/CD28. The pre-activated mTregs were not restimulated. Data are representative of three independent experiments. Asterisk (*) represents p < 0.05 for the difference between the black and gray bars in each group.
To eliminate the possibility that the abrogation of suppression by anti-hCD11a or -hCD18 mAbs was secondary to a mAb induced negative signal, we also tested Tregs from leukocyte adhesion deficiency type-1 (LAD-1) patients, who have mutations in the CD18 gene that result in a complete deficiency of LFA-1 expression. Fresh (Fig. 3A) and pre-activated (Fig. 3B) Tregs from LAD-1 patients, that expressed levels of FOXP3 comparable to normal controls, failed to suppress the proliferation of mouse responders. Moreover, Tregs from LAD-1 patients are significantly less suppressive on human CD4+CD25− (Fig. 3C) or CD8+ T cells (Fig. 3D) when compared to Tregs from healthy donors. The residual suppression by the Tregs from the LAD-1 patients may indicate that other cell interaction pathways (CD2/CD58, VLA-4/VCAM-1, etc) may be operative in the interaction of hTreg with hCD4+CD25− T cells or APC, that a soluble molecule may mediate suppression, or that IL-2 consumption may be playing a role.
Figure 3.
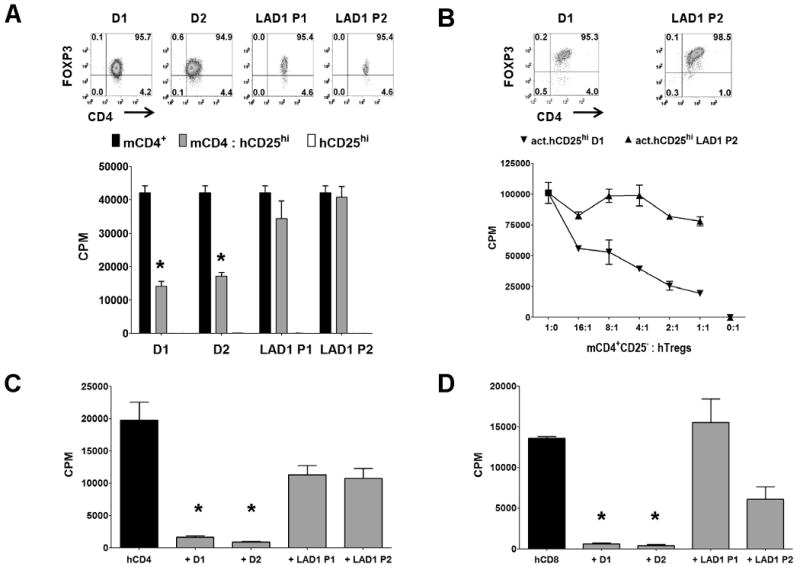
Human Tregs from LAD-1 patients lack suppressive function. A, Proliferation of mouse CD4+CD25− T cells alone (black bar), with 1:1 ratio of fresh hTregs (hCD25hi, gray bar) from healthy donors (D1/D2), or LAD-1 patients (P1/P2). White bar is the hTregs stimulated alone. Upper panel represents post-sort flow cytometric FOXP3 staining on the hTregs. Asterisk (*) represents p < 0.05 for the difference between the black and gray bars in each group. B, Proliferation of mouse CD4+CD25− T cells alone, or cultured with varying numbers of pre-activated hTregs (act.hCD25hi) from healthy donor D1, or LAD-1 patient P1. Upper panel represents FOXP3 staining of 48 h pre-activated hTregs. In these assays, the mouse responders were stimulated with mouse APCs and soluble anti-mCD3 while the fresh hTregs were optimally activated with plate-bound anti-hCD3/CD28. The pre-activated hTregs were not restimulated. C, Proliferation of CD4+CD25− or D, CD8+CD25− T cells from a normal donor alone (black bar) or with 1:1 ratio of fresh hTregs (gray bar) from healthy donors (D1/D2), or LAD-1 patients (P1/P2). In this assay, the human responders and Tregs were stimulated with human APCs and soluble anti-hCD3. Data are representative of two independent experiments. Asterisk (*) represents p < 0.05 for the differences in suppression between healthy donors and LAD-1 patients.
Human Tregs mediate their suppressive function by targeting the mouse DCs via an hLFA-1/mICAM-1 dependent interaction
The cell(s) targeted by Treg-mediated suppression either in vitro or in vivo remains unclear. In the widely used in vitro suppression assays, it has been difficult to determine if the Tregs acted on the APCs, the CD4+ T cells or both. Some studies with mouse Tregs (33) and with human Tregs (19) have demonstrated suppressor activity when anti-CD3/CD28 beads, plate-bound mAbs, or peptide-MHC tetramers were used as stimuli in APC-free systems. The requirement for an LFA-1/ICAM-1 interaction in the human-mouse suppression assay permits the opportunity to pinpoint the target cell for the suppressor effects of the Tregs. To address whether the hTregs targeted mCD4+ T cells or the mAPCs, we cultured CD4+CD25− T cells from ICAM-1−/− mice in the presence of wild-type T-depleted splenocytes (Fig. 4A) or purified splenic DCs (Fig. 4B) and hTregs. The proliferative responses of CD4+CD25− T cells from ICAM-1−/− mice were still inhibited by the hTregs and abrogated by anti-hCD11a. Suppression also was maintained in the presence of a blocking mAb to mICAM-2 that could potentially interact with human LFA-1 in place of mICAM-1 (Fig. 4, A and B). Suppression in this culture system can therefore be mediated solely by an interaction of hLFA-1 expressed on hTregs with mICAM-1 expressed on mDCs, and not the mouse responder CD4+ T cells deficient in ICAM-1. We were not able to directly use ICAM-1−/− mDCs, since LFA-1/ICAM-1 interactions between the mDCs and mCD4+ T cells were required for activation of the mouse T cells.
