

Summary
Combined deficiency of factor V (FV) and factor VIII (FVIII) (F5F8D) is a genetic disorder characterized by mild-to-moderate bleeding and coordinate reduction in plasma FV and FVIII levels, as well as platelet FV level. Recent studies identified mutations in two genes (LMAN1 and MCFD2) as the cause of F5F8D. Though clinically indistinguishable, MCFD2 mutations generally exhibit lower levels of FV and FVIII than LMAN1 mutations. LMAN1 is a mannose-specific lectin that cycles between the endoplasmic reticulum (ER) and the ER-Golgi intermediate compartment. MCFD2 is an EF-hand domain protein that forms a calcium-dependent heteromeric complex with LMAN1 in cells. Missense mutations in the EF-hand domains of MCFD2 abolish the interaction with LMAN1. The LMAN1-MCFD2 complex may serve as a cargo receptor for the ER-to-Golgi transport of FV and FVIII, and perhaps a number of other glycoproteins. The B domain of FVIII may be important in mediating its interaction with the LMAN1-MCFD2 complex.
Keywords: bleeding disorders, blood coagulation, factor VIII, F5F8D
Coagulation factor V (FV) and factor VIII (FVIII) are essential cofactors for the proteases factor X and factor IX, respectively, in the blood clotting cascade. Combined deficiency of FV and FVIII (F5F8D, OMIM 227300) is an autosomal recessive disorder characterized by the simultaneous decrease of FV and FVIII levels in plasma. F5F8D is distinct from chance inheritance of haemophilia A (FVIII deficiency) and parahaemophilia (FV deficiency). F5F8D cases are usually first brought to light by mild-to-moderate bleeding beginning in childhood associated with prolonged prothrombin time (PT) and partial thromboplastin time (PTT). FV and FVIII activities are generally in the range of 5–30% of normal (Fig 1A), but have been reported to be as low as less than 1% or as high as almost 50%. Bleeding symptoms vary in reported cases, and are generally comparable to those observed in patients with single deficiencies of FV or FVIII (Seligsohn et al, 1982; Peyvandi et al, 1998). Spontaneous bleeding symptoms include easy bruising, epistaxis and menorrhagia. Excessive bleeding is particularly common during or after trauma, tooth extraction, surgery or labour. No consistent abnormalities outside of these haemostatic changes have been identified in F5F8D. Obligatory carriers have plasma levels of FV and FVIII in the normal range. Although there are occasional reports of bleeding symptoms of unaffected family members, these are probably unrelated to FV and FVIII. Fresh frozen plasma corrects both FV and FVIII deficiencies, and has been used alone or in combination with plasma-derived or recombinant FVIII concentrate to effectively treat bleeding episodes and manage surgical procedures of F5F8D patients (Zhang & Ginsburg, 2006; Spreafico & Peyvandi, 2008). The molecular mechanism of F5F8D was last reviewed in 2004 (Zhang & Ginsburg, 2004). This review will focus on recent developments in our understanding of the disorder.
Fig 1.

Genotype–phenotype correlation in F5F8D. (A) Correlation of LMAN1/MCFD2 mutations and FV/FVIII levels. The short bars and numbers indicate the average values. (B) Pearson correlation analysis of FV and FVIII levels in LMAN1 mutation group and MCFD2 mutation group of patients. This research was originally published in Zhang et al (2008), © The American Society of Haematology.
Mutations in LMAN1 and MCFD2 account for all F5F8D patients
F5F8D has been reported throughout the world. As is the case for rare autosomal recessive disorders, F5F8D is often associated with consanguineous marriages. The highest reported occurrence of F5F8D is among Middle Eastern Jews and non-Jewish Iranians, estimated at c. 1:100 000 (Seligsohn et al, 1982; Peyvandi et al, 1998; Mansouritorgabeh et al, 2004). The actual prevalence of the disorder in various populations may be difficult to gauge because of ascertainment/publication bias. For example, F5F8D may be significantly under-diagnosed because of the often mild bleeding symptoms, or misdiagnosed as single factor deficiencies in many countries with limited haematology/genetics infrastructure. Using homozygosity mapping and positional cloning approaches, the first gene for F5F8D was identified in 1998 (Nichols et al, 1998) as LMAN1 (lectin, mannose-specific, also known as ERGIC-53) of previously unknown function. However, mutation analysis identified a subset of F5F8D families that have two normal LMAN1 alleles (Neerman-Arbez et al, 1999; Nichols et al, 1999). In 2003, the second cause of F5F8D was identified in these families as mutations in a novel gene that was named MCFD2 (multiple coagulation factor deficiency gene 2) (Zhang et al, 2003). Together, mutations in LMAN1 and MCFD2 account for nearly all cases of F5F8D, with c. 70% of F5F8D families attributable to LMAN1 mutations and c. 30% to MCFD2 mutations (Zhang et al, 2006, 2008).
At least 32 LMAN1 mutations have been reported throughout all 13 exons of the gene. Most are either non-sense or frameshift mutations whose truncated protein products would be predicted to lack the normal LMAN1 function. There are also reports of patients who are deficient in LMAN1 but with no mutations identified in the exons and exon-intron junctions, suggesting the presence of mutations in regulatory regions of the gene (Zhang et al, 2006, 2008). Only two missense mutations have been reported in LMAN1. The first mutation is a substitution of a threonine for the initiator methionine, predicted to abolish the translation and result in the absence of a protein product. The second missense mutation substitutes an arginine for a cysteine near the transmembrane (TM) domain. This cysteine residue had previously been shown to be involved in forming an intermolecular disulfide bond important for the oligomerization of LMAN1 (Nufer et al, 2003). In lymphoblasts derived from the proband, the endogenous mutant protein failed to accumulate in the cell, suggesting that this mutation results in an unstable protein (Zhang et al, 2006).
Sixteen MCFD2 mutations have been reported to date. Among these, nine are deletion or splicing mutations predicted to result in truncated protein products. In contrast to LMAN1, six missense mutations were identified in MCFD2 (Ivaskevicius et al, 2008; Zhang et al, 2008), all of which change amino acid residues that are highly conserved among MCFD2 orthologs of vertebrate species. In one patient, a non-sense mutation was identified that results in the deletion of the last three amino acids of MCFD2 and disrupts the structural integrity of the protein (Nyfeler et al, 2007). In addition, this same patient was found to be a compound heterozygote for a second allele with an 8·5-kb deletion in the promoter region of the MCFD2 gene (Nyfeler et al, 2007).
