

Abstract
The design and synthesis of two new heterofunctional hexagons containing both redox-active ferrocenyl and host—guest crown ether functionalities has been achieved via coordination-driven self-assembly. The size and relative distribution of functional groups on the supramolecular metallacycles can be precisely controlled. The host—guest recognition properties of the crown ether moieties and their ability to complex cationic guests to form tris[2]pseudorotaxane complexes have been investigated. The electronic properties of the ferrocenyl moieties have been obtained through electrochemical studies. The functional moieties are shown to operate orthogonally, resulting in discrete supramolecular hexagons that are capable of carrying out a variety of functions both simultaneously and independently.
Introduction
The natural world presents itself to us with a great multiplicity of forms, yet something in the human mind has sought to tame this great diversity and reduce its order of complexity to a few general principles. The design and creation of regularly shaped nanoscale objects, which can serve as the building blocks of supramolecular materials, is an extremely important goal in material science. Controlling the spatial organization of such objects could deliver materials with defined chemical and topographic nanoscale features, potentially leading to novel physical properties or the integration of several properties into a single molecule or supramolecule.1
Currently, there are a number of important transformations taking place in the field of chemical nanoscience as researchers attempt to take advantage of the benefits of nanoscale materials. One such transformation is developing rational synthetic protocols to control not only the size but also the shape of both organic as well as inorganic objects.2 Another important transformation is the increasing interest in connecting structure to function by designing and incorporating functional moieties into dynamic nanostructures.3 The interest in function has also motivated a growing interest in developing approaches to the patterning of nanostructures in devices, using either top down or bottom up approaches. We have recently introduced a variety of functionalized 120° platinum acceptors and 120° organic donors and have quantitatively assembled them to form molecular hexagons with six functional groups on their periphery.4 The introduction of multiple functional groups onto/into discrete supramolecular systems in a controlled manner can be a major challenge. Coordination-driven self-assembly, however, is a technique that can be used to provide significant structural control and yield products of high purity and functional versatility. Ease of synthesis and the ability to precisely place functional groups throughout a structure, to prepare well-defined cavity-cored discrete supramolecules, and to control the number of functional groups are among the most attractive features of using coordination-driven self-assembly to introduce functional units in nanoscale supramolecular systems.
Furthermore, it is desirable that a supramolecule can demonstrate a combination of multiple properties/functions in a nondestructive manner,5 i.e., to construct multifunctional systems and demonstrate the orthogonality of their functional properties. The challenge is developing a means to “produce” supramolecules that contain multiple different functionalities and being able to independently investigate their properties in a straightforward way. Alternatively, it may be desirable to develop multifunctional systems wherein the properties of two or more functionalities interact in such a way that they lead to new, emergent phenomena. A third possibility is that the multiple functional groups interact destructively such that neither moiety is able to carry out its intended function. This last possibility is, of course, the least desirable.
We envisioned that acceptor—donor-based coordination-driven self-assembly is additionally useful because of the ability to modify functional groups at both the donor and acceptor units. An appropriate variation of the functional groups enables accurate control over the exact number and placement of different peripheral functional groups, thus leading to the concept of a discrete supramolecular analogue of a “copolymer”-type structure. Here we employ 120° acceptor units decorated with one functional group to self-assemble with 120° donor units decorated with another functional group to form supramolecular hexagons with six heterofunctional groups. The redox and recognition properties of these heterofunctional hexagons have been investigated.
Results and Discussions
With the 120° crown ether and ferrocene functionalized precursors in hand, in both donor and acceptor analogues, the self-assembly of heterofunctional hexagons was investigated. Upon mixing 120° ferrocenyl donor unit 14d with 120° crown ether functionalized unit 24e in CH2Cl2, heterofunctional hexagon 5 was obtained (Scheme 1). In a complementary manner, stirring 120° ferrocenyl acceptor unit 34f with an equimolar amount of 120° crown ether donor unit 44c in CH2Cl2 results in heterofunctional hexagon 6.
Scheme 1. Self-Assembly of Heterofunctional Hexagons.