Figure 4.
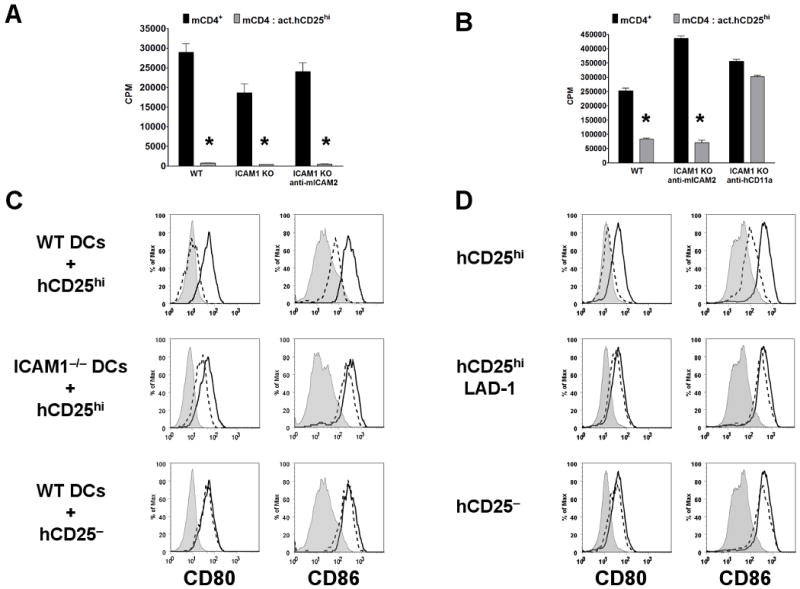
Human Tregs target mouse DCs and not T cells via an LFA-1/ICAM-1 dependent interaction. A, Proliferation of CD4+CD25− T cells from wild-type (WT) C57BL/6 or ICAM-1−/− (KO) mice stimulated with soluble anti-mCD3 and T-depleted splenocytes or B, splenic DCs alone (black bar) or cocultured with 1:1 ratio of pre-activated hTregs (act.hCD25hi, gray bar) with or without blocking anti-mICAM-2 or -hCD11a (efalizumab) mAbs. The pre-activated hTregs were not restimulated. Asterisk (*) represents p < 0.05 for the difference between the black and gray bars in each group. C, Level of CD80/CD86 expression on splenic DCs from wild-type (WT) C57BL/6 or ICAM-1−/− mice at 0 h (shaded histogram) and after 18 h cultured alone (opened histogram) or with pre-activated human (dashed histogram) hCD25hi or hCD25− T cells from healthy donor. D, CD80/CD86 expression on splenic DCs from wild-type (WT) C57BL/6 mice at 0 h (shaded histogram) and after 18 h cultured alone (opened histogram) or with pre-activated human (dashed histogram) hCD25hi from healthy donor, hCD25hi from LAD-1 patient or hCD25− T cells from healthy donor. Data are representative of three (A, B and C) and two (D) independent experiments.
To determine whether the interaction of the hTregs on mDCs modulated their function, we cultured the mouse splenic DCs for 18 h in the presence or absence of pre-activated hTregs and measured the expression of CD80/CD86 on the DCs. Mouse DCs from wild-type, but not ICAM-1−/−, mice cultured with pre-activated hTregs failed to upregulate CD80/CD86 expression (Fig. 4C). Pre-activated human CD4+CD25− T cells had no effect on the expression of CD80/CD86 on mDCs. Furthermore, preactivated hTregs from LAD-1 patients also failed to suppress the upregulation of CD80/CD86 expression on wild-type mDCs (Fig. 4D). No changes in the levels of CD40 or MHC class II expression were seen when the mDCs were cultured with the preactivated hTregs (data not shown). Similarly, preactivated hTregs from healthy donors, but not LAD-1 patients, were able to suppress the upregulation of CD80/CD86 on human CD19+ B cells stimulated for 36 h with lipopolysaccharides (Supplementary Fig. 1). In order to test whether the mDC functions were impaired by their interaction with hTregs, they were purified following the 18 h culture with the different human T cell populations, and tested for their ability to present peptide antigen to mouse CD4+CD25− responder T cells from mice expressing a transgenic TCR specific for a peptide from influenza hemagglutinin (HA). Mouse DCs that had been precultured with hTregs from healthy donors were significantly less efficient at stimulating proliferation and inducing upregulation of CD69 and CD25 expression on mouse HA-TCR Tg responder T cells as compared to the mDCs cultured alone (Fig. 5, A and B). On the other hand, the mDCs cultured with hCD4+CD25− T cells from healthy donors or hTregs from LAD-1 patients had enhanced activity and increased capacity to activate mouse responders. Taken together, these results indicate that hTregs target mDCs via a critical LFA-1/ICAM-1 dependent interaction resulting in the inhibition of CD80/CD86 upregulation and decreased capacity to present antigens to responder T cells.
