

Abstract
Saccharomyces boulardii (Sb) is a probiotic yeast with anti-inflammatory and antimicrobial activities and has been used for decades in the prevention and treatment of a variety of human gastrointestinal disorders. We reported previously that Sb modulates host inflammatory responses through down regulation of Erk1/2 MAP kinase activities both in vitro and in vivo. The aim of this study was to identify upstream mediators responsible for Erk1/2 inactivation and to examine the effects of Sb on tumor development in ApcMin mice. We found that the EGF receptor was deactivated upon exposure to Sb leading to inactivation of both the EGFR-Erk and EGFR-Akt pathways. In human colonic cancer cells, Sb prevented EGF induced proliferation, reduced cell colony formation and promoted apoptosis. HER-2, HER-3 and IGF-1R were also found to be inactivated by Sb. Oral intake of Sb reduced intestinal tumor growth and dysplasia in C57BL/6J Min/+ (ApcMin) mice. Thus, in addition to its anti-inflammatory effects, S. boulardii inhibits EGFR and other receptor tyrosine kinase signaling and thereby may also serve a novel therapeutic or prophylactic role in intestinal neoplasia.
Introduction
Saccharomyces boulardii (Sb) is a non-pathogenic yeast that has been used for many years as a probiotic agent to prevent or treat a wide variety of human gastrointestinal disorders of diverse etiologies1-3. Preclinical and experimental studies of S. boulardii have demonstrated anti-inflammatory, antimicrobial, enzymatic, metabolic and anti-toxin activity4-7. S. boulardii appears to exert its therapeutic effect by multiple mechanisms and to influence several important facets of intestinal host-pathogen interaction. We and others have reported that Saccharomyces boulardii acts through modulation of host signaling pathways that regulate the intestinal mucosal inflammatory response. Particularly, the Erk1/2 MAP kinase pathway is down-regulated by Sb both in vitro and in vivo 8, 9.
MAP kinase pathways are located downstream of many growth-factor receptors, including the Epidermal Growth Factor Receptor (EGFR), through the Raf /MEK /ERK cascade 10. Therefore, we hypothesized that Sb inhibited Erk activation via an upstream effect on EGFR or other receptor tyrosine kinase signaling. Our data in this study suggested that EGFR signaling is inactivated by Sb. It has been well characterized that activation of EGFR results in enhanced cell proliferation, invasion and tumor metastasis, as well as inhibition of apoptosis 11. Signaling pathways that emerge from EGFR activation are critical in colon cancer biology 12 13. EGFR inhibitors have demonstrated clinical benefit for colorectal cancer treatment 14, 15. Since Sb has shown beneficial effects in a series of gastrointestinal disorders, we wanted to know if Sb has potential anti-cancer properties by examining whether the effects of Sb on EGFR signaling can influence proliferation and apoptosis of colon cancer cells in vitro, as well as intestinal tumor growth in ApcMin mice in vivo.
Results
SbS inactivates EGFR-Mek-Erk pathway signaling in colonic cancer cells
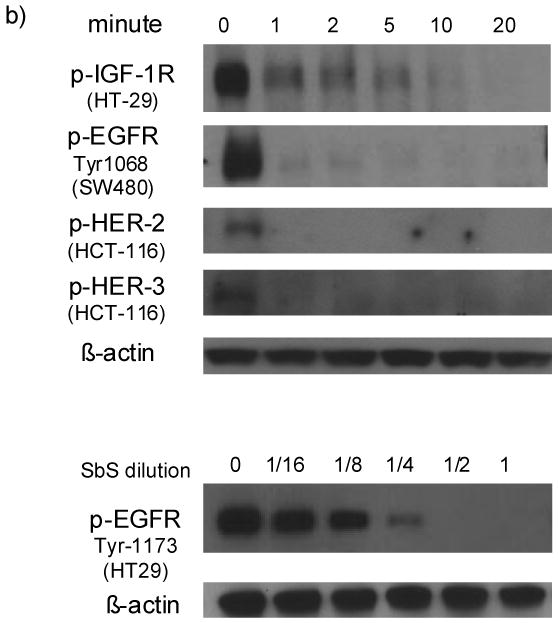
We have reported that Erk1/2 MAP kinase activation is inhibited by SbS in vitro and in vivo 9. We hypothesized that EGFR signaling might be affected upstream to Erk. After varying periods of time (0 to 180 minutes) of exposure to SbS, HT29 cells were harvested, cell extracts prepared and Western blotting performed using either phospho- specific or total EGFR, MEK1/2, ERK1/2 antibodies. We found that SbS markedly reduced EGFR phosphorylation (Tyr1173) with a rapid onset of effect (1 minute). Reversibility of the SbS inhibitory effect was apparent with some return of EGFR phosphorylation becoming evident at later time points (Figure 1a). Phospho-Mek1/2, a downstream signaling molecule to EGFR but an upstream kinase to Erk1/2, also lost much of its activity after 5 minutes of SbS exposure. As expected p-Erk1/2 activity was also significantly decreased following de-phosphorylation of EGFR and MEK1/2 (Figure 1a). These findings are consistent with the theory that phospho-Erk1/2 is reduced by SbS through effects on the EGFR-Mek-Erk pathway. This provided a mechanism to account for the effects of SbS on Erk1/2 MAP kinase activity as described in our previous studies 9. As other receptor tyrosine kinases such as insulin-like growth factor-1 receptor (IGF-1R) and ErbB family members including HER-2 and HER-3 also stimulate MAP kinase signaling, we examined HER-2, HER-3 and IGF-1R activation level in SbS treated HCT-116 or HT29 cells. We found p-HER-2, p-HER-3 and p-IGF-1R, are all inactivated by SbS in similar fashion as p-EGFR (Figure 1b). Other signaling kinases, such as phospho-CamKII (Figure 1a) and PKC family members including phsphorylated forms of PKC-alpha, PKC-delta, PKC-theta and PKCmu were not affected by SbS (data not shown), indicating an effect on receptor tyrosine kinases including EGFR, HER-2, Her-3 and IGF-1R rather than global dephosphorylation and inactivation.
Figure 1. SbS inactivates EGFR MEK ERK signaling in colon cancer cells.