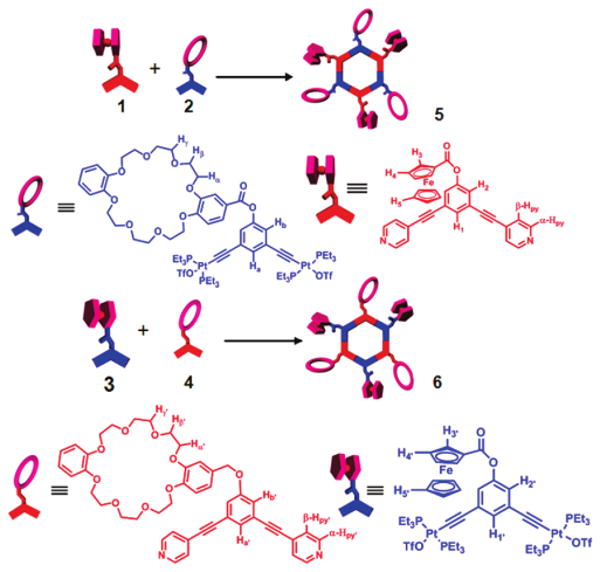
The reaction progress is easily followed with 31P {1H} and 1H NMR. The 31P spectrum of 5 displayed a singlet (ca. 18.3 ppm), which suggests the formation of a discrete, highly symmetric supramolecular assembly. The peak is shifted upfield from the starting platinum donor 2 by 4.3 ppm. This change, as well as the decrease in coupling of the flanking 195Pt satellites (Δ1JPPt = ca. −70.1 Hz), is consistent with back-donation from the platinum atoms. In the corresponding 1H NMR spectra, the β-protons showed a relatively dramatic upfield shift (0.41 ppm) as well as a slight upfield shift of the α-protons (0.04 ppm) due to electron density transfer from the pyridyl donor to the metal acceptor (Figure 2C).
Figure 2.
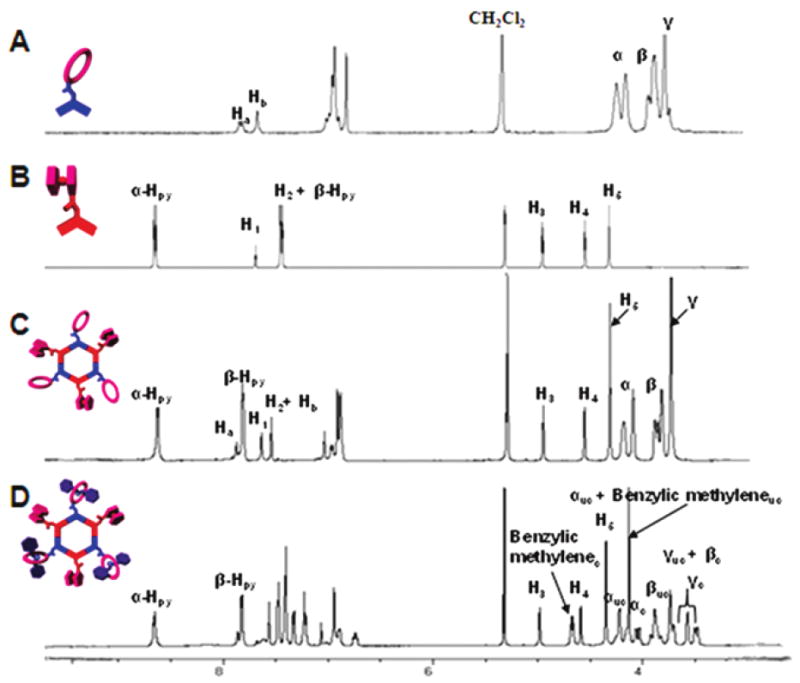
Partial 1H NMR spectra of crown ether acceptor 2 (A), ferrocenyl donor 1 (B), mixed hexagon 5 (C), and pseudorotaxane 8 (D).
The structure of 5 was confirmed by ESI mass spectrometry (Figure 1A) and elemental analysis. In the ESI mass spectra of heterofunctionalized hexagon 5, a peak attributable to the loss of four triflate counterions, [M − 4OTf]4+ where M represents the intact assembly, was observed at m/z = 1563.9. The peak was isotopically resolved and agrees very well with the theoretical distribution.
Figure 1.