Figure 5.
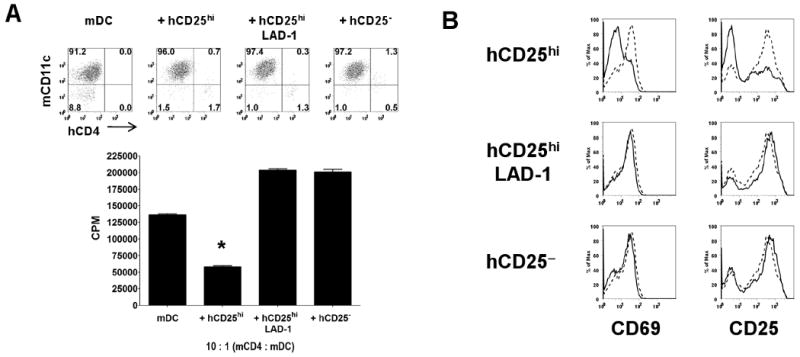
Human Tregs modulate splenic DCs to have decreased capacity to present antigen and activate mouse responder T cells. A, Proliferation of mouse CD4+CD25− T cells from HA-TCR Tg mice in the presence of HA peptide and re-isolated splenic DCs that had been previously cultured for 18 h alone (mDC), with Tregs from healthy donor (hCD25hi), with Tregs from LAD-1 patient (hCD25hi LAD-1), or with CD4+CD25− T cells from healthy donor (hCD25−). Upper panel represents the purity of splenic DCs repurified from the cocultures based on staining with mouse CD11c and human CD4. B, Expression of CD69 and CD25 on mouse CD4+CD25− from the cocultures stimulated with splenic DCs that had been cultured for 18 h alone (dashed histogram) or with the three human T cell populations (solid histograms). Data are representative of two independent experiments. Asterisk (*) represents p < 0.05 for the differences in suppression between mDCs from hCD25hi vs. mDCs from hCD25hi LAD-1 or hCD25−.
IL-2 is required for activation of hTreg suppressor function under suboptimal stimulatory conditions, but IL-2 consumption plays no role in hTreg-mediated suppression
Our previous study had demonstrated that IL-2 production by responder T cells is critical for activation of mouse Treg suppressor function (34). As both IL-2 and anti-CD25 blocking antibodies are currently being used in certain human diseases, it is imperative to address the function of IL-2 signaling in hTregs. Under suboptimal stimulatory condition with anti-hCD3/CD28, highly purified hTregs failed to suppress mouse responders unless a small amount of exogenous human IL-2 (0.5 U/ml) was added to the coculture (Fig. 6A). However, under the same stimulatory conditions, a less purified hTreg population was able to suppress the mouse responders presumably due to the production of IL-2 by the contaminating human CD4+CD25+FOXP3− population. This suppression was abrogated by a blocking anti-human CD25 (Fig. 6B). In contrast, IL-2 was not required for hTreg suppressor function when the hTregs were activated with optimal anti-hCD3/CD28 stimulation (Fig. 6C) and suppression was not reversed by the addition of anti-CD25.
Figure 6.
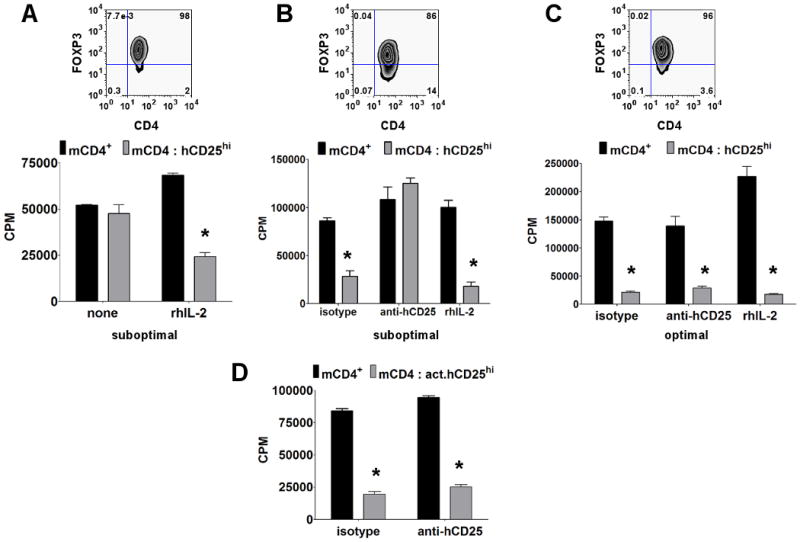
IL-2 is required for Treg suppressor function under suboptimal stimulatory conditions, but IL-2 consumption plays no role in hTreg-mediated suppression. A, Proliferation of mouse CD4+CD25− T cells alone (black bar) or with 1:1 ratio of fresh > 95% FOXP3+ hTregs (hCD25hi, gray bar) in the absence (none) or presence of 0.5 U/ml of recombinant human IL-2 (rhIL-2) under suboptimal stimulatory condition for hTregs. B, Proliferation of mouse CD4+CD25− T cells alone (black bar) or with 1:1 ratio of fresh < 90% FOXP3+ hTregs (hCD25hi, gray bar) in the presence of isotype control, daclizumab (anti-hCD25) or 0.5 U/ml of human IL-2 under suboptimal stimulatory condition. C, Proliferation of mouse CD4+CD25− T cells alone (black bar) or with 1:1 ratio of fresh > 95% FOXP3+ hTregs (hCD25hi, gray bar) in the presence of isotype, daclizumab or 0.5 U/ml of rhIL-2 under optimal stimulatory condition. Top panel represents post-sort FOXP3 purity for hTregs. D, Proliferation of mouse CD4+CD25− T cells alone (black bar) or with 1:1 ratio of pre-activated hTregs (act.hCD25hi, gray bar) in the presence of isotype or daclizumab (anti-hCD25). In these assays, the mouse responders were stimulated with mouse APCs and soluble anti-mCD3, while the fresh hTregs were stimulated either under suboptimal (3 μg/ml anti-hCD3 and 1 μg/ml anti-hCD28 plate-bound mAbs) or optimal stimulatory condition (5 μg/ml anti-hCD3 and 2.5 μg/ml anti-hCD28 plate-bound mAbs) in the cocultures. The pre-activated hTregs were not restimulated in the cocultures. Data are representative of three independent experiments. Asterisk (*) represents p < 0.05 for the difference between the black and gray bars in each group.