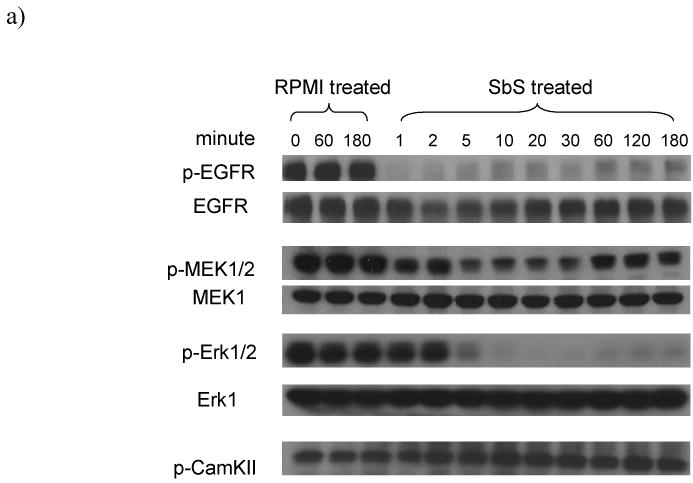
a) HT29 cells grown in complete media with serum were exposed to SbS for varying periods (0 to 180 minutes) and were then harvested. Cell extracts were prepared and Western blotting was performed using either phospho- specific or total EGFR, MEK1/2, ERK1/2, or CamKII antibodies. Unless otherwise noted, antibodies against p-EGFR(Tyr1173) used.
b) HT29, SW480 or HCT-116 cells were exposed to SbS at various time points (0, 1, 2, 5, 10 and 20 minutes), p-IGF-1R p-HER-2&3, and p-EGFR(Tyr1068) were blotted with specific antibodies. Series of SbS dilutions (1, 1/2, 1/4, 1/8, 1/16) were incubated with HT29 cells for 5 minutes, p-EGFR(Tyr1173) were blotted.
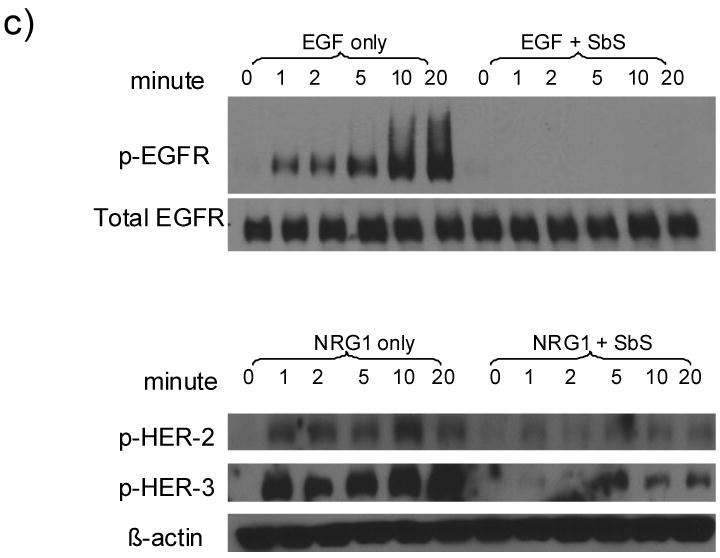
c) SW480 or HCT-116 cells starved overnight in serum-free media were treated by 10ng/ml recombinant EGF or 100ng/ml NRG1 ligand with or without the presence of SbS at various time points (0, 1, 2, 5, 10 and 20 minutes). p-EGFR (Tyr1173) and p-HER-2 and p-HER-3 were blotted using specific antibodies.
All results are representative of at least three independent experiments.
To examine if SbS inhibits ligand-induced activation of these receptor tyrosine kinases, SW480 or HCT-116 cells were treated with 10ng/ml recombinant EGF ligand or 100 ng/ml recombinant Neuregulin (NRG1) ligand, with or without the presence of SbS at various time points. As shown in Figure 1c, SbS completely blocked EGF-induced EGFR activation and attenuated NRG1-induced HER-2 and HER-3 activation.
SbS inhibits colon cancer cell proliferation and cell colony formation
The effect of SbS on EGFR signaling led us to examine whether cancer cell proliferation may be altered as a consequence of EGFR deactivation. Using the MTS assay, semi-confluent colonic adenocarcinoma HT29 cells were stimulated with 10ng/ml EGF in the presence or absence of SbS for 24 hours. We found that SbS abolished EGF-stimulated relative cell number increase (p<0.001) (Figure 2a). Furthermore, BrdU labeling was used in colorimetric immunoassay for the quantification of cell proliferation. As shown in Figure 2b, SbS significantly reduced EGF induced cell proliferation (p<0.01) quantified by incorporation of BrdU.
Figure 2. SbS inhibits EGF-induced cell proliferation (a&b) and prevents HT29 cell colony formation (c).

a&b) semi-confluent HT29 cells are stimulated with 10ng/ml EGF in the presence or absence of SbS for 24 hours, followed by MTS assay and colorimetric BrdU assay. Bars represent mean ± S.E.
c) Cell survival was determined by colony formation assay. HT29 cells were grown in DMEM containing 10% serum in the absence or presence of different concentrations of SbS for 10-20 days. Colonies of 30 cells or more were scored as survivors. Results are from one of two independent experiments.
We next determined the effect of SbS in cell survival using the colony formation assay. HT29 cells were grown in DMEM containing 10% serum in the absence or presence of different concentrations of SbS for 10-20 days. SbS showed a dose dependent inhibition of HT29 cell colony formation. At dilutions as low as 1/64, this inhibitory effect was still evident and statistically significant (Figure 2b). Thus, SbS reduces proliferation and colony formation of HT29 cells in vitro. This inhibition was almost identically repeated in SW480 cells treated by the same dilutions of SbS (data not shown).
SbS inactivates phopho-Akt and induces cancer cell apoptosis
Since Akt is a key signaling molecule for cell survival and is also a downstream molecule to the EGFR, we next tested whether Akt was also affected by SbS. After varying periods (0 - 240 min) of exposure to SbS, cells were lysed and samples tested by Western blotting for Akt phosphorylation. After 15 minutes of exposure to SbS and thereafter Akt phosphorylation was markedly decreased whereas total Akt remain unchanged (Figure 3a).
Figure 3. SbS inactivates phopho-Akt and induces HT29 cell apoptosis.