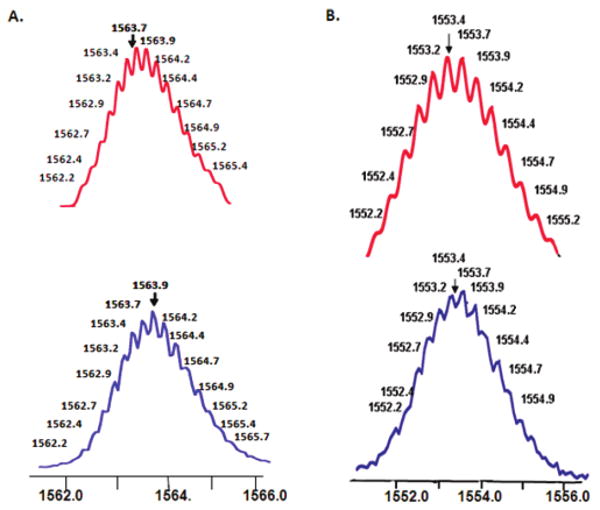
Calculated (top) and experimental (bottom) ESI-MS spectra of hexagon 5 (A) and 6 (B).
Similarly, 31P {1H} NMR analysis of the reaction mixture composed of 3 and 4 is consistent with the formation of a single, highly symmetrical species (6) by the appearance of a sharp singlet (ca. 16.6 ppm) with concomitant 195Pt satellites, shifted 6.1 ppm upfield (Δδ) relative to 3 (Δ1JPPt = −64 Hz). As expected, in the 1H NMR spectrum of hexagon 6 the β-hydrogen nuclei of the pyridine rings exhibited 0.41 ppm downfield shifts. The ESI/MS spectra of 6 (Figure 1B) exhibited characteristics very similar to those of 5. The ESI/MS spectra showed one charged state at m/z = 1553.4 corresponding to the [M − 4OTf]4+ species, and its isotopic resolution is in excellent agreement with the theoretical distribution.
Electrochemical Study of 5 and 6
Cyclic voltammetry (CV) investigation7a of heterofunctional hexagonal assemblies 5 and 6 (see Supporting Information, Figures S10A and S11A) were performed in a CH2Cl2 solution containing 0.10 M nBu4N+PF6− as the supporting electrolyte at a 1.0 mm2 Pt disk electrode. The CVs of both complexes showed nearly identical cathodic and anodic peak currents, as well as nearly scan-rate-independent peak potentials, suggesting the oxidation of the ferrocene moieties in each complex is reversible. The potential difference between cathodic and anodic peak potentials (ΔEp) measured at different scan rates (ν = 50, 100, 150, and 200 mV/s) ranged from 56.0 to 64.2 mV for compound 5 and from 60.4 to 68.4 mV for compound 6, very close to the theoretical value for an ideal reversible redox system (59 mV at 25 °C). Table 1 summarizes some electrochemical parameters (half-wave potential (E1/2) and ΔEp) for complexes 5 and 6.
Table 1. Electrochemical Parameters for Compounds 5 and 6a.
| E1/2 (V vs Ag/AgCl) | ΔEp (V)b | D (10−6 cm2/s) | |
|---|---|---|---|
| 5 | 0.79 ± 0.01 | 0.059 ± 0.001 | 2.9 ± 0.1 |
| 6 | 0.80 ± 0.01 | 0.066 ± 0.001 | 2.0 ± 0.2 |
CH2Cl2 with 0.1 M nBu4NBF6, 25°C.
Scan rate = 100 mV/s.
The steady-state limiting currents (ilim) (see Supporting Information, Figures S10B and S11B) obtained with Pt-microdisk electrodes (with the diameter of 25 μm) were used to predict diffusion coefficients (D) of ferrocene-attached complexes by the following equation:7 ilim = 4nFDaCθ, where n is the number of electrons transferred per ferrocene (= 1), F is the Faraday constant, C is the bulk concentration of redox species (= 0.2 mM), θ is the number of reacting ferrocenyl sites per redox molecule (= 3 for both 5 and 6), and a is the radius of the electrode (= 12.5 μm). The calculated diffusion coefficients of compound 5 and 6 are 2.9 × 10−6 and 2.0 × 10−6 cm2/s, respectively (also presented in Table 1). The D value of 5 is surprisingly larger than that of 6, as the simulated sizes for these two complexes are nearly identical (3.1 nm).
The above calculation assumes that each ferrocene group is oxidized independently, i.e., multiferrocenyl assemblies lack strong intramolecular coupling between ferrocene groups, as observed in our previous studies4d,f of other ferrocene-functionalized supramolecules. This assumption was verified by plotting E versus log[(ilim − i)/i], where E is the applied electrode potential, i is the measured current, and ilim is the limiting current determined by the mass transport of redox species. The slopes of the plots are −62 and −60 mV/dec for compound 5 and 6, respectively (see Supporting Information Figures S10C and S11C). These values are very close to the ideal value (−59 mV/n with n = 1), indicating that the redox species react independently of one another. Here, the presence of the crown ether does not appear to have any electrochemical influence on the physical properties of the ferrocenyl units.