Although most studies demonstrate that the major effect of Tregs on responder CD4+CD25− T cells is to inhibit their capacity to produce IL-2 (5, 35), one of the long standing controversies concerning the Treg suppression assay is whether some of the suppression seen in the cocultures is secondary to absorption and consumption of IL-2 by the Tregs (17) resulting in cytokine deprivation of the responder CD4+ T cells (16). As both fresh and pre-activated hTregs expressed very high levels of CD25, it was important to determine the potential contribution of the consumption of mIL-2 to the suppressor function of the hTregs in our cocultures. Although it is generally believed that human T cells cannot utilize mIL-2, human T cells can respond to mIL-2 albeit with 6-180 fold lower efficiency than hIL-2 (36). The hTreg-mCD4+CD25− coculture system allows us to directly evaluate the role of consumption of mouse IL-2 as a major mechanism of Treg suppression. The addition of a high concentration of daclizumab, a humanized mAb to CD25 that blocks the binding of IL-2 to its receptor, failed to reverse the suppressive functions of the optimally activated fresh (Fig. 6C) and pre-activated (Fig. 6D) hTregs. The concentrations of daclizumab used were shown to be highly effective at blocking the activation of human T blasts to stimulation by exogenous IL-2 (data not shown). Since activated mouse T cells can use hIL-2 almost as efficiently as activated human T cells, we determined the capacity of fresh or pre-activated mTregs, activated with plate-bound anti-mCD3/CD28, to suppress hCD4+CD25− T cells stimulated by hAPCs and anti-hCD3. Although hTregs readily suppressed human responders, we did not observe any appreciable suppression of human CD4+ T cells by mTregs (Fig. 2D). This result is consistent with our anti-hCD25 blocking studies and directly demonstrates that IL-2 consumption is not a major component of human Treg-mediated suppression.
Discussion
Most studies of Treg function in vitro have suggested that a physical interaction between either the Treg and the responder T cell or the APC was required for suppression, yet little data has been obtained as to the nature of the cell surface antigens involved in these interactions. One of the difficulties in the analysis of this aspect of Treg function in vitro is that many of the potential cellular interaction molecules are also involved in the activation of the T effector cells and it has been difficult to separate out the Treg-specific components. To address this issue, we have developed a novel cross-species suppression assay in which we have shown that hTregs are quite efficient in their ability to suppress mouse responder T cells in the presence of mouse APCs. This assay required cell contact between the hTregs and the mouse cells, as it was not seen across a transwell (data not shown). The major advantage of this model is that we could target reagents to the hTregs that would not interfere with the activation of the mouse effector cells. Although we initially tested a large panel of anti-human mAbs for their ability to reverse the suppressor function of hTregs, only mAbs against the hLFA-1 heterodimer (CD11a/CD18) reproducibly abrogated suppression. It is unlikely that hLFA-1/hICAM interactions were involved in this assay as the hTregs were activated with plate-bound antibodies and anti-hICAM reagents did not reverse suppression. We then took advantage of two different genetic deficiencies, ICAM-1−/− T cells from mice and hTregs from LAD-1 patients with mutations in CD18. Human Tregs readily inhibited the activation of CD4+ T cells from ICAM-1−/− mice in the presence of wild type APCs and hTregs from LAD-1 patients failed to inhibit the activation of responder T cells and APCs from wild type mice. We do not believe that the Tregs from LAD-1 patients are defective during development in the thymus and periphery, which would negatively affect their intrinsic suppressive function. While there appears to be a decreased in T cell in LFA-1 knockout mice (29), LAD-1 patients have an increased numbers of T cells, particularly Tregs. While CD4+ T cells from healthy donors contain 5-10% FOXP3+ cells, CD4+ T cells from LAD-1 patients have 15-50% FOXP3+ T cells in their peripheral blood (Supplementary Fig. 2). While the Treg phenotype of the FOXP3+ cells from LAD-1 patients appears to be similar to that of healthy donors with high expression of CD25 and CTLA-4 and low expression of CD127, their inability to suppress may also be secondary to factors other than the absence of LFA-1. Taken together, these studies demonstrate that hTregs mediate the suppression of mouse T cell activation by targeting mouse DCs. We confirmed this finding by demonstrating that culture of pre-activated hTregs with mouse DCs in the absence of responder T cells impaired the ability of the DCs to upregulate the costimulatory molecules, CD80/CD86, and markedly diminished the subsequent capacity of the hTreg exposed mDCs to activate responder T cells.