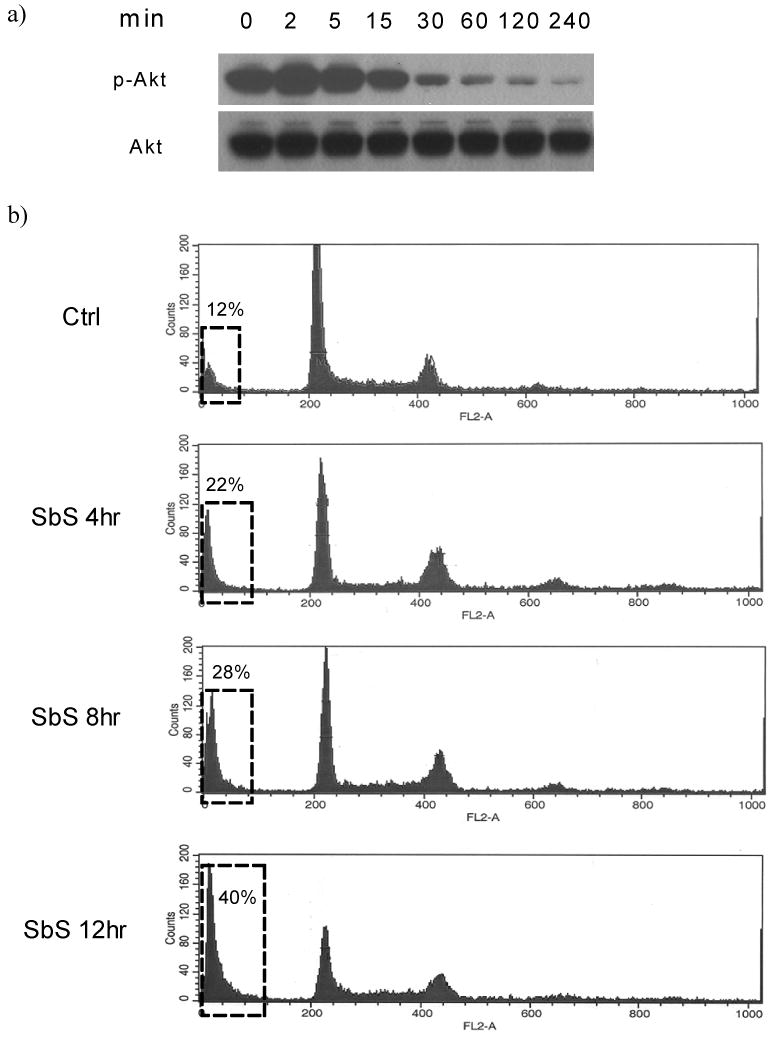
a) After varying periods of time (0 - 240 min) of exposure to SbS, HT29 cells were lysed and samples tested by Western blotting using antibodies against total and phopho-Akt.
b) Human colonic cancer cells were treated with SbS for different time periods (4, 8, 12 hours), harvested and stained by propidium iodide followed by flow cytometric analysis. Cells treated with RPMI for 12 hours were used as a control. Apoptosis was determined by the sub-G1 fraction (in rectangular boxes in figure). Results are from one of two independent experiments.
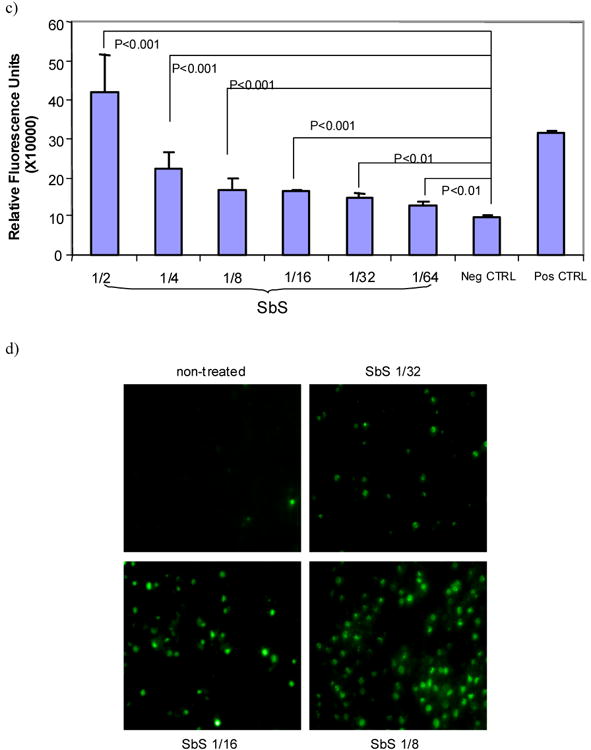
c) HT29 cells treated with SbS for 4 hours at varying dilutions (1/2 to 1/64) were then tested using homogenous caspase assay kit (Roche Applied Science). Cells treated with vehicle serves as negative control. Cells treated with 4 μg/ml camptothecin serve as positive control. Bars represent mean ± S.E. p < 0.01 between SbS treated groups (at all tested doses) versus negative control.
d) HT29 cells treated with SbS for 12 hours at varying dilutions (1/8, 1/16, 1/32) were tested for apoptosis by tunel assay using TUNEL apoptosis detection kit. Cells labeled with green fluorescence are those undergoing apoptosis. The average percentage of positively stained cells in those treated with SbS (1/8 dilution) is 32.0±8.5%, vs. control cells 1.5±0.5%, (p<0.01). Representative images are shown in 3d.
