

Isolated from the tropical interstitial ciliate Euplotes vannus strains Si121 and BUN3, vannusals A and B were assigned structures 1 and 2, respectively (Figure 1).[1, 2] These novel and challenging molecular architectures fascinated scientists since their disclosure in 1999 and stood defiant to chemical synthesis until 2008, when we reported the first total synthesis of the originally assigned structure of vannusal B (2) and proved it to be wrong.[3] The puzzle of the correct structure of vannusal B was complicated by the scarcity of the natural product and its unprecedented carbon framework, leaving the challenge of its solution to chemical synthesis. In this and the following communication,[4] we report our investigations that led to the total synthesis of several suspected stereoisomers of this molecule and the eventual elucidation of its true structure (and that of its sibling, vannusal A) through its total synthesis.
Figure 1.

Originally assigned structures of vannusals A (1) and B (2) and initially targeted stereoisomers 3 [C21-epi-2] and 4 [C21-epi, C25-epi-2].
Based on the interplay between total synthesis and NMR spectroscopy, the journey to the true structure of vannusal B was long and arduous. It became urgent and was initiated immediately upon completion of the total synthesis of its originally assigned structure (2).[3] In the following description, we unravel the logical evolution of events that led to the emergence of useful intelligence that allowed the eventual solution of the vannusal conundrum. Thus, on comparison of the NMR spectral data of natural vannusal B and synthetic 2, it became apparent that the most striking differences were located in the “northeastern” region of the molecule, particularly around rings D and E. Strong NMR spectroscopic evidence (see Figure 2) indicated that stereocenters C26 (w-coupling, JH26,15left = 1.6 Hz; NOE, H26/H27) and C18 (NOE, H25/H16) were likely correct, leaving C25 and C21 as the most logical positions to start our structural modifications. This narrowed our choice to four diastereomeric structures, one of which (i.e. 2) we had already synthesized.[3] From the remaining three, we selected the C21-epi diastereomer of 2, structure 3 (Figure 1), as our next target molecule based on a subtle and intriguing observation: that inversion of stereochemistry at C21 would bring the “northeastern” domain of vannusal B in line with the proposed biosynthetic hypothesis that postulated a dimerization of two identical monomeric units (prevannusal, naturally occurring)[2] as the biosynthetic precursors to vannusals A and B.
Figure 2.

Key 1H NMR coupling constant (w-JH26, 15left = 1.6 Hz) and NOE exhibited by both the originally assigned structure (synthetic, 2) and natural vannusal B.
The strategy for the total synthesis of the targeted vannusal B diastereomer 3 relied on the retrosynthetic analysis outlined in Figure 3. Thus, based on our experience in the total synthesis of the originally assigned structure of vannusal B (2),[3] we dissected structure 3 at the indicated bonds through (a) a Li-mediated coupling reaction (C11-C12, originally as a C-C bond and eventually as a C=C bond), and (b) a SmI2-based[5] cyclization (C10-C28 bond). Accompanied by appropriate functional group modifications, these disconnections revealed vinyl iodide 5 and aldehyde 6 as potential key building blocks for the proposed construction.
Figure 3.

Retrosynthetic analysis of vannusal B stereoisomer 3 [C21-epi-2]. BOM = benzyloxymethyl; SEM = trimethylsilylethoxymethyl; TIPS = triisopropylsilyl.
With vinyl iodide (−)-5 already in hand in its enantiopure form,[3] we proceeded to devise a synthesis for racemic aldehyde 6, the other required fragment for the construction of structure 3. This objective demanded different chemistry from that employed in the construction of its C21-epi counterpart[3] used to synthesize the originally assigned vannusal B structure (2). Thus, and as shown in Scheme 1, epoxide 9 was synthesized through vanadium-catalyzed epoxidation [tBuOOH, VO(acac)2 (cat.), 90 % yield] of homoallylic alcohol 8, prepared from racemic 7[3] by treatment with Martin’s sulfurane (87 % yield). Epoxide 9 was formed as a single diastereomer through the exquisite stereocontrol exerted by the free homoallylic hydroxyl group within substrate 8. The subsequent task of installing the intended nitrile moiety, through the use of the Nagata reagent (Et2AlCN), however, required protection of this hydroxyl group as an acetate (Ac2O, Et3N, 4-DMAP, 90 % yield).[6] Upon exposure to Et2AlCN, the latter compound afforded the targeted trans hydroxy nitrile 11, as expected, in 81 % yield. Removal of the acetate group from 11 (K2CO3, MeOH) furnished the required dihydroxy nitrile 12, in 100 % yield. The structure of 12 was secured unambiguously by X-ray crystalographic analysis[7] (see ORTEP drawing, Scheme 1) of its crystalline acetonide derivative 13 (mp 87–88 °C, hexanes) prepared by exposure of 12 to 2,2-dimethoxypropane in the presence of PPTS (cat.) (89 % yield) as shown in Scheme 1.