The diverse nature of the mutations suggests multiple independent genetic origins. Haplotype analyses performed on two MCFD2 mutations (c.149+5G>A and c.407T>C) that are identified in multiple distinct populations indicate that these mutations arose independently (Zhang et al, 2006). Of note, the c.149+5G>A mutation is by far the most common MCFD2 mutation identified, probably due to a mutation hotspot at a CpG dinucleotide. Some mutations are uniquely associated with certain populations, suggesting that founder mutations may account for all or the majority of F5F8D cases in some isolated populations. For example, two mutations in LMAN1 account for all Jewish F5F8D patients. One of these mutations is prevalent in Jews originating from the island of Djerba in Tunisia, but not found among North African Jews (Segal et al, 2004). The second LMAN1 mutation is unique to Middle Eastern Jews (Nichols et al, 1998). Another notable mutation is the missense mutation abolishing the initiation codon (c.2T>C) of LMAN1, which has only been reported in families of Italian origin.
Phenotypic differences between LMAN1 mutations and MCFD2 mutations in F5F8D
Although patients with LMAN1 mutations and patients with MCFD2 mutations are considered clinically indistinguishable, a genotype–phenotype correlation became evident with the comparison of a large number of patients with known mutations in either of the genes. A small but statistically significant difference in the distribution of factor levels can be observed between these two classes of patients (Zhang et al, 2008). Patients with MCFD2 mutations tend to have FV and FVIII levels at a lower range than patients with LMAN1 mutations (Fig 1A). The mean levels of plasma FV and FVIII in patients with MCFD2 mutations are significantly lower than the corresponding levels in patients with LMAN1 mutations. It should be noted that there is considerable overlap between the two groups of patients. Thus, FV and FVIII levels by themselves cannot be used to predict which gene mutation an individual patient may have. The normal plasma concentration of FVIII is two orders of magnitude lower than that of FV. If the observed variations in factor levels are the results of biological differences in the effect of loss of function for MCFD2 versus LMAN1, then plasma FV levels might be expected to correlate with FVIII levels for each individual patient. Indeed, a Pearson correlation analysis indicated a significant correlation between FV and FVIII levels in both patients with LMAN1 mutations and those with MCFD2 mutations (Fig 1B), suggesting that deficiencies in either LMAN1 or MCFD2 exert a similar impact on the secretion of both FV and FVIII (Zhang et al, 2008).
Circulating FV exists in both plasma and the alpha granules of platelets as a 330 kDa single chain polypeptide. In humans, the platelet FV pool is thought to originate from endocytosis of plasma FV (Camire et al, 1998; Gould et al, 2005), whereas in mice, platelet FV is derived exclusively from biosynthesis within the megakaryocytes (Sun et al, 2003; Yang et al, 2003). Comparison of platelet FV levels in F5F8D patients and normal controls indicates that platelet FV and plasma FV are reduced to the same extent for both the LMAN1 group and the MCFD2 group (Zhang et al, 2008). Therefore, mutations in LMAN1 or MCFD2 similarly affect the steady-state levels of FV in both pools.
Functions of LMAN1 and MCFD2 in FV and FVIII biosynthesis
The early secretory pathway
Proper intracellular trafficking and localization of proteins are fundamentally important processes in the normal homeostasis of cells. Proteins that traverse the secretory pathway are collectively referred to as ‘cargo proteins’. The early secretory pathway refers to the transport of proteins from the endoplasmic reticulum (ER) to the Golgi. Defects in the early secretory pathway are becoming increasingly recognized as pathological mechanisms of human disease, as a number of congenital disorders have recently been attributed to defects in genes that function in ER-to-Golgi transport (Gedeon et al, 1999; Jones et al, 2003; Zhang et al, 2003; Boyadjiev et al, 2006; Fromme et al, 2007). Cargo proteins exit the ER in coat protein complex-II (COPII) vesicles (Bonifacino & Glick, 2004; Lee & Miller, 2007). COPII is composed of a Guanosine triphosphate-binding protein, Sar1, and two cytosolic protein complexes, Sec23-Sec24 and Sec31-31. Sar1 in its GTP-bound form initiates membrane curvature (Lee et al, 2005) and stimulates the recruitment of Sec23-24 heterodimers to form a prebudding complex (Bi et al, 2002). Sec13/31 heterotetramers subsequently bind to the prebudding complex to form a flexible coat cage that can accommodate various sizes of vesicles (Fath et al, 2007; Stagg et al, 2008). Upon exiting the ER, COPII vesicles fuse with each other to form the ER-Golgi intermediate compartment (ERGIC) (Appenzeller-Herzog & Hauri, 2006), a structure located between the ER and Golgi that is unique to higher eukaryotic cells. In the ERGIC, cargo proteins are separated from ER resident proteins and transported to the cis-Golgi along microtubules (Palmer et al, 2005; Watson et al, 2005). ER resident proteins and cargo receptors that display ER-retrieval signals are returned to the ER via coat protein complex-I (COPI) vesicles (Lippincott-Schwartz & Liu, 2006).
How cargo proteins are recruited into COPII vesicles is not entirely clear. Some TM cargo proteins display specific sorting signals on the cytoplasmic side of the proteins that directly interact with the Sec24 subunit of COPII (Miller et al, 2003; Mossessova et al, 2003). The recruitment of soluble cargo proteins may be explained by two competing models (Barlowe, 2003; Baines & Zhang, 2007): bulk flow (passive diffusion into budding vesicles) and receptor-mediated (selective packaging of soluble cargo through specific interaction with a TM cargo receptor). Bulk flow has been observed in the transport of some highly abundant soluble cargo proteins (Martinez-Menarguez et al, 1999; Oprins et al, 2001). Less abundant soluble cargo proteins are generally thought to require cargo receptors for efficient export. However, cargo receptors have proven elusive to identify, with only a few characterized to date, mostly in yeast (Barlowe, 2003; Baines & Zhang, 2007). Studies of F5F8D unexpectedly provided direct evidence for the existence of mammalian cargo receptors.