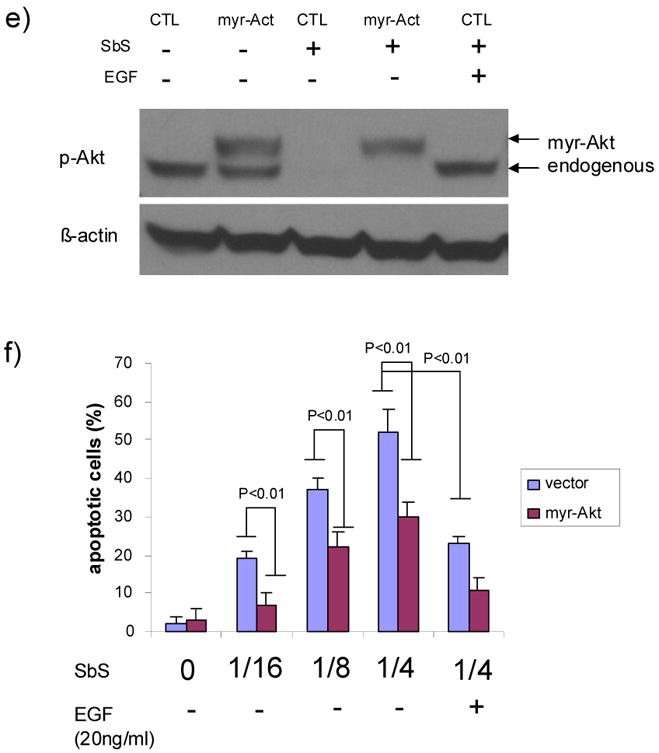
e) 24 hours after HT29 cells were transfected with pUSE empty vector or pUSE-Akt, cells were treated with SbS at 1/2 dilution for 30mins then lysed for Western blotting using p-Akt. SbS decreased endogenous p-Akt signal whereas ectopic myr-Akt remained intact. Co-treatment of cells with EGF (20ng/ml) reversed the p-Akt signal reduction induced by SbS
f) HT 29 cells transiently transfected with vector or myr-Akt were treated with different dilutions of SbS for 12 hours. Apoptotic cells were then analyzed by Tunel staining. The percentage of Tunel positive cells were averaged in three independent experiments. Both ectopic myr-Akt expression and stimulation by 20ng/ml EGF ligand reduced SbS-induced apoptosis. Bars represent mean ± S.E.
In view of these effects on Akt activations we next determined whether SbS affects cell apoptosis. We evaluated apoptosis by flow cytometry, caspase assay and tunnel staining. For flow cytometry, HT29 cells were treated with or without SbS for different time periods (4, 8, 12 hours), and then stained by propidium iodide followed by flow cytometric analysis. We found that SbS treatment increased the sub-G1 cell fraction in a time-dependent manner (control 12%, SbS treated 4 hr 22%, 8 hr 28%, 12 hr 40%), suggesting that SbS promoted HT29 cancer cell death (Figure 3b).
HT29 cells treated with SbS for 4 hours at varying dilutions (1/2 to 1/64) were then tested using a homogenous caspase assay kit (Roche Diagnostics). We found that SbS induced caspase activity in a dose-dependent fashion. Even at the 1/64 dilution, the induction of caspase activity was significantly higher compared to negative control cells (Figure 3c). Furthermore, we demonstrated that SbS induces apoptosis by tunnel staining as shown in Figure 3d. The average percentage of positively stained cells in those treated with SbS (1/8 dilution) is 32.0±8.5%, vs. control cells 1.5±0.5%, (p<0.01). These data indicate that SbS inactivates phospho-Akt and induces cancer cell apoptosis.
To confirm the role of SbS-regulated EGFR and Akt in apoptosis, we transiently transfected HT29 cells with constitutively active myr-Akt using the empty pUSE vector as control. Treatment of transected cells by SbS at 1/2 dilution for 30 minutes decreased endogenous p-Akt signal whereas ectopic myr-Akt remained intact. Adding EGF (20ng/ml) to SbS treated cells reversed the p-Akt signal reduction induced by SbS (Figure 3e). HT 29 cells transiently transfected with vector or myr-Akt were treated with different dilutions of SbS for 12 hours and the apoptotic rate of cells was analyzed by Tunel staining,. As shown in Figure 3f, ectopic myr-Akt expression inhibited Sb-mediated apoptosis at all SbS dilutions. (P<0.01). Stimulation by 20ng/ml EGF ligand also significantly reduced SbS-induced apoptosis at 1/4 dilution of SbS. (P<0.01). (Figure 3f)
Oral intake of Sb inhibited tumor growth in ApcMin mice
To test whether Sb inhibits cancer growth in vivo, we administrated Sb to C57BL6/min/+ (ApcMin) mice daily from age 7 weeks until 16 weeks of age. Mice were then sacrificed and the 10 cm section of the distal small intestine was harvested, from which we measured the number, size and grade of dysplasia in intestinal tumors. We found that orally administered Sb led to a significant reduction in the number (mean ± SE: 29.7 ± 4.8 for control (n=6) versus 16.4 ± 3.0 for Sb treated (n=8), p=0.03), diameter (3.3 ± 0.2 mm versus 2.7 ± 0.2 mm, p=0.03) and total surface area (102 ± 21.7 mm2 versus 44.5 ± 9.4 mm2, p=0.02) of intestinal tumors in the ApcMin mice (Figure 4a).
Figure 4. Oral intake of Sb inhibited tumor growth in ApcMin mice.
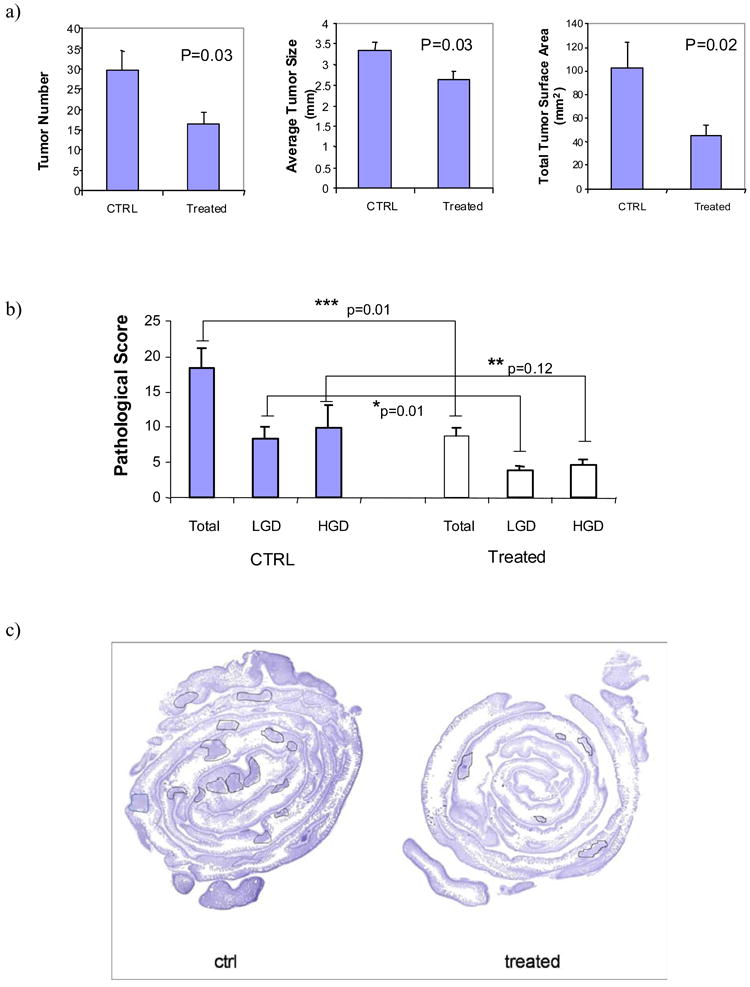
a) After 9 weeks of oral administration of Sb, ApcMin mice were sacrificed and the 10 cm section of the distal small intestine was harvested, from which we measured the number and size of intestinal tumors. Tumor number is significantly reduced in ApcMin mice treated with Sb (N=8) compared to control untreated ApcMin mice (N=6, p=0.03). Tumor size is also significantly reduced in the treated group (p=0.03), as well as total tumor surface area (p=0.02). Bars represent mean ± S.E.