Scheme 1.

Construction of nitrile 12 (top) and X-ray derived ORTEP drawing of 13 (bottom). Reagents and conditions: (a) Martin’s sulfurane (1.1 equiv), Et3N (10 equiv), CH2Cl2, 25 °C, 5 h, 87 %; (b) tBuOOH (3.0 equiv), VO(acac)2 (0.2 equiv), PhH, 25 °C, 6 h, 90 %; (c) Ac2O (10 equiv), Et3N (30 equiv), 4-DMAP (1.0 equiv), CH2Cl2, 4 h, 25 °C, 90 %; (d) Et2AlCN (10 equiv), PhMe, −78→−20 °C, 19 h, 81 %; (e) K2CO3 (1.0 equiv), MeOH, 25 °C, 2 h, 100 %; (f) DMF/2,2-dimethoxypropane (1:1), PPTS (1.0 equiv), 24 h, 89 %.
The elaboration of dihydroxy nitrile 12 to aldehyde 6 is summarized in Scheme 2. Thus, protection of the hydroxy groups of 12 with SEM moieties (SEMCl, iPr2NEt, 90 % yield) led to bis–SEM derivative 14, which was then converted to tertiary alcohol 15 through a four-step sequence [DIBAL-H reduction, MeMgBr addition, NMO-TPAP (cat.) oxidation, and MeMgBr addition, 72 % overall yield for four steps]. Capping the newly generated tertiary hydroxyl group within 15 required more forcing conditions (SEMCl, KHMDS, THF, −78→25 °C, 92 % yield), and led to the expected tri-SEM derivative 16. The remaining steps to the desired aldehyde 6 followed our previously developed strategy[3] which required initial ozonolysis of 16 (89 % yield) and subsequent enolization/O-alkylation of the resulting aldehyde (KH, allyl chloride) to afford allyl enol ether 17 (91 % yield). Heating of the latter under μ–wave conditions (200 °C) effected the desired Claisen rearrangement, and NaBH4 reduction converted the resulting aldehyde to the primary alcohol 18 (91 % overall yield for the two steps). Subsequent protection of the primary hydroxyl group within the latter (BOMCl, iPr2NEt, nBu4NI) followed by ozonolysis (O3; Ph3P) led to 19 (92 % overall yield for two steps), which was finally converted to (±)-6 through silyl enol ether formation (DBU, TBSCl) and another ozonolysis (O3; Ph3P), in 92 % overall yield for the two steps.
Scheme 2.

Construction of aldehyde (±)-6. Reagents and conditions: (a) SEMCl (10 equiv), iPr2NEt (30 equiv), CH2Cl2, 50 °C, 48 h, 90 %; (b) DIBAL-H (1.1 equiv), PhMe, −78→30 °C, 1 h; then 0.1 M HCl, 25 °C, 20 min; (c) MeMgBr (10 equiv), THF, 0 °C, 30 min; (d) NMO (2.0 equiv), TPAP (0.05 equiv), CH2Cl2/CH3CN (7:1), 25 °C, 3 h; (e) MeMgBr (10 equiv), THF, −10 °C, 20 min, 72 % for four steps; (f) KHMDS (2.0 equiv), SEMCl (5.0 equiv), Et3N (10 equiv), 1 h, 92 %; (g) O3, py (1.0 equiv), CH2Cl2:MeOH (1:1), −78 °C; then Ph3P (5.0 equiv), −78→25 °C, 1 h, 89 %; (h) KH (10 equiv), allyl chloride (30 equiv), HMPA (10 equiv), DME, 25 °C, 12 h, 91 %; (i) iPr2NEt (1.0 equiv), o-dichlorobenzene, 200 °C (μ-waves), 20 min; then NaBH4 (10 equiv), MeOH, 1 h, 25 °C, 91 % for two steps; (j) BOMCl (10 equiv), iPr2NEt (30 equiv), nBu4NI (1.0 equiv), CH2Cl2, 50 °C 12 h; (k) O3, py (1.0 equiv), CH2Cl2:MeOH (1:1), −78 °C; then Ph3P (5.0 equiv), − 78→25 °C, 1 h, 92 % for two steps; (l) TBSCl (10 equiv), DBU (20 equiv), CH2Cl2, 25 °C, 48 h; (m) O3, py (1.0 equiv), CH2Cl2:MeOH (1:1), −78 °C; then Ph3P (5.0 equiv), −78→25 °C, 1 h, 92 % for two steps. HMPA = hexamethylphosphoramide, DBU = 1,8-diazoicyclo[5.4.0]undec-7-ene.