Structure of MCFD2
MCFD2 is a 16-kDa soluble protein with two Ca2+-binding motifs known as EF-hand domains at the C-terminus (Fig 2A). The protein sequence is conserved in vertebrates from mammals to fish (Zhang et al, 2003). In contrast to LMAN1, MCFD2 is a monomeric protein (Guy et al, 2008; Kawasaki et al, 2008). Recently, the MCFD2 structure was determined by nuclear magnetic resonance (NMR) (Guy et al, 2008). It was revealed that in the presence of calcium ions, the EF-hand domains of the C-terminus of MCFD2 fold from a disordered apo state into a structure similar to that of calmodulin. However, the MCFD2 EF-hand domains do not form a typical protein-binding motif commonly observed in other EF-hand domain proteins. A notable feature of the MCFD2 structure is that the N-terminus of the molecule remains disordered even in the Ca2+-bound state (Fig 2B). The linker region between the two EF-hand domains is also largely unstructured. However, these disordered regions could become structured upon interacting with other proteins and could be important in forming protein binding sites in vivo (Guy et al, 2008).
Fig 2.
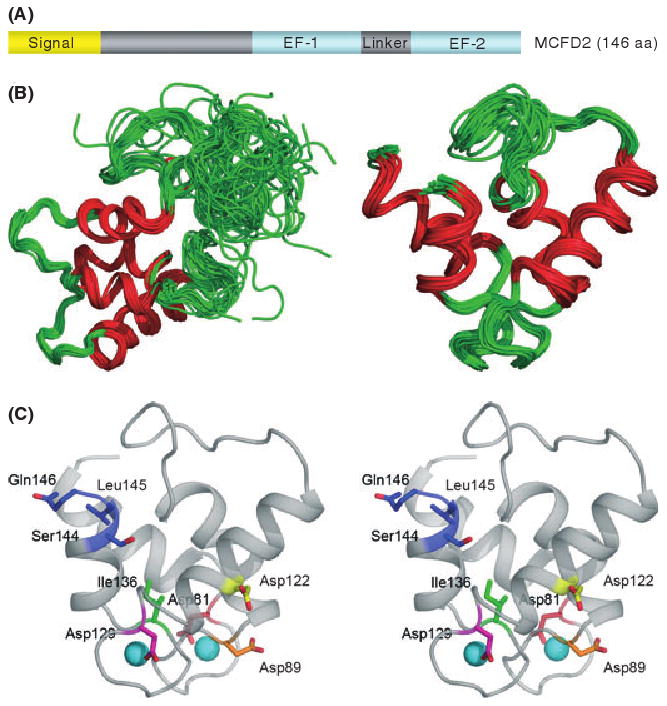
Structure of MCFD2 and locations of missense mutations. (A) MCFD2 primary sequence and domain structure. (B) The NMR structure of MCFD2 showing the complete structure (left) and only the ordered EF-hand domains (right). Red lines represent the alpha helices of the EF-hands. (C) A stereo view showing the positions of MCFD2 missense and C-terminal deletion mutations in the ordered structure of wild-type EF-hand domains (shown in colour). The two spheres represent calcium ions. Adapted from Guy et al (2008), with permission from Elsevier.
Structure of LMAN1
LMAN1 is a type-I integral membrane protein that was first described as a 53-kDa marker of the ERGIC (Hauri et al, 2000). The protein sequence is conserved in both vertebrates and some invertebrates, such as Caenorhabditis elegans and Drosophila melanogaster. Although the majority of LMAN1 is localized to the ERGIC at steady state, it cycles between the ER and ERGIC in live cells. This is achieved mainly through a di-phenylalanine (FF) ER exit motif and a di-lysine (KK) ER retrieval signal located at the C-terminal end of the protein that interact with COPII and COPI respectively (Nufer et al, 2002). The importance of the C-terminal sequence is underscored by a disease-causing mutation of a single nucleotide deletion (c.1519delA) that replaces the last three amino acids of the protein with a random sequence of 17 amino acids. LMAN1 also contains a short TM domain, a luminally-oriented carbohydrate recognition domain (CRD), and a series of four α-helices predicted to form a coiled-coil domain (Fig 3A). Two luminal cysteines near the TM domain form inter-molecular disulfide bonds, which, along with structural determinants in both the luminal and TM domains, lead to the oligomerization of LMAN1 (Nufer et al, 2003). Two homo-hexameric forms of LMAN1 exist within the cell, the first composed of six covalently linked monomers, and the second composed of three disulfide-linked dimers (Neve et al, 2005).
Fig 3.

Structure of LMAN1 and the LMAN1-MCFD2 protein complex. (A) LMAN1 primary sequence and domain structure. Arrowheads indicate the locations of two cysteines that are important in oligomerization of LMAN1. (B) Organization of the LMAN1-MCFD2 complex and the interaction with FVIII. Hexameric LMAN1 is recruited into budding vesicles via a C-terminal diphenolalanine motif (FF) that binds to COPII, and recycled back to the ER via a dilysine (KK) motif. MCFD2 interacts with LMAN1 in the ER lumen in a 1:1 stoichiometry. FVIII interacts with LMAN1 and MCFD2 in a tertiary complex. B domain-deleted FVIII binds only weakly to the LMAN1-MCFD2 complex. Dark grey ovals represent MCFD2 and light grey cylinders represent LMAN1. FVIII B domain is coloured purple. The fork-shaped structures on FVIII represent glycosylation sites. Adapted from Baines and Zhang (2007), with permission from Elsevier.
LMAN1 is a member of the family of L-type animal lectins that bear significant structural homology to leguminous lectins. All three members of the L-type lectins (LMAN1, VIP36 and VIPL) are localized to the early secretory pathway. The CRD of LMAN1 binds to high-mannose oligosaccharides in a Ca2+-dependent manner (Appenzeller-Herzog et al, 2004; Kamiya et al, 2008). The crystal structure of the CRD has been solved in the absence (Velloso et al, 2002) or presence (Velloso et al, 2003) of Ca2+. The CRD of LMAN1 is similar to Ca2+-dependent leguminous lectins and the ER chaperone calnexin, with two Ca2+-binding sites. Binding of Ca2+ induces a localized conformational change that enables an open carbohydrate-binding site capable of accommodating highly branched sugar structures found in glycoproteins in the ER. On the opposite side of the carbohydrate-binding site, there is a large surface patch with conserved residues that may form a protein-binding site.