b) Using a system based on human colonic carcinoma profiles, we categorized the intestinal tumors into low grade dysplasia (LGD), high grade dysplasia (HGD) and total pathological score (LGD + HGD). Sb-treated ApcMin mice had a significantly lower LGD compared to control mice (p=0.01). Scores for high grade dysplasia were also lower than in the control group but this difference did not reach statistical significance (p=0.12). Total dysplasia score is significantly lower in the Sb-treated mice (p=0.01).
c) The proximal sections of the small intestines were H&E stained. Representative images illustrates the contrast between Sb-treated ApcMin mice and control ApcMin mice. Visible tumors were outlined by circles.
The tumors in the ApcMin mice were categorized as showing low or high grade dysplasia, using a system based on human colonic carcinoma profiles. Sb-treated mice had a significantly lower score for low-grade dysplasia compared to control mice (p=0.01). Scores for high grade dysplasia were also lower than in the control group but this difference did not reach statistical significance (p=0.12, Figure 4b). Representative images from H&E stained small intestines demonstrated reduced numbers of polyps (Figure 4c).
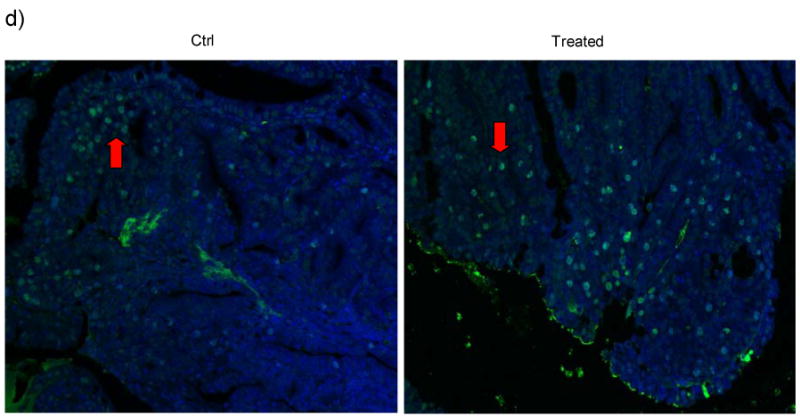
Immunochemistry using antibodies against PCNA, a cell proliferation marker, showed that Sb-treated ApcMin mice had fewer proliferative cells in their tumors (PCNA positive staining) compared to control mice. Figure 5 shows representative images from both Sb treated ApcMin mice and control mice. PCNA positive cells within the tumor are 66% in control mice, compared to 37% in Sb-treated mice (66 ± 14% vs. 37 ± 20 %, p<0.01), indicating that cell proliferation is attenuated in tumors of Sb-treated mice (Figure 5a). The average percentage of phospho-EGFR (Tyr1173) positive cells per high power field in Sb-treated mice vs. control mice are 6.5 ± 2.8 % vs. 17 ± 5.5 %, p<0.05. The average percentage of p-Akt strongly stained cells per high power field from Sb-treated mice vs. control mice are 49.6 ± 5.4% vs. 70.4 ± 4.5%, p=0.01. Representative images of p-EGFR and p-Akt in Figure 5b&c showed that both activation molecules are less evident in treated mice, indicating that the effects of Sb on phospho-EGFR and phospho-Akt is also evident in vivo, consistent with the results of our in vitro experiments with HT-29 cells (Figure 1 and 3a). Tunel staining of intestinal tissues and control Apcmin mice demonstrated a significant increase of apoptotic cells from Sb-treated mice. The average percentages of apoptotic cells per high power field are 15 ± 3.0 % in Sb-treated mice, vs. 4.5 ± 2.3 % in control mice respectively. p<0.01. Representative images are shown in Figure 5d.
Figure 5. Immunohistochemical staining showing the in vivo effect of Sb on EGFR and Akt phosphorylation and on cell proliferation in the intestinal tumors of ApcMin mice.
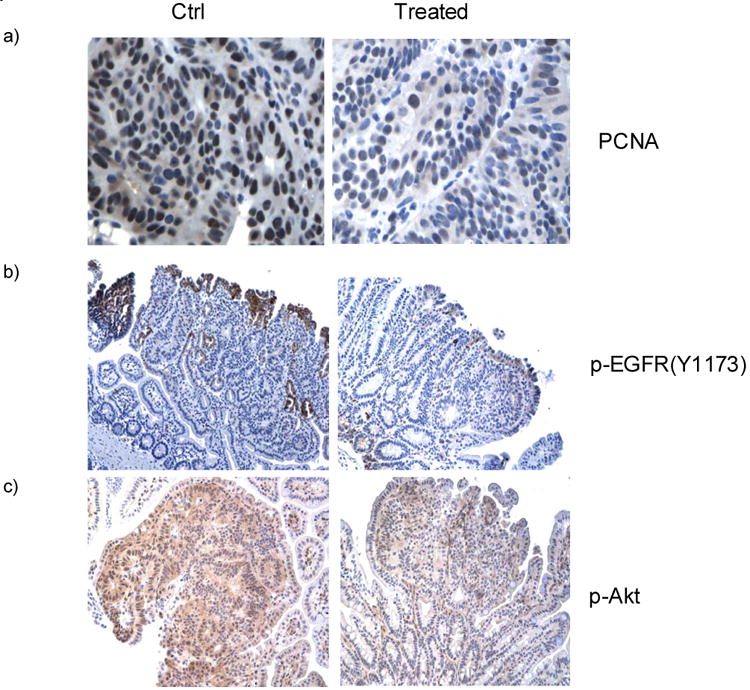
a) The murine small intestines were stained using PCNA antibodies and counter-stained with haematoxylin. Dark/black colored nuclear staining indicates PCNA positive cells. Cells with blue colored nuclei are PCNA negative. Semi-quantative analysis using 5 representative images from each group showed that tumors from ApcMin mice treated with Sb possess fewer proliferative cells compared to those from control mice (37 ± 20 % vs. 66 ± 14%, p<0.01).