With both key building blocks (−)-5 and (±)-6 in hand, we proceeded with their union and further elaboration of the desired diastereomeric coupling product to its final destination, vannusal B structure 3, as shown in Scheme 3. Lithium–iodide exchange within (−)-5 (tBuLi, THF, −78→ −40 °C) followed by addition of (±)-6 led to two coupling products (ca. 1:1 dr), which, after removal of the TIPS group (TBAF, 25 °C), were chromatographically separated, providing 20 (41 % yield for two steps) and its diastereomer (d–20, not shown, 42 % yield for two steps). Diastereomer 20 was converted to cyclization precursor aldehyde carbonate 21 through a four-step sequence involving temporary TES protection of the primary hydroxyl group (TESCl, imid.), installation of a carbonate moiety at C12 (ClCO2Me, Et3N), removal of the TES group [HF•py/py (1:4), 79 % over the three steps], and oxidation of the regenerated primary alcohol [PhI(OAc)2-AZADO (cat.),[8] 95 % yield]. With precursor 21 at hand, we were then in a position to attempt the crucial SmI2-induced ring closure reaction that would forge the entire carbon skeleton of our target molecule, a process whose efficiency and stereochemical outcome we found to be dependent on the nature of the protecting groups residing on the C26, C25 and C22 oxygen residues, as well as the relative stereochemistry of the two domains of the molecule (i.e. 20 vs d-20). In this instance, the SEM groups on these positions in precursor 21 proved co-operative, facilitating its intended cyclization (SmI2, HMPA, THF, − 10→25 °C) to afford two chromatographically separable diastereomers (22β, 33 % yield, 22α, 21 % yield).[9] Both diastereomers could be easily converted into the same conjugated diene 23 through previously developed procedures[3] as a prelude to correcting their stereochemistry. Treatment of 22α with POCl3 and pyridine led to the formation of 23 in 72 % yield, while conversion of 22β into 23 proceeded through a xanthate formation (NaH, CS2; MeI) and Chugaev syn-elimination (microwave heating, 185 °C, 86 % yield for the two steps). Conjugated diene 23 was transformed regio- and stereoselectively to intermediate 24, possessing the inverted and desired stereochemistry at C10 and C28, by sequential hydroboration/oxidation (ThexBH2; BH3•THF; H2O2, 70 % yield) and phenylselenenylation/syn elimination (oNO2C6H4SeCN, nBu3P; H2O2, 68 % overall yield). The final drive from 24 to vannusal B structure 3 proceeded through intermediate 25 (Scheme 3) and required installment of a TES group at C28 (TESCl, KHMDS, 89 % yield), removal of the BOM groups (LiDBB, 83 % yield), selective oxidation of the primary alcohol over the secondary [PhI(OAc)2, 1-Me-AZADO (cat.)],[8] acetylation (Ac2O, 4-DMAP, 87 % yield for two steps), and, finally, aq. HF-induced global deprotection [aq. HF:THF (1:3), 77 %]. The AZADO and 1-Me-AZADO catalysts[8] (see structures, Scheme 3) proved to be superior to TEMPO in these studies, and were subsequently employed with success in several other sequences instead of the latter. Although consistent with its structure, the NMR spectroscopic data of synthetic vannusal B structure 3 did not match those reported for the natural product, sending us back to the drawing board to contemplate our next move. Disappointing as they were, these data, however, pointed to a new line of investigation. Specifically, the rather large coupling constant between H25 and H21 (JH25, 21 = 8.5 Hz, see Figure 4) exhibited in the 1H NMR spectrum of 3 (a C25/C21 trans structure) seemed to suggest that the true structure of vannusal B (exhibiting JH25, 21 = 1.6 Hz) possessed a C25/C21 cis arrangement rather than a trans relationship.
Scheme 3.