LMAN1-MCFD2 interaction
Unlike LMAN1, MCFD2 contains no identifiable sorting signals other than an N-terminal signal peptide that directs it into the lumen of the ER, and in fact, proper localization of MCFD2 to ERGIC is dependent upon LMAN1 (Zhang et al, 2003, 2008), suggesting a direct interaction between the two proteins. In the absence of LMAN1, the majority of MCFD2 is secreted in an O-glycosylated form (Zhang et al, 2005) and only trace amounts of the protein can be detected within the cell (Zhang et al, 2006). Evidence for a direct molecular interaction between LMAN1 and MCFD2 comes from the observations that these two proteins can be immunoprecipitated in a complex from cells (Zhang et al, 2003, 2005). In addition, both exhibit similar half-lives and appear to form a Ca2+-dependent complex with a 1:1 stoichiometry. This interaction can also be directly visualized inside the live cells (Nyfeler et al, 2005, 2006). Taken together, these results suggest that each subunit of the hexameric LMAN1 is stably associated with one MCFD2 molecule in cells (Fig 3B). The LMAN1-MCFD2 complex can be reconstituted in vitro using the purified CRD of LMAN1 and MCFD2, suggesting that the CRD also contains the MCFD2 binding domain (Kawasaki et al, 2008). Structural details of the LMAN1-MCFD2 interaction remain to be elucidated.
Missense mutations in both EF-hand domains of MCFD2 disrupt LMAN1-MCFD2 interaction
Missense mutations identified in MCFD2 are localized to both the first EF-hand domain (D81Y and D89A) and the second EF-hand domain (D122V, D129E, Y135N and I136T). Five of these mutations have been expressed in cultured cells and were found to abolish the binding of MCFD2 to LMAN1 (Zhang et al, 2003, 2008). Consistent with this finding, the endogenous MCFD2 level in lymphoblasts isolated from a patient with the I136T mutation was markedly reduced (Zhang et al, 2006). In vitro studies demonstrate that the affinity of MCFD2 mutants (D129E and I136T) to the CRD of LMAN1 is reduced by 4–5 orders of magnitude (Kawasaki et al, 2008). Because MCFD2 lacks the ER exit and retrieval signals, it requires LMAN1 binding for proper intracellular localization. Whereas the wild-type MCFD2 co-localizes with LMAN1 in the ERGIC, MCFD2 mutant proteins exhibit a largely ER staining pattern (Zhang et al, 2003, 2008).
Each EF-hand domain contains six calcium coordinating residues that interact with the calcium ion (Gifford et al, 2007). All but two (D122 and I136) of the amino acid residues mutated in F5F8D patients are involved in chelating Ca2+ (Fig 2C). Ca2+ is required for the EF-hand domains of MCFD2 to assume a folded state. Although I136 is not directly involved in interacting with Ca2+, it is part of a so-called ‘β scaffold’ that is essential for calcium binding. MCFD2 with either D129E or I136T mutation demonstrates a profound change in the NMR spectra and altered circular dichroism (CD) spectra reminiscent of the calcium-free structure of MCFD2 (Guy et al, 2008), indicating that these mutations abolish the tertiary structure of the protein. D122V is the only mutation that alters an amino acid residue not directly involved in Ca2+ binding function of the EF-hand. Though not directly studied, this mutation could be predicted to also compromise the structural integrity of MCFD2 (Guy et al, 2008). Another interesting mutation is a non-sense mutation that results in deletion of the last three amino acids of MCFD2 (ΔSLQ). When expressed in cells, this truncated protein showed markedly reduced binding to LMAN1 (Nyfeler et al, 2007). Similarly, CD spectra suggest that this nonsense mutation causes severe disruption of the tertiary structure of MCFD2 (Nyfeler et al, 2007). Taken together, these studies suggest that the EF-hand domains of MCFD2 are essential for the LMAN1 interaction. Disruption of the LMAN1-MCFD2 interaction is thus the underlying mechanism for these missense and non-sense mutations in causing F5F8D.
The LMAN1-MCFD2 complex is an ER-to-Golgi transport receptor for FV and FVIII
The localization of the LMAN1-MCFD2 complex to the ERGIC suggests a cargo receptor function. Cargo receptors are thought to recruit cargo proteins into the budding vesicles by directly binding them in the ER. Though the stoichiometric interaction between LMAN1 and MCFD2 was readily observed, a consistent interaction of either protein or the complex with FV or FVIII was only reliably detected by treating cells with a chemical crosslinker (Zhang et al, 2005) that covalently links two proteins in close proximity, suggesting that these interactions are either low affinity or transient in nature. FVIII was shown to specifically co-immunoprecipitate with both anti-LMAN1 and anti-MCFD2 antibodies in extracts of crosslinker-treated cells. Additional experiments indicated that the co-immunoprecipitation of FVIII with LMAN1 was markedly reduced when cells were depleted of Ca2+ in the ER lumen (Zhang et al, 2005). On the other hand, treatment of cells with an N-linked glycosylation inhibitor had no significant effect on the amount of FVIII that crosslinked to LMAN1 or MCFD2. These results suggest that, while the interaction of FVIII with the LMAN1/MCFD2 complex is Ca2+-dependent, it is not dependent on the glycosylation state of FVIII.
Other potential cargo proteins of LMAN1
FV and FVIII are the only cargo proteins known to be affected in patients with F5F8D. Given the ubiquitous expression pattern of LMAN1 and MCFD2 and the presence of LMAN1 and MCFD2 orthologous in lower eukaryotes without a blood clotting system, additional cargo proteins that are targeted by this complex probably also exist. However, aside from mild-to-moderate bleeding symptoms, patients with F5F8D do not exhibit any overt clinical phenotypes that could be attributed to deficiencies of other cargo proteins, suggesting that transport deficiency in other proteins is likely to be at a level insufficient to produce a clinical phenotype. Studies in a cell culture system identified the lysosomal protease precursors pro-cathepsin C (pro-CatC) and pro-cathepsin Z (pro-CatZ), as well as α1-antitrypsin (A1AT) as potential cargo for LMAN1 (Vollenweider et al, 1998; Appenzeller et al, 1999; Nyfeler et al, 2008). In addition, Nicastrin (a component of the γ-secretase complex), SUMF1 (the formylglicine generating enzyme) and a mutant immunoglobulin heavy chain have also been reported to interact with LMAN1 (Mattioli et al, 2006; Morais et al, 2006; Anelli et al, 2007).