We have not yet excluded the possibility that hTregs are also capable of interacting with mouse responder T cells in addition to mAPCs. This problem is difficult to address because the activation of most T cell responses requires an LFA-1/ICAM-1-dependent interaction between the responder T cells and APCs. We have previously shown that mTregs can inhibit the activation of mouse CD8+ T cells activated by peptide-MHC class I tetramers (18) in the absence of mAPCs, but it remains unclear whether the tetramers directly activate the CD8+ T cells or whether activation of the responder CD8+ T cells also involves a cell-cell interaction. In preliminary studies, the responses of mouse CD8+ T cells to tetramer stimulation were inhibited by pre-activated hTregs, but also inhibited by anti-mLFA-1 or anti-mICAM-1 strongly suggesting that cell-cell interactions are involved in the response to tetramer stimulation (data not shown). The mechanism of suppression of CD8+ T cells may be different from that of CD4+ T cells. We have previously demonstrated that mTregs fail to inhibit the responses of mouse CD4+ T cells to stimulation by plate-bound anti-CD3 mAb (23). Other studies of both mouse and human Tregs have shown that responses to solid-phase anti-CD3 in the absence of APCs can be inhibited by Tregs, but in general significant suppression is only observed at high (1:1) Treg to responder ratios. While hTregs were able to suppress mouse responders stimulated by mAPCs, the hTregs failed to suppress mouse responders in an APC-free system stimulated by anti-mCD3/CD28 conjugated beads (Supplementary Fig. 3). In this APC-free system, we did observe some suppression by mTregs but only at 1:1 and 2:1 ratio of responder to suppressor. It remains possible that Tregs utilize a completely distinct mechanism to directly suppress responder cells under these conditions of stimulation. IL-2 consumption by the Tregs may be very important when T cells are stimulated in the absence of APCs and may ultimately lead to the death of the responder T cells due to cytokine deprivation (16). However, IL-2 consumption does not appear to play a significant role in the ability of hTregs to suppress mouse responder T cells stimulated by soluble anti-CD3 in the presence of mAPCs, as anti-hCD25 had no effect on Treg suppression.
Tregs have been reported to inhibit the maturation of bone-marrow derived or splenic DCs by modulating the expression of costimulatory molecules (37, 38) or the induction of inhibitory molecules (13, 39), but also have been shown to inhibit T cell responses by fully mature DCs both in vivo (40) and in vitro (41). Antigen-specific Tregs have been shown to prevent the interaction between effector T cells and DCs (20, 42). Antigen-specific TGFb-induced Tregs also have been shown to inhibit the expansion of autoreactive effector T cells by acting on DCs and by reducing their ability to present autoantigen in vivo and in vitro (21). We observed similar results when we cultured mouse DCs with pre-activated hTregs. The hTregs prevented the upregulation of CD80/CD86 and reduced the ability of the DCs to present peptide antigen to responder T cells. Suppression of DC maturation also required an hLFA-1/mICAM-1 interaction as it was blocked by anti-hCD11a and hTregs from LAD-1 patients did not suppress. The molecular basis for the suppression remains to be elucidated, but it can clearly cross species. Our data demonstrate that the first step in the process is likely to be an LFA-1/ICAM-1 interaction. The critical function of LFA-1 on Tregs and their interaction with DCs are supported by two recent publications demonstrating the importance of LFA-1 on Tregs in mouse studies (43, 44).
From a practical standpoint, we believe that the human-mouse suppression assay has the potential of being more reproducible than the standard human-human in vitro suppression assay for evaluating the function of hTregs, particularly in diseases where the immune system has been perturbed. It allows one to specifically assay hTreg function in the absence of potentially hyperactivated responder T cells that can be present in patients with autoimmune diseases. As IL-2 consumption plays no role in the assay, it also avoids the potential consumption of IL-2 by CD4+CD25+FOXP3− effector T cells that can contaminate CD4+CD25+FOXP3+ isolated from patients with ongoing inflammatory processes. A limitation of this assay is that potential restrictions may exist in the participation of other receptor/counter-receptor molecules between species in this system. Thus, we have not evaluated the contribution of other adhesion molecules such as CD2/CD58 or CD49d (or others) that may also play prominent roles in the interaction of Tregs and APCs (or between responder cells and Tregs) within the species in addition to LFA-1. Therefore, the simultaneous use of the hybrid and the conventional suppression assays might be a better method of evaluating the function of human Tregs.