b&c) Antibodies against Phospho-EGFR(Y1173) and Phospho-Akt were used to stain the intestinal tumors counter-stained with haemotoxylin. From microscopic views of 10 tumors from each group, the percentage of phospho-EGFR (Tyr1173) positive cells per high power field in Sb-treated mice vs. control mice are 6.5 ± 2.8 % vs. 17 ± 5.5 %, (mean ± S.E., p<0.05). The average percentage of p-Akt strongly stained cells per high power field from Sb-treated mice vs. control mice are 49.6 ± 5.4% vs. 70.4 ± 4.5%, p=0.01. Representative images are shown to compare the different staining intensity for p-EGFR and p-Akt between treated and control ApcMin mice. (Brown color = Phospho-EGFR or Phospho-Akt).
d) Tunel staining of intestinal tissues from Sb-treated and control Apcmin mice. FITC (green) labeled cells are apoptotic tumor cells with DAPI (blue) staining as background. From 10 microscopic views each from the treated and control groups, the average percentages of apoptotic cells per high power field are 15 ± 3.0% in Sb-treated mice, vs. 4.5 ± 2.3 % in control mice respectively, (mean ± S.E., p<0.01). Representative tunnel staining images are shown Figure 5d with red arrow pointing to apoptotic cells within the tumors.
Discussion
One of the beneficial effects of Saccharomyces boulardii is its ability to reduce intestinal inflammatory responses. Erk1/2 MAP kinases, a major modulator of host inflammatory responses, are negatively regulated by Sb 5, 9. Here we report that Sb inhibits Erk kinase activity through effects on the EGFR/Mek pathway. The active (phosphorylated) form of EGFR rapidly loses its activity upon exposure to SbS, which leads to the downstream inactivation of p-Mek and p-Erk as well as p-Akt. Dephosphorylation of p-EGFR is not due to a toxic effect of SbS since, as we previously reported, the inhibitory effect of SbS is fully reversible16. The fact that p-EGFR, p-Mek1/2 and p-Erk1/2 slowly recover their activities, as indicated by our Western blot data, is entirely consistent with this finding.
SbS prevented cancer cell colony formation, reduced EGF-mediated cell proliferation and increased apoptosis. The in vitro effects are consistent with inhibition of the EGFR and Akt pathways. The inhibitory effect of Sb on p-EGFR and p-Akt appears to occur in vivo also. Immunostaining intensity of both p-EGFR and p-Akt decreased in Sb treated ApcMin mice. Furthermore, ApcMin mice treated with Sb demonstrated beneficial effects compared to non-treated mice by showing a 50% decrease in tumor number and a substantial drop in tumor volume. This suggests that S. boulardii may have a beneficial role in preventing or treating intestinal adenomatous polyps and/or adenocarcinoma.
Aberrant activation of EGFR has been shown to be critical for the maintenance of malignancy in a number of solid tumors, including colorectal cancer 17, 18. Interference with the activation of EGFR and other growth factor receptors represents a promising strategy for novel and selective anticancer therapies 19. In the ApcMin mouse model, it has been shown that EGFR activity is important in the establishment of intestinal tumors and Apc deficiency is associated with increased EGFR activity 20, 21. Our in vitro cell signaling studies indicated SbS inactivates other receptor tyrosine kinases including HER-2, HER-3 and IGF-1R besides EGFR, which are all involved in cell proliferation and apoptosis, and commonly overexpressed in many cancers and play important roles in neoplasia 22-24. In our hands, immunohistochemical staining of phospho-IGF-1R yielded no staining pattern in tumors of Apcmin mice. However, phospho-EGFR showed clear differences between Sb-treated and control mice. Our data also suggested both EGF ligand stimulation and constitutively active Akt expression inhibit the pro-apoptotic effect of SbS, indicating the direct involvement of EGFR-Akt pathway in Sb-mediated apoptosis. The roles of IGF-1R and HER-2 & 3 in Sb-mediated cell proliferation and apoptosis, as well as tumor reduction in Apcmin mice need to be further addressed in our future studies. However, the inhibitory effect of Sb on EGFR activity, with downstream effects on Akt, at least in part explains its beneficial effect in reducing tumor growth.
As yet, the detailed mechanism of how SbS inactivates both basal and ligand induced activation of EGFR and HER-2 & 3 is unknown. Compartmentalization of receptor tyrosine kinases including EGFR into sub-domains of the cell membrane is an important control mechanism for signaling 25-27. One hypothesis is that SbS may alter the membrane localization of receptor tyrosine kinases and their association with lipid rafts. This may explain the effects of Sb on a variety of receptor tyrosine kinases. Nonetheless, our findings indicate that the yeast S. boulardii can exert anti-cancer effects through inactivation of growth receptors. Receptor tyrosine kinases are key molecular targets for cancer therapy and, to our knowledge, this is the first report of major receptor tyrosine kinase inactivation by a probiotic microorganism. Several animal studies showed a reduction in chemically induced colorectal tumor incidence accompanying probiotic bacteria administration, however the mechanisms were not well understood 28-30. Our findings that Sb inhibits EGFR signaling and reduces tumor growth in ApcMin mice provide a novel mechanism for probiotic actions against cancer. S. boulardii appears to exert its beneficial effects by multiple potential mechanisms of action 31, including neutralization of bacterial virulence factors 4, 32, interference with bacterial adhesion 33, strengthening of enterocyte tight junctions 34, enhancement of the mucosal immune response 35, 36, altering immune cell redistribution 37 and modulating inflammatory signaling pathways of the host 8, 9. Given the complicated picture of probiotic actions, we cannot exclude other potential mechanisms that may explain the anti-cancer phenomenon of Sb. Besides receptor tyrosine kinase inactivation, the anti-inflammatory properties of Sb or possible gut flora changes due to Sb administration in Apcmin mice may also impact intestinal tumor development. Further studies are needed to explore these potential mechanisms. Nevertheless, with decades of usage profile in treating gastrointestinal disorders, the clinical effects of Saccharomyces boulardii on prevention and treatment of colon polyps and colon cancer clearly warrant further investigation.