In a pulse-chase experiment, CatC secretion was delayed in cells expressing a dominant-negative mutant of LMAN1 that is retained in the ER (Vollenweider et al, 1998). Pro-CatZ was identified as a protein that interacts with LMAN1 in a chemical cross-linking study (Appenzeller et al, 1999). Interactions between pro-CatC/pro-CatZ and LMAN1 were also observed in live cells using a protein fluorescence complementation assay (PCA) (Nyfeler et al, 2005). Using a PCA-based screen, α1-antitrypsin (A1AT) was recently identified as an interaction partner of LMAN1. A kinetic delay in ER exit of A1AT was observed in Lman1-deficient mouse embryonic fibroblasts (Nyfeler et al, 2008). It is not clear whether a kinetic delay in ER exit will translate to a decrease in steady-state level of the protein in LMAN1 deficient patients. The levels of CatC, CatZ, A1AT and other potential cargo proteins have not been reported in F5F8D patients.
Cargo recognition by the LMAN1-MCFD2 receptor complex
What are the unique features of FV and FVIII that enable them to both interact with the LMAN1-MCFD2 complex? FV and FVIII are two large plasma glycoproteins that function as essential cofactors for the serine proteases FX and FIX respectively. FV and FVIII circulate as inactive precursors that are activated through limited proteolysis by thrombin. FV and FVIII share a similar domain structure (A1-A2-B-A3-C1-C2), and undergo similar extensive post-translational modifications (Kaufman, 1998). The A and C domains of FV and FVIII are highly similar to each other. On the other hand, the functionally dispensable B domains of FV and FVIII share little sequence similarities, but are both heavily glycosylated. B-domain deleted FVIII is poorly secreted compared with full-length FVIII and FVIII variants that retain small B domain fragments (Miao et al, 2004). B-domain deleted FVIII exhibits markedly reduced binding to the LMAN1-MCFD2 complex (Moussalli et al, 1999; Zhang et al, 2005), suggesting an interaction between the lectin LMAN1 and sugar side chains of the heavily glycosylated B domains of FV and FVIII (Fig 3B). However, the essential role of MCFD2 and the observation that unglycosylated FVIII can still interact with the LMAN1-MCFD2 complex (Zhang et al, 2005) suggest that protein-protein interactions are also involved (Cunningham et al, 2003). The interaction of pro-CatZ with LMAN1 requires an N-linked high-mannose glycan located in a β-hairpin loop of pro-CatZ (Appenzeller-Herzog et al, 2005). This binding motif is conformation-based, presumably ensuring that only correctly folded proteins can access their cargo receptor. The recognition of pro-CatZ/pro-CatC by LMAN1 may be different from the recognition of FV/FVIII by the LMAN1-MCFD2 complex, because the two groups of cargo proteins share no significant sequence homology.
LMAN1 and MCFD2 should act in concert to facilitate the transport of FV and FVIII, since mutations in either gene lead to impaired secretion of FV and FVIII. However, the precise role of MCFD2 in cargo receptor function is not clear. The observation that patients with MCFD2 mutations generally exhibit FV and FVIII levels at lower ranges than patients with LMAN1 mutations suggest that MCFD2 may play a more direct role in recruiting FV and FVIII. A MCFD2 missense mutant that cannot bind LMAN1 can still interact with FVIII (Zhang et al, 2005), suggesting that the interaction between MCFD2 and FVIII is, at least in part, independent of LMAN1. It is not clear whether LMAN1 can bind to FVIII independent of MCFD2. MCFD2 appears to be dispensable for the interaction between LMAN1 and pro-CatC and pro-CatZ, suggesting that MCFD2 is specifically required for the recruitment of FV and FVIII, thereby conferring cargo selectivity to LMAN1 (Nyfeler et al, 2006). The in vitro sugar binding ability of the CRD of LMAN1 is enhanced upon binding to MCFD2 (Kawasaki et al, 2008). However, it is not clear whether this is also true in vivo, and if so, whether this is a mechanism of cargo selectivity.
Conclusion
First reported in 1954 (Oeri et al, 1954), the molecular mechanism of F5F8D began to be understood over the last decade with the identification of LMAN1 and MCFD2 as the disease genes (Seligsohn & Ginsburg, 2006). Recent studies uncovered the early secretory pathway as an important step in the regulation of the in vivo biosynthesis of FV and FVIII. At present, the LMAN1-MCFD2 protein complex is the only confirmed mammalian cargo receptor for soluble cargo proteins. The requirement of both a TM component and a soluble cofactor is a unique feature among the currently known cargo receptors. The detailed organization of the LMAN1-MCFD2 receptor complex, and how it recognizes, transports, and releases FV and FVIII represent important aspects for future study. Better understanding of this secretory pathway should provide new insights into the general mechanisms that control protein transport from the ER to Gogi. F5 R506Q (FV Leiden) and increased FVIII activities are two important risk factors for venous thrombosis. The relatively mild symptoms associated with F5F8D suggest that inhibiting this secretary pathway might provide a safe alternative strategy for antithrombotic therapy.
Acknowledgments
The author thanks Dr David Ginsburg and Andrea Baines for critical reading of the manuscript. Research at the author's laboratory has been supported by the National Haemophilia Foundation, the March of Dimes Foundation (#5-FY08-86) and the Cleveland Clinic Foundation.