From a therapeutic standpoint, the human-mouse assay allows for the development of specific reagents that specifically target hTregs. We have already screened a panel of antibodies to human cell surface antigens (OX-40, GITR, ICOS or 4-1BB) that have been purported to abrogate the function of Tregs, yet none had any effect on the ability of human Tregs to suppress mouse responders (data not shown). While anti-hCD11a or -hCD18 abrogated suppression by hTregs, blocking human CTLA-4, CD2/CD58, and FAS ligand, or neutralizing human TGFb and IL-10 had no effect. Our studies showing a critical role for CD11a/CD18 for the function of hTregs is supported by a recent publication describing three LAD-1 patients with reversion mutations that resulted in a selective expression of LFA-1 only in their CD8+ T cells (45). Curiously, all three patients developed chronic inflammatory bowel disease. Typically LAD-1 patients do not develop autoimmunity even though their Tregs are deficient in LFA-1 because the rest of the T cells also lack LFA-1 and are unable to mount a productive immune response. The development of autoimmunity in these LAD-1 patients with reversion mutations might be due to the inability of the Tregs that lack LFA-1 to suppress the CD8+ T cells that can be fully activated because of their selective expression of LFA-1. Biologics such as efalizumab, a humanized monoclonal anti-CD11a approved for the treatment of psoriasis and in clinical trials for other autoimmune diseases, might have a detrimental effect in some patients depending on the dosage by providing greater inhibition of Treg function over effector T cell activation. Evidence for this hypothesis is demonstrated by a recent publication (44) showing that diminished CD18 expression on mouse Tregs resulted in impaired cell-cell contact between Tregs and DCs. These dysfunctional Tregs failed to suppress the pathogenic T cells and promoted the onset and severity of psoriasis.
Supplementary Material
Supplementary Figure 1. Tregs from LAD-1 patients fail to modulate the expression of CD80/86 on human LPS-stimulated B cells. Flow cytometric analysis of CD80/CD86 expression on human CD19+ B cells at 0 h (shaded histogram) and after 36 h of stimulation with 1 μg/ml LPS alone (opened histogram) or with pre-activated human (dashed histogram) hCD25hi from healthy donors (D1/D2) or from LAD-1 patients (LAD1 P1/P2).
Supplementary Figure 2. Phenotype and frequency of Tregs in LAD-1 patients. Flow cytometric analysis of CD4+ T cells in peripheral blood of healthy donor (HD) and LAD-1 patients (P1/P2). The cells were surface stained with CD25, CD127 or CD18 and intracellular stained with FOXP3 (clone 236A/E7) and CD152. Percentage is based on CD4 gating.
Supplementary Figure 3. Suppressive functions of human and mouse Tregs in the presence and absence of APCs. In vitro suppression of CD4+CD25− T cells from BALB/c mice with FACS-sorted human hCD25hi or mouse mCD25+ Tregs stimulated for 3 days with A, soluble anti-mCD3 and mouse T-depleted splenocytes or B, with anti-mCD3/CD28 conjugated Dynabeads at 10:1 cell to bead ratio. The fresh human Tregs were activated under optimal conditions with plate-bound anti-hCD3 (5 μg/ml) and anti-hCD28 (2.5 μg/ml). Data are representative of three independent experiments.
Acknowledgments
We thank the NIAID Flow Cytometry Section, particularly Carol Henry, Elina Stregevsky, Tom Moyer and Calvin Eigsti, for their help in sorting our cells. We also thank Cynthia Matthews and Rosemary Werden in the Department of Transfusion Medicine for providing us with the healthy donors' samples.
Footnotes
Disclosures
The authors have no financial conflict of interest.
This work was funded by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases.