Materials and Method
Cells and Reagent
HT29, SW480 and HCT-116 cells were obtained from ATCC. HT29 and SW 480 cells were cultured in DMEM, whereas HCT-116 in McCoy's 5A modified media, supplemented with 10% (vol/vol) FBS, 2 mM L-glutamine (GIBCO), 100 U/ml penicillin, and 100 μg/ml streptomycin in a 5% CO2 incubator at 37°C. Recombinant human (rh) Epidermal Growth Factor (EGF), recombinant human Neuregulin (NRG-1) were purchased from R & D Systems (Minneapolis, MN). Antibodies against Erk, MEK, CamKII kinases, PKCs, EGFR, IGF-1R, HER-2, HER-3 and Akt, phosphorylated and/or non-phosphorylated forms, were purchased from Cell Signaling Technology (Beverly, MA). DNA constructs myr-Akt-pUSE and pUSE empty vector were gifts from Dr. Grant D Stewart from University of Edinburgh. Preparation of Saccharomyces boulardii culture supernatant (SbS) was done as previously described 9. Briefly, lyophilized Sb (Biocodex Laboratories, France) was cultured in RPMI 1640 cell culture medium (100mg/ml) for 24 hours in 37°C. The suspension was then centrifuged at 9000g for 15 minutes and the supernatant collected. The supernatant was then passed through a 0.22 mm filter (Fisher Scientific) and then a 10 kDa cutoff filter (Millipore, MA).
Western blot analysis
HT29, SW480 or HCT-116 cells were incubated with SbS at 1:1 dilution for different time periods at different conditions. Treated cells were then lysed in a lysis buffer (62.5 mM Tris-HCl, 10% glycerol, 2% SDS, 0.01% bromphenol blue, and 1% 2-mercaptoethanol). Equal amounts of cell extract were fractionated by 10% SDS-PAGE, and proteins were transferred onto nitrocellulose membranes (Bio-Rad) at 300 mA for 3 h. Membranes were blocked in 5% nonfat dried milk in TBST (50 mM Tris, pH 7.5, 0.15 M NaCl, 0.05% Tween 20) and then incubated with antibodies directed against phopho- or nonphosphorylated forms of ERK1/2, MEK1/2, HER-2, HER-3, IGF-1R (Cell Signaling, MA) and EGFR (Santa Cruz Technology). Membranes were washed with TBST and incubated with horseradish peroxidase-labeled secondary antibodies for 1 h. The peroxidase signal was detected by Supersignal chemiluminescent substrate (Pierce), and the image of the signal was recorded by exposure to x-ray film (Fujifilm, Tokyo, Japan).
Colony formation assay and cell proliferation assays
HT29 and SW480 cells were seeded at 1,000 cells per well in six-well plates and allowed to attach for 48 h. SbS was diluted in DMEM at different concentrations and added directly to cell culture wells. Cultures were observed daily for 10–20 days and then were fixed and stained with modified Wright-Giemsa stain (Sigma). Colonies of 30 cells were scored as survivors 38. Cells were maintained at 37°C in 5% CO2 in complete humidity. HT29 cells were cultured in DMEM supplemented with 10% (vol/vol) FBS and 2 mM L-glutamine (GIBCO). Cell proliferation assays were carried out using both MTS assay kit (Promega) and BrdU colorimetric kit (Roche Applied Science) following manufacture's instructions.
Cell transfection
90-100% confluent HT29 cells grown in 12-well dishes or 4 Chamber Polystyrene Vessel Glass Slides were transfected with pUSE empty vector or pUSE-myr-Akt plasmids using Lipofecatmine 2000 (Invitrogen, Inc.). 24 hour after transfection, cells were treated with SbS for 30 mins before being lysed for Western blot analysis, or treated over night for TUNEL staining.
Apoptosis assays
Tunel assay was carried out in both HT29 cell culture and intestinal tissues using TUNEL Apoptosis Detection Kit (Upstate Biotechnology Inc., Lake Placid, NY) according to the manufacturer's instructions. For flow cytometry, adherent HT29 cells were released by treatment with 0.25% trypsin. Each sample was fixed overnight with 70% ethanol at 4°C. Cells were rehydrated with phosphate-buffered saline (PBS) and then stained with 10% propidium iodide (PI) in 100 U/mL of ribonuclease in PBS for 60 minutes at room temperature. Cells were filtered through 35-μm filters before analysis. The flow cytometer was configured to track the number of events with the FL2 parameter (FACScan; Becton Dickinson, Lincoln Park, NJ). The DNA content was analyzed using a nonlinear least-squares algorithm. Caspase activation was measured with a fluorimetric homogeneous caspase assay kit (Roche Applied Science).
ApcMin mice
C57BL/6J Min/+ (ApcMin) mice were obtained from the Jackson Laboratory (Bar Harbor, ME) at age 7 weeks. Sb was administrated daily in their drinking water 3×108 CFU per ml and 3 times each week by oral gavage at a dose of 6×108 CFU for 9 weeks. Mice were sacrificed at 16 weeks of age. Intestinal tumor number was counted in the distal 10 cm of the small intestine using Methylene Blue in PBS (0.05%). Tumor sizes were measured by external caliper and taken as diameter to calculate total areas. Proximal sections of the rest of the small intestine (remaining section after usage of 10cm distal part) were fixed and used for immunohistochemistry. Grade of dysplasia was measured by a blinded pathologist using a simple grading system based on the criteria for adenomatous change in the human colon: high-grade vs. low-grade dysplasia, where low-grade dysplasia showed nuclear elongation with a sessile or villous architecture, and high-grade dysplasia showed a cribriform growth pattern with loss of nuclear polarity and nuclear rounding. Relevant tissues were fixed overnight in 10% neutral-buffered formalin for immunohistochemistry. Animal studies were approved by the Institutional Animal Care and Use Committee.