Tris[2]Pseudorotaxanes 8 and 9
With these novel, heterofunctionalized crown ether derivatives in hand, an investigation of the self-assembly of tris[2]pseudorotaxanes was carried out to investigate any effect(s) of ferrocene substituents in the binding of crown ether units. Within 15 min of adding 3 equiv of dibenzylammonium triflate salt 7 to a solution of hexagonal metallacycles 5 and 6, tris[2]pseudorotaxanes 8 and 9, respectively, were obtained (Scheme 2).
Scheme 2. Entry to Hexagonal Cavity-Cored Tris[2]pseudorotaxanes.
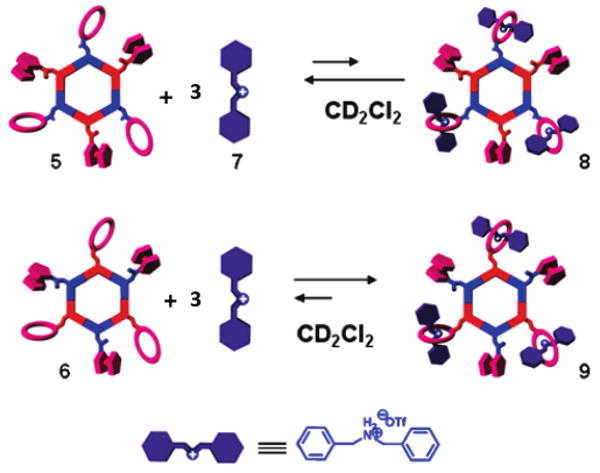
Compared to the spectra of free hosts 5 and 6, the 31P NMR spectrum of each complex does not exhibit any significant change, indicating that the threading of the dibenzylammonium ions does not substantially change the chemical environment of the phosphorus atoms present in the hexagons. The 1H NMR spectra of 8 and 9, however, displayed characteristic shifts associated with the complexation of dibenzylammonium ion 7 by a dibenzo[24]crown-8 (DB24C8) macrocycle (Figure 2D). A 0.45 ppm downfield shift of the signal for the benzylic methylene protons adjacent to the NH2+ center was observed, and protons Hα, Hβ, and Hγ of of the crown ether moiety exhibited upfield shifts. The crown ether moieties of tris-host 5 are directly attached to the di-Pt(II) acceptor building blocks, whereas in tris-host 6, the crown ether moieties are covalently linked to the bispyridyl donor building blocks. These structural differences in turn result in differing abilities of 5 and 6 to complex 7. In the case of tris[2]pseudorotaxane 8, the equilibrium ratio of complexed to uncomplexed species was observed to be somewhat less than 1:1, indicating a slight excess of uncomplexed species. This observation is analogous to previous assemblies composed of di-Pt(II) crown ether acceptors4e and further supports the conclusion that covalent attachment of the crown ether derivatives to di-Pt(II) acceptors lowers the binding abilities of the typically electron-rich macrocycles.
Further characterization with 2-D spectroscopic techniques (1H –1H COSY and NOESY) is in agreement with the formation of the hexagonal cavity-cored tris[2] pseudorotaxanes. For example, through-space interactions between the α-H proton of the complexed crown ether and the benzylic methylene protons were observed in the NOESY spectrum of each complex (see Supporting Information).
The self-assembly of hexagonal cavity-cored tris[2]pseudorotaxanes 8 and 9 was also confirmed by ESI-MS spectrometry. Two peaks at m/z = 1429.4 and 1824.8 were observed, corresponding to [M − 5OTf]5+ and [M − 4OTf]4+, respectively, for 8, as were peaks attributable to [M − 5OTf]5+ and [M − 4OTf]4+ of 9 at m/z = 1421.1 and 1813.4, respectively. All peaks were isotopically resolved and agree very well with their theoretical distribution (Supporting Information, Figure S8).