Synthesis of vannusal B structure 3 [C21-epi-2]. Reagents and conditions: (a) (−)-5 (1.3 equiv), tBuLi (2.5 equiv), THF, −78→−40 °C, 50 min; then (±)-6 (1.0 equiv), −40→0 °C, 20 min; (b) TBAF (2.0 equiv), THF, 25 °C, 6 h, 20, 41% for two steps, d-20, 42 % for two steps; (c) TESCl (2.0 equiv), imid (10 equiv), CH2Cl2, 25 °C, 5 h; (d) KHMDS (5.0 equiv), ClCO2Me (10.0 equiv), Et3N (10 equiv), THF, −78→25 °C, 1 h; (e) HF•py/py (1:4), 0→25 °C, 12 h, 79 % for three steps; (f) PhI(OAc)2 (2.0 equiv), AZADO (0.1 equiv), CH2Cl2, 25 °C, 24 h, 95 %; (g) SmI2 (0.1 M in THF, 4.0 equiv), HMPA (12 equiv), THF, −20→25 °C, 3.5 h, 22β, 33 % yield, 22α, 21 %; (h) NaH (15 equiv), CS2 (30 equiv), THF, 0→25 °C, 30 min; then MeI (45 equiv), 25 °C, 24 h; then 185 °C, μ-waves), o-dichlorobenzene, 15 min, 86 % for two steps; (i) POCl3, pyridine, 72 % (j) ThexBH2 (5.0 equiv), THF, − 10→25 °C, 0.5 h; then BH3•THF (15 equiv), 25 °C, 1 h; then 30 % H2O2/3 N NaOH (1:1 dr), 0→45 °C, 1 h; 70 %; (k) oNO2C6H4SeCN (3.0 equiv), nBu3P (6.0 equiv), py (9.0 equiv), THF, 25 °C; then 30 % H2O2, 0→45 °C, 68 %; (l) KHMDS (6.0 equiv), TESCl (4.0 equiv), Et3N (8.0 equiv), THF, −50→25 °C, 30 min, 89 %; (m) LiDBB (excess), THF, −78→−50 °C, 1 h, 83 %; (n) PhI(OAc)2 (2.0 equiv), 1-Me-AZADO (0.2 equiv), CH2Cl2, 25 °C, 22 h; (o) Ac2O (30 equiv), Et3N (90 equiv), 4-DMAP (2.0 equiv), CH2Cl2, 25 °C, 36 h, 87 % for two steps; (p) 48 % aq. HF:THF (1:3), 25 °C, 7 h, 77 %. KHMDS = potassium hexamethyldisilyazide, Thexyl = thexylborane, LiDBB = Lithium di-tert-butylbiphenyl.
Figure 4.
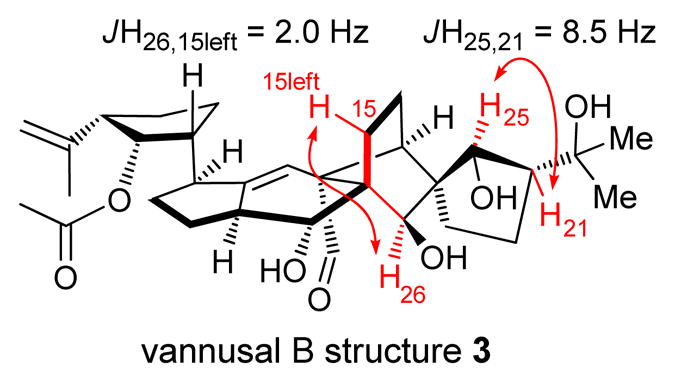
Key 1H NMR coupling constants of vannusal B structure 3 (JH25,21 = 8.5 Hz, w-JH26,15left = 2.0 Hz)
Having excluded structure 3 [C21-epi-2] as the true structure of vannusal B, we then moved to our second target, diastereomer 4 [C21-epi, C25-epi-2], possessing a C25/C21 cis relationship, as a possibility for the coveted vannusal B structure. The required aldehyde building block 30 for this construction was synthesized from diketone 26[3] as summarized in Scheme 4. Thus, lithium enolate generation from 26 (LDA, −78→−40 °C) followed by addition of acetone led to a diastereomeric mixture of aldol products in which the β-stereoisomer was predominating (ca. 3:1 dr). TES protection (TESOTf, 2,6-lut.) followed by chromatographic separation furnished isomerically pure diketone 27 (66 % overall yield for the two steps), which was selectively reduced from the α-face with NaBH4 (THF:MeOH, 1:1, −10→25 °C) at both carbonyl sites to afford, after removal of the TES group (PPTS, EtOH), triol 28 in 91% yield. Regioselective acetonide formation within the latter intermediate [(MeO)2CMe2, PPTS, 100 % yield], followed by SEM installation (SEMCl, iPr2NEt, nBu4NI, 97 % yield] led to intermediate 29. The latter was converted to the desired aldehyde, (±)-30, by the same route and in similar yields, as the one described above for the conversion of 16 to (±)-6 (see Scheme 2), as summarized in Scheme 4.