References
- 1.Shevach EM, DiPaolo RA, Andersson J, Zhao DM, Stephens GL, Thornton AM. The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol Rev. 2006;212:60–73. doi: 10.1111/j.0105-2896.2006.00415.x. [DOI] [PubMed] [Google Scholar]
- 2.Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 3.Shevach EM. From vanilla to 28 flavors: multiple varieties of T regulatory cells. Immunity. 2006;25:195–201. doi: 10.1016/j.immuni.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. 2008;9:239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi T, Kuniyasu Y, Toda M, Sakaguchi N, Itoh M, Iwata M, Shimizu J, Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10:1969–1980. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali AD. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 9.Veldhoen M, Moncrieffe H, Hocking RJ, Atkins CJ, Stockinger B. Modulation of dendritic cell function by naive and regulatory CD4+ T cells. J Immunol. 2006;176:6202–6210. doi: 10.4049/jimmunol.176.10.6202. [DOI] [PubMed] [Google Scholar]
- 10.Tang Q, Boden EK, Henriksen KJ, Bour-Jordan H, Bi M, Bluestone JA. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur J Immunol. 2004;34:2996–3005. doi: 10.1002/eji.200425143. [DOI] [PubMed] [Google Scholar]
- 11.Paust S, Lu L, McCarty N, Cantor H. Engagement of B7 on effector T cells by regulatory T cells prevents autoimmune disease. Proc Natl Acad Sci U S A. 2004;101:10398–10403. doi: 10.1073/pnas.0403342101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oderup C, Cederbom L, Makowska A, Cilio CM, Ivars F. Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology. 2006;118:240–249. doi: 10.1111/j.1365-2567.2006.02362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, Belladonna ML, Fioretti MC, Alegre ML, Puccetti P. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4:1206–1212. doi: 10.1038/ni1003. [DOI] [PubMed] [Google Scholar]
- 14.Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, Heib V, Becker M, Kubach J, Schmitt S, Stoll S, Schild H, Staege MS, Stassen M, Jonuleit H, Schmitt E. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med. 2007;204:1303–1310. doi: 10.1084/jem.20062129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M, Kuchroo VK, Strom TB, Robson SC. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4(+)CD25(+)Foxp3(+) regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4(+) T cells. Nat Immunol. 2007;8:1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- 17.Scheffold A, Huhn J, Hofer T. Regulation of CD4+CD25+ regulatory T cell activity: it takes (IL-)two to tango. Eur J Immunol. 2005;35:1336–1341. doi: 10.1002/eji.200425887. [DOI] [PubMed] [Google Scholar]
- 18.Piccirillo CA, Shevach ME. Cutting edge: control of CD8+ T cell activation by CD4+CD25+ immunoregulatory cells. J Immunol. 2001;167:1137–1140. doi: 10.4049/jimmunol.167.3.1137. [DOI] [PubMed] [Google Scholar]
- 19.Dieckmann D, Plottner H, Berchtold S, Berger T, Schuler G. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J Exp Med. 2001;193:1303–1310. doi: 10.1084/jem.193.11.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, Santamaria P, Locksley RM, Krummel MF, Bluestone JA. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol. 2006;7:83–92. doi: 10.1038/ni1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DiPaolo RJ, Brinster C, Davidson TS, Andersson J, Glass D, Shevach ME. Autoantigen-specific TGFbeta-induced Foxp3+ regulatory T cells prevent autoimmunity by inhibiting dendritic cells from activating autoreactive T cells. J Immunol. 2007;179:4685–4693. doi: 10.4049/jimmunol.179.7.4685. [DOI] [PubMed] [Google Scholar]
- 22.Klein L, Khazaie K, von Boehmer H. In vivo dynamics of antigen-specific regulatory T cells not predicted from behavior in vitro. Proc Natl Acad Sci U S A. 2003;100:8886–8891. doi: 10.1073/pnas.1533365100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thornton AM, Shevach ME. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164:183–190. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- 24.Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H, Strober W. TGF-beta 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J Immunol. 2004;172:834–842. doi: 10.4049/jimmunol.172.2.834. [DOI] [PubMed] [Google Scholar]
- 25.Blazar BR, Taylor PA, Linsley PS, Vallera AD. In vivo blockade of CD28/CTLA4: B7/BB1 interaction with CTLA4-Ig reduces lethal murine graft-versus-host disease across the major histocompatibility complex barrier in mice. Blood. 1994;83:3815–3825. [PubMed] [Google Scholar]
- 26.Annunziato F, Cosmi L, Liotta F, Lazzeri E, Manetti R, Vanini V, Romagnani P, Maggi E, Romagnani S. Phenotype, localization, and mechanism of suppression of CD4(+)CD25(+) human thymocytes. J Exp Med. 2002;196:379–387. doi: 10.1084/jem.20020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Springer TA. Adhesion receptors of the immune system. Nature. 1990;346:425–434. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- 28.Patarroyo M, Prieto J, Rincon J, Timonen T, Lundberg C, Lindbom L, Asjo B, Gahmberg GC. Leukocyte-cell adhesion: a molecular process fundamental in leukocyte physiology. Immunol Rev. 1990;114:67–108. doi: 10.1111/j.1600-065x.1990.tb00562.x. [DOI] [PubMed] [Google Scholar]
- 29.Marski M, Kandula S, Turner JR, Abraham C. CD18 is required for optimal development and function of CD4+CD25+ T regulatory cells. J Immunol. 2005;175:7889–7897. doi: 10.4049/jimmunol.175.12.7889. [DOI] [PubMed] [Google Scholar]