It is possible that the binding of dibenzyl ammonium salt 7 by 5 and 6 may influence the electrochemical properties of the ferrocenyl moieties of 8 and 9. To investigate this possibility, cyclic voltametric studies of pseudorotaxanes 8 and 9 were carried out (see Supporting Information, Figures S12 and S13). No significant change in E1/2 was observed, indicating that the crown ether and ferrocene moieties are noninteracting. Nevertheless, the absolute currents of the voltammetric waves were both slightly decreased for 8 (Figure S12) and 9 (Figure S13), as compared to 5 (Figure S10A) and 6 (Figure S11A), respectively. The larger sizes and higher charges of 8 and 9, in relation to 5 and 6, most likely result in smaller values of D (1.8 × 10−6 cm2/s for 8, 1.7 × 10−6 cm2/s for 9), which is reflected in the decreased currents. There have also been literature reports indicating a slight decrease in the diffusion coefficient of a rotaxanated polymer compared to its lone macrocycle.8 It should be noted that the electrochemical experiments were carried out in the presence of an excess of nBu4N+PF6− as the supporting electrolyte. Therefore, the guest species are present in solution as ion pairs with PF6− anions. Investigations of 5 and 6 and nBu4N+PF6− solutions revealed no changes in the 1H NMR spectra under experimental conditions, indicating no interactions between macrocycle and the salt.
Pseudorotaxane Stoichiometry and Binding Constant
The host:guest stoichiometry for heterofunctional supramolecular assemblies 8 and 9 was established using the nonlinear least-squares fit method based 1H NMR titration experiments. Fitting the data to a 1:3 binding mode for the hosts gave host—guest association constants (Table 2). These values are on the same order as those obtained for the related homo tris-crown ether hosts that have been previously studied.4c,e The results support the conclusion that placement of the derivatized crown ether macrocycles on acceptor building blocks (i.e., 2) results in a loss of electron density and, thus, binding ability. From the ratios of binding constants, it is clear that the crown ether sites act more or less independently of each other and pseudorotaxane formation is not influenced by any cooperative effects from the presence of the other crown ether hosts or the ferrocenyl moieties. Rather, the decrease in the efficiency of the threading process may be attributed to the entropic cost of forming higher-order pseudorotaxanes or, potentially, the result of steric effects imposed by the ferrocene units on the approaching dibenzyl ammonium salt to the crown ether unit.
Table 2. Thermodynamic Binding Constants of Poly[2]pseudorotaxanes 8 and 9a.
| Ks.1 (M−1) | Ks.2 (M−1) | Ks.3 (M−1) | |
|---|---|---|---|
| 8 | (2.45 ± 0.44) × 103 | (1.66 ± 0.19) × 103 | (4.02 ± 0.19) × 102 |
| 9 | (2.09 ± 0.33) × 104 | (1.83 ± 0.06) × 103 | (7.83 ± 0.40) × 101 |
CD2Cl2, 298 K.
Molecular Modeling Studies
Molecular force-field simulations were used to gain further insight into the structural characteristics of heterofunctional hexagons 5 and 6 as well as cavity-cored tris[2] pseudorotaxanes 8 and 9. A 1.0 ns molecular dynamics simulation (OPLS 2003 force field) was used to equilibrate each supramolecule 5, 6, 8, and 9, followed by energy minimization of the resulting structures to full convergence. The modeled structures of hexa-functionalized hexagons 6 and tris[2] pseudorotaxane 9, for example, are shown in Figure 4 (for compounds 5 and 8, see Supporting Information). Molecular simulations reveal that the addition of dibenzylammonium to DB24C8 hosts does not disrupt the underlying polygonal scaffolds, as ammonium salts are complexed by their pendant DB24C8 macrocycles. Furthermore, the different functional groups are observed to be distant from one another, supportive of the observation that the two different functionalities are orthogonal and do not influence each other. In both cases, the underlying rigid nature of the 2-D polygonal cavity is retained, while the flexibility of each crown ether is reduced as a result of host—guest complexation.
Figure 4.
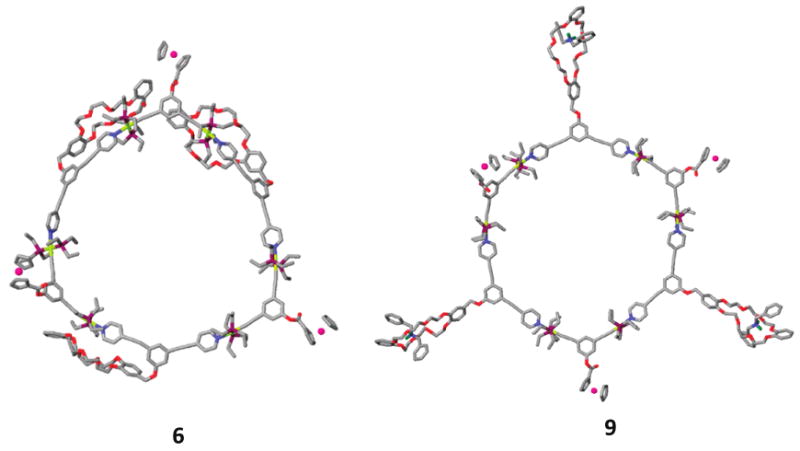
Simulated molecular models of 6 and 9, optimized with the molecular mechanics force field. Color scheme: C, gray; O, red; N, blue; P, purple; Pt, yellow; Fe, pink; and R2NH2+ hydrogen atoms, green (all other hydrogen atoms have been removed for clarity).