Scheme 4.

Synthesis of aldehyde (±)-30 and vannusal B structure 4. Reagents and conditions: (a) LDA [generated from iPr2NH (5.0 equiv), nBuLi (2.5 M in haxanes, 5.0 equiv)], THF, −78→−40 °C; then acetone (20 equiv), −40→25 °C, 1 h, (3:1 dr); (b) TESOTf (2.0 equiv), 2,6-lut. (5.0 equiv), −78→−40 °C, 1 h, 66 % for two steps; (c) NaBH4 (20 equiv), THF:MeOH (1:1), −10→25 °C, 4 h; (d) EtOH, PPTS (0.10 equiv), 25 °C, 2 h, 91 % for two steps; (e) (MeO)2C(Me)2:DMF (1:1), PPTS (1.0 equiv), 25 °C, 48 h, 100 %; (f) SEMCl (5.0 equiv), iPr2NEt (15 equiv), nBu4NI (1.0 equiv), CH2Cl2, 50 °C, 24 h, 97 %;
The total synthesis of vannusal B diastereomeric structure 4 [C21-epi, C25-epi-2] from (−)-5 and (±)-30 proceeded through similar intermediates and along the same lines as the route to vannusal B structure 3 [C21-epi-2] from (−)-5 and (±)-6 discussed above (see Scheme 3). Notable differences between the two routes were the higher yield obtained in the SmI2-mediated ring closure step (74 %), which was most likely a consequence of the use of the acetonide moiety at the C25/C21 site, and the isolation of only one diastereomer at this stage, that corresponding to 22β (Scheme 3). Again, the NMR spectroscopic data of synthetic structure 4 were disappointing in that they did not match those of the natural vannusal B. However, we were encouraged by the 1H NMR spectrum of this structure, which revealed much closer chemical shifts for H17 and H26 (δH17 = 2.50; δH26 = 4.05 ppm) to those exhibited by the natural product (δH17 = 2.48 ppm; δH26 = 3.95 ppm), than the originally assigned structure 2, whose chemical shifts for these protons were far from close (δH17 = 2.24 ppm; δH26 = 4.53 ppm) to those of the natural product (see Figure 5). Based on these observations, we surmised that the “northeastern” domain (i.e. ring E) of the true structure of vannusal B possessed the stereochemistry shown in structure 4 with the cis C25/C21 stereochemical arrangement (Figure 1). At this point, we turned our attention to the “southwestern” part of the molecule (i.e. ring A), aiming at stereochemical changes in that region to define our next targets.
Figure 5.

Key 1H NMR coupling constants (w-JH26, 15left = 1.5 Hz, JH25, 21 = 3.5 Hz) and selected chemical shifts for vannusal B structure 4 and comparisons with those of natural vannusal B and its originally assigned structure (2).