References
- Anelli T, Ceppi S, Bergamelli L, Cortini M, Masciarelli S, Valetti C, Sitia R. Sequential steps and checkpoints in the early exocytic compartment during secretory IgM biogenesis. EMBO Journal. 2007;26:4177–4188. doi: 10.1038/sj.emboj.7601844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appenzeller C, Andersson H, Kappeler F, Hauri HP. The lectin ERGIC-53 is a cargo transport receptor for glycoproteins. Nature Cell Biology. 1999;1:330–334. doi: 10.1038/14020. [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Hauri HP. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. Journal of Cell Sciences. 2006;119:2173–2183. doi: 10.1242/jcs.03019. [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Roche AC, Nufer O, Hauri HP. pH-induced conversion of the transport lectin ERGIC-53 triggers glycoprotein release. Journal of Biological Chemistry. 2004;279:12943–12950. doi: 10.1074/jbc.M313245200. [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Nyfeler B, Burkhard P, Santamaria I, Lopez-Otin C, Hauri HP. Carbohydrate- and conformation-dependent cargo capture for ER-exit. Molecular Biology of the Cell. 2005;16:1258–1267. doi: 10.1091/mbc.E04-08-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines AC, Zhang B. Receptor-mediated protein transport in the early secretory pathway. Trends in Biochemical Sciences. 2007;32:381–388. doi: 10.1016/j.tibs.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Barlowe C. Signals for COPII-dependent export from the ER: what's the ticket out? Trends in Cell Biology. 2003;13:295–300. doi: 10.1016/s0962-8924(03)00082-5. [DOI] [PubMed] [Google Scholar]
- Bi X, Corpina RA, Goldberg J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419:271–277. doi: 10.1038/nature01040. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- Boyadjiev SA, Fromme JC, Ben J, Chong SS, Nauta C, Hur DJ, Zhang G, Hamamoto S, Schekman R, Ravazzola M, Orci L, Eyaid W. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulumto-Golgi trafficking. Nature Genetics. 2006;38:1192–1197. doi: 10.1038/ng1876. [DOI] [PubMed] [Google Scholar]
- Camire RM, Pollak ES, Kaushansky K, Tracy PB. Secretable human platelet-derived factor V originates from the plasma pool. Blood. 1998;92:3035–3041. [PubMed] [Google Scholar]
- Cunningham MA, Pipe SW, Zhang B, Hauri HP, Ginsburg D, Kaufman RJ. LMAN1 is a molecular chaperone for the secretion of coagulation factor VIII. Journal of Thrombosis and Haemostasis. 2003;1:2360–2367. doi: 10.1046/j.1538-7836.2003.00415.x. [DOI] [PubMed] [Google Scholar]
- Fath S, Mancias JD, Bi X, Goldberg J. Structure and organization of coat proteins in the COPII cage. Cell. 2007;129:1325–1336. doi: 10.1016/j.cell.2007.05.036. [DOI] [PubMed] [Google Scholar]
- Fromme JC, Ravazzola M, Hamamoto S, Al-Balwi M, Eyaid W, Boyadjiev SA, Cosson P, Schekman R, Orci L. The genetic basis of a craniofacial disease provides insight into COPII coat assembly. Developmental Cell. 2007;13:623–634. doi: 10.1016/j.devcel.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedeon AK, Colley A, Jamieson R, Thompson EM, Rogers J, Sillence D, Tiller GE, Mulley JC, Gecz J. Identification of the gene (SEDL) causing X-linked spondyloepiphyseal dysplasia tarda. Nature Genetics. 1999;22:400–404. doi: 10.1038/11976. [DOI] [PubMed] [Google Scholar]
- Gifford JL, Walsh MP, Vogel HJ. Structures and metalion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochemical Journal. 2007;405:199–221. doi: 10.1042/BJ20070255. [DOI] [PubMed] [Google Scholar]
- Gould WR, Simioni P, Silveira JR, Tormene D, Kalafatis M, Tracy PB. Megakaryocytes endocytose and subsequently modify human factor V in vivo to form the entire pool of a unique platelet-derived cofactor. Journal of Thrombosis and Haemostasis. 2005;3:450–456. doi: 10.1111/j.1538-7836.2005.01157.x. [DOI] [PubMed] [Google Scholar]
- Guy JE, Wigren E, Svard M, Hard T, Lindqvist Y. New insights into multiple coagulation factor deficiency from the solution structure of human MCFD2. Journal of Molecular Biology. 2008;381:941–955. doi: 10.1016/j.jmb.2008.06.042. [DOI] [PubMed] [Google Scholar]
- Hauri HP, Kappeler F, Andersson H, Appenzeller C. ERGIC-53 and traffic in the secretory pathway. Journal of Cell Sciences. 2000;113(Pt 4):587–596. doi: 10.1242/jcs.113.4.587. [DOI] [PubMed] [Google Scholar]
- Ivaskevicius V, Windyga J, Baran B, Bykowska K, Daugela L, Watzka M, Seifried E, Oldenburg J. The first case of combined coagulation factor V and coagulation factor VIII deficiency in Poland due to a novel p.Tyr135Asn missense mutation in the MCFD2 gene. Blood Coagulation and Fibrinolysis. 2008;19:531–534. doi: 10.1097/MBC.0b013e3283061103. [DOI] [PubMed] [Google Scholar]
- Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR, Eichenbaum-Voline S, Rudling M, Myrdal U, Annesi G, Naik S, Meadovvs N, Quattrone A, Islam SA, Naoumova RP, Angelin B, Infante R, Levy E, Roy CC, Freemont PS, Scott J, Shoulders CC. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nature Genetics. 2003;34:29–31. doi: 10.1038/ng1145. [DOI] [PubMed] [Google Scholar]
- Kamiya Y, Kamiya D, Yamamoto K, Nyfeler B, Hauri HP, Kato K. Molecular basis of sugar recognition by the human L-type lectins ERGIC-53, VIPL, and VIP36. Journal of Biological Chemistry. 2008;283:1857–1861. doi: 10.1074/jbc.M709384200. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Post-translational modifications required for coagulation factor secretion and function. Thrombosis and Haemostasis. 1998;79:1068–1079. [PubMed] [Google Scholar]
- Kawasaki N, Ichikawa Y, Matsuo I, Totani K, Matsumoto N, Ito Y, Yamamoto K. The sugar-binding ability of ERGIC-53 is enhanced by its interaction with MCFD2. Blood. 2008;111:1972–1979. doi: 10.1182/blood-2007-06-097022. [DOI] [PubMed] [Google Scholar]