Conclusion
In summary, we have provided two complementary approaches to generate two new heterofunctional hexagons via coordination-driven self-assembly from a ferrocenyl 120° di-Pt(II) acceptor/donor and a complementary 120° crown ether decorated donor/acceptor unit while exhibiting precise control of size and the distribution of the ferrocene and crown ether moieties. The work presented here shows a simple, highly efficient approach to the construction, via coordination-driven self-assembly, of heterofunctional moieties possessing well-defined hexagonal cavities. These heterofunctional hexagons demonstrate that they can retain their individual functions in the multifunctional metallacycles without interfering with each other or the underlying metallacycle structure.
These heterofunctional hexagons are unique in not only their discrete structures but also the presence of chemically different units, since each unit introduces into the supramolecular structure its own “pieces of information” (in the form of specific properties such as recognition properties, redox levels, etc.) that are preserved faithfully in the structure. By the choice of functional units it is possible to obtain compounds where different functional units are located in the vertex of hexagons. Further investigations aimed at expanding the orthogonality of functional units along these lines are currently underway.
Experimental Section
General Procedure for the Preparation of Hexagonal Assemblies 5 and 6
To a 0.5 mL dichloromethane-d2 solution of triflate 2, 3 was added a 0.5 mL dichloromethane-d2 solution of the appropriate donors 1 and 4 drop by drop with continuous stirring (10 min). The reaction mixture was stirred 30 min at room temperature. The solution was evaporated to dryness, and the product was collected.
Data for 5
Yield: 96%. 1H NMR (CD2Cl2, 500 MHz): δ 8.65 (d, J = 6.5 Hz, 12H, Hα-Py), 7.88 (s, 3H), 7.86 (d, J = 5.7 Hz, 12H, Hβ-Py), 7.65 (s, 6H, ArH), 7.56 (m, 3H, ArH), 7.06 (s, 6H, ArH), 6.99–6.93 (m, 21H, PhH), 4.98 (m, 6H, Fc), 4.59 (m, 6H, Fc), 4.35 (m, 15H, Fc), 4.22 (m, 12H, α-CH2), 4.13 (m, 12H, α-CH2), 3.93–3.85 (m, 24H, β-CH2), 3.84 (m, 24H, γ-CH2), 1.82 (m, 72H, PCH2CH3), 1.21 (m, 108H, PCH2CH3). 31P{1H} NMR (CD2Cl2, 121.4 MHz): δ 18.3 (s, 1JPt-P = 2304.1 Hz). Anal. Calcd for C276H342N6O54Fe3F18P12Pt6S6 + CH2Cl2: C, 47.97; H, 4.98; N, 1.21. Found: C, 47.81; H, 5.10; N, 1.25.
Data for 6
Yield: 97%. 1H NMR (CD2Cl2, 500 MHz): δ 8.63 (d, J = 5.7 Hz, 12H, Hα-Py), 7.82 (d, J = 5.7 Hz, 12H, Hβ-Py), 7.32 (s, 6H, ArH), 7.05 (s, 12H, ArH), 6.90 (b, 21H, ArH, PhH) 5.06 (s, 6H, PhCH2), 4.95 (m, 6H, Fc), 4.93 (m, 6H, Fc) 4.56 (m, 15H, Fc), 4.13 (m, 24H, α-CH2), 3.87(m, 24H, β-CH2), 3.84 (m, 24H, γ-CH2), 1.84 (m, 72H, PCH2CH3), 1.22 (m, 108H, PCH2CH3). 31P {1H} NMR (CD2Cl2, 121.4 MHz): δ 16.3 (s, 1JPt-P = 2305.87 Hz). Anal. Calcd for C276H348Fe3N6O51F18P12 Pt6S6: C, 48.68; H, 5.15; N, 1.23. Found: C, 49.21; H, 4.69; N, 1.27.