Careful consideration of the reported 1H NMR spectral data of both vannusals A and B led us to believe that the relative stereochemistry at C6 with regards to C3, C29, and C7 of the originally assigned structures of the vannusal (1, see Figure 6a) was correct. This assumption was based on a) the rather large 1H NMR coupling constant between H6 and H7 (JH6,7 = 10.0 Hz), which supported the assigned trans-diaxial orientation of these two protons, and b) the observed NOEs between H29 and H8β, and H5α and H12 (see Figure 6a). Indeed, the 6-epi diastereomer of 2 (not shown) is problematic in that it cannot accommodate these observations as supported by model system 31e[10] (Figure 6c), which exhibits rather similar 1H NMR coupling constants between H6 and H7 (JH6,7 = 12.0 Hz), H6 and H29 (JH6,29 = 3.5 Hz), and H3 and H29 (JH3,29 = 3.5 Hz) as those exhibited by the natural product (JH6,7 = 10.0 Hz; JH6,29 = 3.5 Hz; JH3,29 = 3.5 Hz), but no NOE between H29 and neither of the two H8 protons. This conclusion left C3 and C29 as the possible sites of structural misassignment in the original report.[1] In order to elucidate this point, we synthesized all four possible diastereomers of the model AB ring system (compounds 31a–31d),[10] Figure 6b) and compared their NMR spectral data with those of the natural vannusal A. While all four model systems exhibited the expected NOEs between H29 and H8β, their 1H NMR chemical shifts and coupling constants of H29 were revealing (31a: δ = 5.43 ppm, JH29,3 = 3.5 Hz, JH29,6 = 3.5 Hz; 31b: δ = 5.22 ppm, JH29,3 = 7.8 Hz, JH29,6 = 7.8 Hz; 31c: δ = 5.29 ppm, JH29,3 = 4.5 Hz, JH29,6 = 2.0 Hz; 31d: δ = 5.30 ppm, JH29,3 = 1.8 Hz, JH29,6 = 5.4 Hz) (see Figure 6b). Indeed, the striking resemblance of the 1H NMR spectral data of model system 31a (as opposed to the other three) to those reported for the natural vannusal A (δ = 5.43 ppm, JH29,3 = 3.5 Hz, JH29,6 = 3.5 Hz) were convincing of the correctness of the originally assigned configurations at C3, C29, C6 and C7 of the molecule. It was through this pathpointing intelligence that we returned back to the most “northeastern” ring of the vannusal structure to contemplate the remaining possibilities. In the following communciation, we unravel our next course of action and the events that led to the total synthesis of the true structure of vannusal B, and thereby, the elucidation of its molecular architecture, and that of its sibling, vannusal A.
Figure 6.

Relevant NOE’s and coupling constants (J) of AB ring model compounds 31a–31e. R =TBDPS = tert-butyldiphenylsilyl.
Supplementary Material
Footnotes
We thank Professor Graziano Guella for samples and NMR spectra of vannusals A and B, and for helpful discussions. We also thank Drs. D. H. Huang, G. Siuzdak and R. Chadha for NMR spectroscopic, mass spectroscopic and X-ray crystallographic assistance, respectively. Financial support for this work was provided by the NSF (fellowship to A. Ortiz), the Skaggs Institute for Research, and a grant from the National Institutes of Health (USA).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from authors.
References
- 1.Guella G, Dini F, Pietra F. Angew Chem. 1999;111:1217–1220. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1134::AID-ANIE1134>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 1999;38:1134–1136. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1134::AID-ANIE1134>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 2.Guella G, Callone E, Di Giuseppe G, Frassanito R, Frontini FP, Mancini I, Dini F. Eur J Org Chem. 2007:5226–5234. [Google Scholar]
- 3.Nicolaou KC, Zhang H, Ortiz A, Dagneau P. Angew Chem. 2008;120:8733–8738. doi: 10.1002/anie.200804228. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:8605–8610. doi: 10.1002/anie.200804228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicolaou KC, et al. Angew Chem. 2009 submitted. [Google Scholar]; Angew Chem Int Ed. 2009 submitted, see following communication in this issue. [Google Scholar]
- 5.For reviews on samarium diiodide used in organic synthesis, see: Edmonds DJ, Johnston D, Procter DJ. Chem Rev. 2004;104:3371–3403. doi: 10.1021/cr030017a.Kagan HB. Tetrahedron. 2003;59:103510–10372.Molander GA, Harris CR. Chem Rev. 1996;96:307–308. doi: 10.1021/cr950019y.
- 6.Reaction of hydroxy epoxide 9 with Et2AlCN resulted in the predominant transfer of an Et group to the substrate rather than a CN group, presumably through initial formation of the –OAlEt2 species (C26) followed by intramolecular epoxide activation and delivery of an Et group.
- 7.CCDC–726953 contains the supplementary crystallographic data for 13 and is available free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
- 8.Shibuya M, Tomizawa M, Suzuki I, Iwabuchi Y. J Am Chem Soc. 2006;128:8412–8413. doi: 10.1021/ja0620336. We thank Professor Iwabuchi and Nissan Chemical Industries, Ltd. for generous gifts of AZADO and 1-MeAZADO catalysts. [DOI] [PubMed] [Google Scholar]
- 9.Despite the modest yield, this reaction served us well given the failure of the originally employed C25/C26 acetonide, C21–SEM precursor to give any cyclization product under the same conditions.
- 10.Details of these syntheses will be reported in the full account of this work, selected physical properties of compounds 31a–31e are given in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.