- Lee MC, Miller EA. Molecular mechanisms of COPII vesicle formation. Seminars in Cell and Developmental Biololgy. 2007;18:424–434. doi: 10.1016/j.semcdb.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Lee MC, Orci L, Hamamoto S, Futai E, Ravazzola M, Schekman R. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell. 2005;122:605–617. doi: 10.1016/j.cell.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Liu W. Insights into COPI coat assembly and function in living cells. Trends in Cell Biology. 2006;16:e1–e4. doi: 10.1016/j.tcb.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Mansouritorgabeh H, Rezaieyazdi Z, Pourfathollah AA, Rezai J, Esamaili H. Haemorrhagic symptoms in patients with combined factors V and VIII deficiency in north-eastern Iran. Haemophilia. 2004;10:271–275. doi: 10.1111/j.1365-2516.2004.00890.x. [DOI] [PubMed] [Google Scholar]
- Martinez-Menarguez JA, Geuze HJ, Slot JW, Klumperman J. Vesicular tubular clusters between the ER and Golgi mediate concentration of soluble secretory proteins by exclusion from COPI-coated vesicles. Cell. 1999;98:81–90. doi: 10.1016/S0092-8674(00)80608-X. [DOI] [PubMed] [Google Scholar]
- Mattioli L, Anelli T, Fagioli C, Tacchetti C, Sitia R, Valetti C. ER storage diseases: a role for ERGIC-53 in controlling the formation and shape of Russell bodies. Journal of Cell Sciences. 2006;119:2532–2541. doi: 10.1242/jcs.02977. [DOI] [PubMed] [Google Scholar]
- Miao HZ, Sirachainan N, Palmer L, Kucab P, Cunningham MA, Kaufman RJ, Pipe SW. Bioengineering of coagulation factor VIII for improved secretion. Blood. 2004;103:3412–3419. doi: 10.1182/blood-2003-10-3591. [DOI] [PubMed] [Google Scholar]
- Miller EA, Beilharz TH, Malkus PN, Lee MCS, Hamamoto S, Orci L, Schekman R. Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell. 2003;114:497–509. doi: 10.1016/s0092-8674(03)00609-3. [DOI] [PubMed] [Google Scholar]
- Morais VA, Brito C, Pijak DS, Crystal AS, Fortna RR, Li T, Wong PC, Doms RW, Costa J. N-glycosylation of human nicastrin is required for interaction with the lectins from the secretory pathway calnexin and ERGIC-53. Biochimica et Biophysica Acta. 2006;1762:802–810. doi: 10.1016/j.bbadis.2006.06.018. [DOI] [PubMed] [Google Scholar]
- Mossessova E, Bickford LC, Goldberg J. SNARE selectivity of the COPII coat. Cell. 2003;114:483–495. doi: 10.1016/s0092-8674(03)00608-1. [DOI] [PubMed] [Google Scholar]
- Moussalli M, Pipe SW, Hauri HP, Nichols WC, Ginsburg D, Kaufman RJ. Mannose-dependent endoplasmic reticulum (ER)-Golgi intermediate compartment-53-mediated ER to Golgi trafficking of coagulation factors V and VIII. Journal of Biological Chemistry. 1999;274:32539–32542. doi: 10.1074/jbc.274.46.32539. [DOI] [PubMed] [Google Scholar]
- Neerman-Arbez M, Johnson KM, Morris MA, Mcvey JH, Peyvandi F, Nichols WC, Ginsburg D, Rossier C, Antonarakis SE, Tuddenham EGD. Molecular analysis of the ERGIC-53 gene in 35 families with combined factor V factor VIII deficiency. Blood. 1999;93:2253–2260. [PubMed] [Google Scholar]
- Neve EP, Lahtinen U, Pettersson RF. Oligomerization and interacellular localization of the glycoprotein receptor ERGIC-53 is independent of disulfide bonds. Journal of Molecular Biology. 2005;354:556–568. doi: 10.1016/j.jmb.2005.09.077. [DOI] [PubMed] [Google Scholar]
- Nichols WC, Seligsohn U, Zivelin A, Terry VH, Hertel CE, Wheatley MA, Moussalli MJ, Hauri HP, Ciavarella N, Kaufman RJ, Ginsburg D. Mutations in the ER-Golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation factors V and VIII. Cell. 1998;93:61–70. doi: 10.1016/s0092-8674(00)81146-0. [DOI] [PubMed] [Google Scholar]
- Nichols WC, Terry VH, Wheatley MA, Yang A, Zivelin A, Ciavarella N, Stefanile C, Matsushita T, Saito H, de Bosch NB, Ruiz-Saez A, Torres A, Thompson AR, Feinstein DI, White GC, Negrier C, Vinciguerra C, Aktan M, Kaufman RJ, Ginsburg D, Seligsohn U. ERGIC-53 gene structure and mutation analysis in 19 combined factors V and VIII deficiency families. Blood. 1999;93:2261–2266. [PubMed] [Google Scholar]
- Nufer O, Guldbrandsen S, Degen M, Kappeler F, Paccaud JP, Tani K, Hauri HP. Role of cytoplasmic C-terminal amino acids of membrane proteins in ER export. Journal of Cell Sciences. 2002;115:619–628. doi: 10.1242/jcs.115.3.619. [DOI] [PubMed] [Google Scholar]
- Nufer O, Kappeler F, Guldbrandsen S, Hauri HP. ER export of ERGIC-53 is controlled by cooperation of targeting determinants in all three of its domains. Journal of Cell Sciences. 2003;116:4429–4440. doi: 10.1242/jcs.00759. [DOI] [PubMed] [Google Scholar]
- Nyfeler B, Michnick SW, Hauri HP. Capturing protein interactions in the secretory pathway of living cells. Proceedings of National Academy of Sciences of the United States of America. 2005;102:6350–6355. doi: 10.1073/pnas.0501976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyfeler B, Zhang B, Ginsburg D, Kaufman RJ, Hauri HP. Cargo selectivity of the ERGIC-53/MCFD2 transport receptor complex. Traffic. 2006;7:1473–1481. doi: 10.1111/j.1600-0854.2006.00483.x. [DOI] [PubMed] [Google Scholar]
- Nyfeler B, Kamiya Y, Boehlen F, Yamamoto K, Kato K, de Moerloose P, Hauri HP, Neerman-Arbez M. Deletion of three residues from the C-terminus of MCFD2 affects binding to ERGIC-53 and causes combined factor V and factor VIII deficiency. Blood. 2007;111:1299–1301. doi: 10.1182/blood-2007-09-112854. [DOI] [PubMed] [Google Scholar]
- Nyfeler B, Reiterer V, Wendeler MW, Stefan E, Zhang B, Michnick SW, Hauri HP. Identification of ERGIC-53 as an intracellular transport receptor of a1-antitrypsin. Journal of Cell Biology. 2008;180:705–712. doi: 10.1083/jcb.200709100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeri J, Matter M, Isenschmid H, Hauser F, Koller F. Angeborener mangel an faktor V (parahaemophilie) verbunden mit echter haemophilie A bein zwei brudern. Modern Problems in Paediatrics. 1954;1:575–588. [PubMed] [Google Scholar]