Supplementary Material
Figure 3.
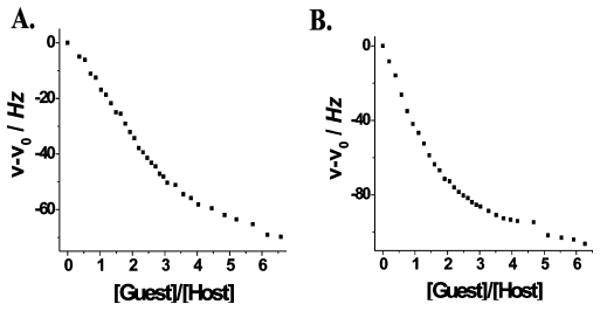
1H NMR titration isotherms of 8 (A) and 9 (B), recorded at 500 MHz in CD2Cl2 at 298 K, indicating the change in chemical shift of the proton signal corresponding to the γ-H of the crown ether.
Acknowledgments
P.J.S. thanks the NIH (Grant GM-057052) for financial support. B.H.N. thanks the NIH (Grant GM-080820) for financial support. We thank Prof. Mei-Xiang Wang and Dr. Han-Yuan Gong for their help with the calculation of thermodynamic binding constants. J.H. thanks the Pao Yu-Kong and Pao Zhao-Long Scholarship for financial support. H.S.W. thanks the NSF (Grant CHE-0616505) for financial support.
Footnotes
Supporting Information Available: 1H and 31P NMR spectra of 5, 6, 8, and 9 and two-dimensional spectra (1H –1H COSY and NOESY) of 8 and 9; experimental data for 1H NMR titration experiments; ESI/MS spectra of assemblies 8 and 9; cyclic voltammetric and all electrochemical calculations; and computational procedures and structures for all self-assemblies obtained from modeling. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Hupp JT, Poeppelmeier KT. Science. 2005;309:2008. doi: 10.1126/science.1117808. [DOI] [PubMed] [Google Scholar]; (b) Brunsveld L, Folmer BJB, Meijer EW, Sijbesma RP. Chem Rev. 2001;101:4071. doi: 10.1021/cr990125q. [DOI] [PubMed] [Google Scholar]; (c) Ikkala O, Brinke G. Science. 2002;295:2407. doi: 10.1126/science.1067794. [DOI] [PubMed] [Google Scholar]; (d) Smith DK. Chem Commun. 2006:34. doi: 10.1039/b507416a. [DOI] [PubMed] [Google Scholar]; (e) Fréchet JMJ. Science. 1994;263:1710. doi: 10.1126/science.8134834. [DOI] [PubMed] [Google Scholar]; (f) Lehn JM. Chem Soc Rev. 2007;36:151. doi: 10.1039/b616752g. [DOI] [PubMed] [Google Scholar]
- 2.(a) Stang PJ, Olenyuk B. Acc Chem Res. 1997;30:502. [Google Scholar]; (b) Leininger S, Olenyuk B, Stang PJ. Chem Rev. 2000;100:853. doi: 10.1021/cr9601324. [DOI] [PubMed] [Google Scholar]; (c) Seidel SR, Stang PJ. Acc Chem Res. 2002;35:972. doi: 10.1021/ar010142d. [DOI] [PubMed] [Google Scholar]; (d) Schwab PFH, Levin MD, Michl J. Chem Rev. 1999;99:1863. doi: 10.1021/cr970070x. [DOI] [PubMed] [Google Scholar]; (e) Gianneschi NC, Masar MS, III, Mirkin CA. Acc Chem Res. 2005;38:825. doi: 10.1021/ar980101q. [DOI] [PubMed] [Google Scholar]; (f) Cotton FA, Lin C, Murillo CA. Acc Chem Res. 2001;34:759. doi: 10.1021/ar010062+. [DOI] [PubMed] [Google Scholar]; (g) Fujita M, Tominaga M, Hori A, Therrien B. Acc Chem Res. 2005;38:369. doi: 10.1021/ar040153h. [DOI] [PubMed] [Google Scholar]; (h) Fiedler D, Leung DH, Bergman RG, Raymond KN. Acc Chem Res. 2005;38:349. doi: 10.1021/ar040152p. [DOI] [PubMed] [Google Scholar]; (i) Steel PJ. Acc Chem Res. 2005;38:243. doi: 10.1021/ar040166v. [DOI] [PubMed] [Google Scholar]