- 30.Landis RC, Bennett RI, Hogg N. A novel LFA-1 activation epitope maps to the I domain. J Cell Biol. 1993;120:1519–1527. doi: 10.1083/jcb.120.6.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnston SC, Dustin ML, Hibbs ML, Springer TA. On the species specificity of the interaction of LFA-1 with intercellular adhesion molecules. J Immunol. 1990;145:1181–1187. [PubMed] [Google Scholar]
- 32.Xu H, Bickford JK, Luther E, Carpenito C, Takei F, Springer AT. Characterization of murine intercellular adhesion molecule-2. J Immunol. 1996;156:4909–4914. [PubMed] [Google Scholar]
- 33.Ermann J, Szanya V, Ford GS, Paragas V, Fathman CG, Lejon K. CD4(+)CD25(+) T cells facilitate the induction of T cell anergy. J Immunol. 2001;167:4271–4275. doi: 10.4049/jimmunol.167.8.4271. [DOI] [PubMed] [Google Scholar]
- 34.Thornton AM, Donovan EE, Piccirillo CA, Shevach ME. Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol. 2004;172:6519–6523. doi: 10.4049/jimmunol.172.11.6519. [DOI] [PubMed] [Google Scholar]
- 35.Oberle N, Eberhardt N, Falk CS, Krammer PH, Suri-Payer E. Rapid suppression of cytokine transcription in human CD4+CD25 T cells by CD4+Foxp3+ regulatory T cells: independence of IL-2 consumption, TGF-beta, and various inhibitors of TCR signaling. J Immunol. 2007;179:3578–3587. doi: 10.4049/jimmunol.179.6.3578. [DOI] [PubMed] [Google Scholar]
- 36.Mosmann TR, Yokota T, Kastelein R, Zurawski SM, Arai N, Takebe Y. Species-specificity of T cell stimulating activities of IL 2 and BSF-1 (IL 4): comparison of normal and recombinant, mouse and human IL 2 and BSF-1 (IL 4) J Immunol. 1987;138:1813–1816. [PubMed] [Google Scholar]
- 37.Cederbom L, Hall H, Ivars F. CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. Eur J Immunol. 2000;30:1538–1543. doi: 10.1002/1521-4141(200006)30:6<1538::AID-IMMU1538>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 38.Misra N, Bayry J, Lacroix-Desmazes S, Kazatchkine MD, Kaveri SV. Cutting edge: human CD4+CD25+ T cells restrain the maturation and antigen-presenting function of dendritic cells. J Immunol. 2004;172:4676–4680. doi: 10.4049/jimmunol.172.8.4676. [DOI] [PubMed] [Google Scholar]
- 39.Vlad G, Cortesini R, Suciu-Foca N. License to heal: bidirectional interaction of antigen-specific regulatory T cells and tolerogenic APC. J Immunol. 2005;174:5907–5914. doi: 10.4049/jimmunol.174.10.5907. [DOI] [PubMed] [Google Scholar]
- 40.Oldenhove G, de Heusch M, Urbain-Vansanten G, Urbain J, Maliszewski C, Leo O, Moser M. CD4+ CD25+ regulatory T cells control T helper cell type 1 responses to foreign antigens induced by mature dendritic cells in vivo. J Exp Med. 2003;198:259–266. doi: 10.1084/jem.20030654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brinster C, Shevach ME. Bone marrow-derived dendritic cells reverse the anergic state of CD4+CD25+ T cells without reversing their suppressive function. J Immunol. 2005;175:7332–7340. doi: 10.4049/jimmunol.175.11.7332. [DOI] [PubMed] [Google Scholar]
- 42.Tadokoro CE, Shakhar G, Shen S, Ding Y, Lino AC, Maraver A, Lafaille JJ, Dustin ML. Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J Exp Med. 2006;203:505–511. doi: 10.1084/jem.20050783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci U S A. 2008;105:10113–10118. doi: 10.1073/pnas.0711106105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang H, Peters T, Sindrilaru A, Kess D, Oreshkova T, Yu XZ, Seier AM, Schreiber H, Wlaschek M, Blakytny R, Rohrbein J, Schulz G, Weiss JM, Scharffetter-Kochanek K. TGF-beta-dependent suppressive function of Tregs requires wild-type levels of CD18 in a mouse model of psoriasis. J Clin Invest. 2008;118:2629–2639. doi: 10.1172/JCI34916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uzel G, Tng E, Rosenzweig SD, Hsu AP, Shaw JM, Horwitz ME, Linton GF, Anderson SM, Kirby MR, Oliveira JB, Brown MR, Fleisher TA, Law SK, Holland MS. Reversion mutations in patients with leukocyte adhesion deficiency type-1 (LAD-1) Blood. 2008;111:209–218. doi: 10.1182/blood-2007-04-082552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Tregs from LAD-1 patients fail to modulate the expression of CD80/86 on human LPS-stimulated B cells. Flow cytometric analysis of CD80/CD86 expression on human CD19+ B cells at 0 h (shaded histogram) and after 36 h of stimulation with 1 μg/ml LPS alone (opened histogram) or with pre-activated human (dashed histogram) hCD25hi from healthy donors (D1/D2) or from LAD-1 patients (LAD1 P1/P2).
Supplementary Figure 2. Phenotype and frequency of Tregs in LAD-1 patients. Flow cytometric analysis of CD4+ T cells in peripheral blood of healthy donor (HD) and LAD-1 patients (P1/P2). The cells were surface stained with CD25, CD127 or CD18 and intracellular stained with FOXP3 (clone 236A/E7) and CD152. Percentage is based on CD4 gating.
Supplementary Figure 3. Suppressive functions of human and mouse Tregs in the presence and absence of APCs. In vitro suppression of CD4+CD25− T cells from BALB/c mice with FACS-sorted human hCD25hi or mouse mCD25+ Tregs stimulated for 3 days with A, soluble anti-mCD3 and mouse T-depleted splenocytes or B, with anti-mCD3/CD28 conjugated Dynabeads at 10:1 cell to bead ratio. The fresh human Tregs were activated under optimal conditions with plate-bound anti-hCD3 (5 μg/ml) and anti-hCD28 (2.5 μg/ml). Data are representative of three independent experiments.