- Oprins A, Rabouille C, Posthuma G, Klumperman J, Geuze HJ, Slot JW. The ER to Golgi interface is the major concentration site of secretory proteins in the exocrine pancreatic cell. Traffic. 2001;2:831–838. doi: 10.1034/j.1600-0854.2001.21112.x. [DOI] [PubMed] [Google Scholar]
- Palmer KJ, Watson P, Stephens DJ. The role of microtubules in transport between the endoplasmic reticulum and Golgi apparatus in mammalian cells. Biochemical Society Symposia. 2005;72:1–13. doi: 10.1042/bss0720001. [DOI] [PubMed] [Google Scholar]
- Peyvandi F, Tuddenham EG, Akhtari AM, Lak M, Mannucci PM. Bleeding symptoms in 27 Iranian patients with the combined deficiency of factor V and factor VIII. British Journal of Haematology. 1998;100:773–776. doi: 10.1046/j.1365-2141.1998.00620.x. [DOI] [PubMed] [Google Scholar]
- Segal A, Zivelin A, Rosenberg N, Ginsburg D, Shpilberg O, Seligsohn U. A mutation in LMAN1 (ERGIC-53) causing combined factor V and factor VIII deficiency is prevalent in Jews originating from the island of Djerba in Tunisia. Blood Coagulation and Fibrinolysis. 2004;15:99–102. doi: 10.1097/00001721-200401000-00016. [DOI] [PubMed] [Google Scholar]
- Seligsohn U, Ginsburg D. Deciphering the mystery of combined factor V and factor VIII deficiency. Journal of Thrombosis and Haemostasis. 2006;4:927–931. doi: 10.1111/j.1538-7836.2006.01939.x. [DOI] [PubMed] [Google Scholar]
- Seligsohn U, Zivelin A, Zwang E. Combined factor V and factor VIII deficiency among non-Ashkenazi Jews. New England Journal of Medicine. 1982;307:1191–1195. doi: 10.1056/NEJM198211043071907. [DOI] [PubMed] [Google Scholar]
- Spreafico M, Peyvandi F. Combined FV and FVIII deficiency. Haemophilia. 2008;14:1201–1208. doi: 10.1111/j.1365-2516.2008.01845.x. [DOI] [PubMed] [Google Scholar]
- Stagg SM, Lapointe P, Razvi A, Gurkan C, Potter CS, Carragher B, Balch WE. Structural basis for cargo regulation of COPII coat assembly. Cell. 2008;134:474–484. doi: 10.1016/j.cell.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Yang TL, Yang A, Wang X, Ginsburg D. The murine platelet and plasma factor V pools are biosynthetically distinct and sufficient for minimal hemostasis. Blood. 2003;102:2856–2861. doi: 10.1182/blood-2003-04-1225. [DOI] [PubMed] [Google Scholar]
- Velloso LM, Svensson K, Schneider G, Pettersson RF, Lindqvist Y. Crystal structure of the carbohydrate recognition domain of p58/ERGIC- 53, a protein involved in glycoprotein export from the endoplasmic reticulum. Journal of Biological Chemistry. 2002;277:15979–15984. doi: 10.1074/jbc.M112098200. [DOI] [PubMed] [Google Scholar]
- Velloso LM, Svensson K, Pettersson RF, Lindqvist Y. The crystal structure of the carbohydrate-recognition domain of the glycoprotein sorting receptor p58/ERGIC-53 reveals an unpredicted metal-binding site and conformational changes associated with calcium ion binding. Journal of Molecular Biology. 2003;334:845–851. doi: 10.1016/j.jmb.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Vollenweider F, Kappeler F, Itin C, Hauri HP. Mistargeting of the lectin ERGIC-53 to the endoplasmic reticulum of HeLa cells impairs the secretion of a lysosomal enzyme. Journal of Cell Biology. 1998;142:377–389. doi: 10.1083/jcb.142.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson P, Forster R, Palmer KJ, Pepperkok R, Stephens DJ. Coupling of ER exit to microtubules through direct interaction of COPII with dynactin. Nature Cell Biology. 2005;7:48–55. doi: 10.1038/ncb1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang TL, Pipe SW, Yang A, Ginsburg D. Biosynthetic origin and functional significance of murine platelet factor V. Blood. 2003;102:2851–2855. doi: 10.1182/blood-2003-04-1224. [DOI] [PubMed] [Google Scholar]
- Zhang B, Ginsburg D. Familial multiple coagulation factor deficiencies: new biologic insight from rare genetic bleeding disorders. Journal of Thrombosis and Haemostasis. 2004;2:1564–1572. doi: 10.1111/j.1538-7836.2004.00857.x. [DOI] [PubMed] [Google Scholar]
- Zhang B, Ginsburg D. Familial multiple coagulation factor deficiencies. In: Colman RW, Marder VJ, Clowes AW, George JN, Goldhaber SZ, editors. Hemostasis and Thrombosis, Basic Principles and Clinical Practice. Lippincott Williams & Wilkins; Philadelphia: 2006. pp. 953–960. [Google Scholar]
- Zhang B, Cunningham MA, Nichols WC, Bernat JA, Seligsohn U, Pipe SW, Mcvey JH, Schulte-Overberg U, de Bosch NB, Ruiz-Saez A, White GC, Tuddenham EGD, Kaufman RJ, Ginsburg D. Bleeding due to disruption of a cargo-specific ER-to-Golgi transport complex. Nature Genetics. 2003;34:220–225. doi: 10.1038/ng1153. [DOI] [PubMed] [Google Scholar]
- Zhang B, Kaufman RJ, Ginsburg D. LMAN1 and MCFD2 form a cargo receptor complex and interact with coagulation factor VIII in the early secretory pathway. Journal of Biological Chemistry. 2005;280:25881–25886. doi: 10.1074/jbc.M502160200. [DOI] [PubMed] [Google Scholar]
- Zhang B, McGee B, Yamaoka JS, Guglielmone H, Downes KA, Minoldo S, Jarchum G, Peyvandi F, de Bosch NB, Ruiz-Saez A, Chatelain B, Olpinski M, Bockenstedt P, Sperl W, Kauman RJ, Nichols WC, Egd T, Ginsburg D. Combined deficiency of factor V and factor VIII is due to mutations in either LMAN1 or MCFD2. Blood. 2006;107:1903–1907. doi: 10.1182/blood-2005-09-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Spreafico M, Yang A, Platzer P, Callaghan MU, Avci Z, Ozbek N, Mahlangu J, Haw T, Kaufman RJ, Marchant K, Tuddenham EG, Seligsohn U, Peyvandi F, Ginsburg D. Genotype-phenotype correlation in combined deficiency of factor V and factor VIII. Blood. 2008;111:5592–5600. doi: 10.1182/blood-2007-10-113951. [DOI] [PMC free article] [PubMed] [Google Scholar]