- 3.(a) Gianneschi NC, Masar MS, III, Mirkin CA. Acc Chem Res. 2005;38:825. doi: 10.1021/ar980101q. [DOI] [PubMed] [Google Scholar]; (c) Kamiya N, Tominaga M, Sato S, Fujita M. J Am Chem Soc. 2007;129:3816. doi: 10.1021/ja0693082. [DOI] [PubMed] [Google Scholar]; (d) Northrop BH, Yang HB, Stang PJ. Chem Commun. 2008:5896. doi: 10.1039/b811712h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tominaga M, Suzuki K, Murase T, Fujita MJ. Am Chem Soc. 2007;129:11950. doi: 10.1021/ja054069o. [DOI] [PubMed] [Google Scholar]
- 4.(a) Yang HB, Das N, Huang F, Hawkridge AM, Muddiman DC, Stang PJ. J Am Chem Soc. 2006;128:10014. doi: 10.1021/ja063377z. [DOI] [PubMed] [Google Scholar]; (b) Yang HB, Hawkridge AM, Huang SD, Das N, Bunge SD, Muddiman DC, Stang PJ. J Am Chem Soc. 2007;129:2120. doi: 10.1021/ja066804h. [DOI] [PubMed] [Google Scholar]; (c) Yang HB, Ghosh K, Northrop BH, Zheng YR, Lyndon MM, Muddiman DC, Stang PJ. J Am Chem Soc. 2007;129:14187. doi: 10.1021/ja073744m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yang HB, Ghosh K, Zhao Y, Northrop BH, Lyndon MM, Muddiman DC, White HS, Stang PJ. J Am Chem Soc. 2008;130:839. doi: 10.1021/ja710349j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ghosh K, Yang HB, Northrop BH, Lyndon MM, Zheng YR, Muddiman DC, Stang PJ. J Am Chem Soc. 2008;130:5320. doi: 10.1021/ja711502t. [DOI] [PubMed] [Google Scholar]; (f) Ghosh K, Zhao Y, Yang HB, Northrop BH, White HS, Stang PJ. J Org Chem. 2008;73:8553. doi: 10.1021/jo801692v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Yang HB, Northrop BH, Zheng YR, Ghosh K, Lyndon MM, Muddiman DC, Stang PJ. J Org Chem. 2009;74:3524. doi: 10.1021/jo900067v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Balzani V, Credi A, Raymo FM, Stoddart JF. Angew Chem, Int Ed. 2000;39:3348. doi: 10.1002/1521-3773(20001002)39:19<3348::aid-anie3348>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]; (b) McNitt KA, Parimal K, Share AI, Fahrenbach AC, Witlicki EH, Pink M, Bediako DK, Plaisier CL, Le N, Heeringa LP, Vander G, Douglas A, Flood AH. J Am Chem Soc. 2009;13:1305. doi: 10.1021/ja8085593. [DOI] [PubMed] [Google Scholar]; (c) Moonen NNP, Flood AH, Fernandez JM, Stoddart JF. Top Curr Chem. 2005;262:99. [Google Scholar]; (d) de Silva AP, Dobbin CM, Vance TP, Wannalerse B. Chem Commun. 2009;11:1386. doi: 10.1039/b822181b. [DOI] [PubMed] [Google Scholar]; (e) Credi A. Angew Chem, Int Ed. 2007;46:5472. doi: 10.1002/anie.200700879. [DOI] [PubMed] [Google Scholar]; (f) Tian H, Wang QC. Chem Soc Rev. 2006;35:361. doi: 10.1039/b512178g. [DOI] [PubMed] [Google Scholar]
- 6.Bard AJ, Faulkner LR. Electrochemical Methods: Fundamentals and Applications. Wiley; New York: 1980. [Google Scholar]
- 7.(a) Denault G, Mirkin M, Bard AJ. J Electroanal Chem. 1991;308:27. [Google Scholar]; (b) Biondi C, Bellugi L. J Electroanal Chem. 1970;24:263. [Google Scholar]; (c) Amatore C, Azzali M, Calas P, Jutand A, Lefrou C, Rollin Y. J Electroanal Chem. 1990;288:45. [Google Scholar]; (d) Mirkin MV, Nilov AP. J Electroanal Chem. 1990;283:35. [Google Scholar]; (e) Nowinski SA, Anjo DM. J Chem Eng Data. 1989;34:265. [Google Scholar]
- 8.Zhao T, Beckham HW, Gibson HW. Macromolecules. 2003;36:4833. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.