Immunohistochemistry and imaging analysis
Immunohistochemical staining was done on fixed intestinal tissues in Beth Israel Deaconess Medical Center Immunohistochemistry Core Facility using specific antibodies against PCNA, phospho-EGFR, and phosphor-Akt and counterstained with hematoxylin. Images were viewed under light microscopy (Eclipse E800, Nikon) by using a plain Apo 40×/0.95 objective, imported via a SPOT Insight camera (Diagnostic Instruments, Sterling Heights, MI), and stored digitally using SPOT software (Diagnostic Instruments). For semi-quantitative analysis of immunohistochemistry imaging, microscopic views of 10 random tumors from each animal group were randomly chosen and examined by a blinded observer. On each microscopic field, cells positively stained by p-EGFR or p-Akt were counted, as well as the total number of cells by haemotoxylin staining. In the case of quantifying Tunel stained colon tumor images, cells positively stained by FITC and total cells by DAPI were counted. The percentage of positively staining cells on each high power field was calculated for both groups and statistically analyzed.
Statistical Analyses
Results were expressed as mean ± S.E. Data were analyzed using the SIGMA-STAT™ professional statistics software program (Jandel Scientific Software, San Rafael, CA). Analyses of variance with protected t test were used for intergroup comparison.
Acknowledgments
This work was supported by NIH grants PO1 DK 33506 (to CPK), and Crohn's & Colitis Foundation of America (research fellowship to XC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Elmer GW, Surawicz CM, McFarland LV. Biotherapeutic agents. A neglected modality for the treatment and prevention of selected intestinal and vaginal infections. Jama. 1996;275:870–6. doi: 10.1001/jama.275.11.870. [DOI] [PubMed] [Google Scholar]
- 2.Sullivan A, Nord CE. The place of probiotics in human intestinal infections. Int J Antimicrob Agents. 2002;20:313–9. doi: 10.1016/s0924-8579(02)00199-1. [DOI] [PubMed] [Google Scholar]
- 3.Guslandi M, Mezzi G, Sorghi M, Testoni PA. Saccharomyces boulardii in maintenance treatment of Crohn's disease. Dig Dis Sci. 2000;45:1462–4. doi: 10.1023/a:1005588911207. [DOI] [PubMed] [Google Scholar]
- 4.Castagliuolo I, LaMont JT, Nikulasson ST, Pothoulakis C. Saccharomyces boulardii protease inhibits Clostridium difficile toxin A effects in the rat ileum. Infect Immun. 1996;64:5225–32. doi: 10.1128/iai.64.12.5225-5232.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Czerucka D, Dahan S, Mograbi B, Rossi B, Rampal P. Saccharomyces boulardii preserves the barrier function and modulates the signal transduction pathway induced in enteropathogenic Escherichia coli-infected T84 cells. Infect Immun. 2000;68:5998–6004. doi: 10.1128/iai.68.10.5998-6004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Czerucka D, Roux I, Rampal P. Saccharomyces boulardii inhibits secretagogue-mediated adenosine 3′,5′-cyclic monophosphate induction in intestinal cells. Gastroenterology. 1994;106:65–72. doi: 10.1016/s0016-5085(94)94403-2. [DOI] [PubMed] [Google Scholar]
- 7.Dalmasso G, Cottrez F, Imbert V, Lagadec P, Peyron JF, Rampal P, Czerucka D, Groux H. Saccharomyces boulardii Inhibits Inflammatory Bowel Disease by Trapping T Cells in Mesenteric Lymph Nodes. Gastroenterology. 2006;131:1812–1825. doi: 10.1053/j.gastro.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Dahan S, Dalmasso G, Imbert V, Peyron JF, Rampal P, Czerucka D. Saccharomyces boulardii interferes with enterohemorrhagic Escherichia coli-induced signaling pathways in T84 cells. Infect Immun. 2003;71:766–73. doi: 10.1128/IAI.71.2.766-773.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen X, Kokkotou EG, Mustafa N, Bhaskar KR, Sougioultzis S, O'Brien M, Pothoulakis C, Kelly CP. Saccharomyces boulardii inhibits ERK1/2 mitogen-activated protein kinase activation both in vitro and in vivo and protects against Clostridium difficile toxin A-induced enteritis. J Biol Chem. 2006;281:24449–54. doi: 10.1074/jbc.M605200200. [DOI] [PubMed] [Google Scholar]
- 10.Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. The Lancet Oncology. 2005;6:322–327. doi: 10.1016/S1470-2045(05)70168-6. [DOI] [PubMed] [Google Scholar]
- 11.Jimeno A, Hidalgo M. Pharmacogenomics of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors. Biochim Biophys Acta. 2006;1766:217–29. doi: 10.1016/j.bbcan.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 12.de Castro-Carpeno J, Belda-Iniesta C, Casado Saenz E, Hernandez Agudo E, Feliu Batlle J, Gonzalez Baron M. EGFR and colon cancer: a clinical view. Clin Transl Oncol. 2008;10:6–13. doi: 10.1007/s12094-008-0147-3. [DOI] [PubMed] [Google Scholar]
- 13.Calonghi N, Pagnotta E, Parolin C, Mangano C, Bolognesi ML, Melchiorre C, Masotti L. A new EGFR inhibitor induces apoptosis in colon cancer cells. Biochem Biophys Res Commun. 2007;354:409–13. doi: 10.1016/j.bbrc.2006.12.214. [DOI] [PubMed] [Google Scholar]
- 14.Ponz-Sarvise M, Rodriguez J, Viudez A, Chopitea A, Calvo A, Garcia-Foncillas J, Gil-Bazo I. Epidermal growth factor receptor inhibitors in colorectal cancer treatment: what's new? World J Gastroenterol. 2007;13:5877–87. doi: 10.3748/wjg.v13.i44.5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marshall J. Clinical implications of the mechanism of epidermal growth factor receptor inhibitors. Cancer. 2006;107:1207–18. doi: 10.1002/cncr.22133. [DOI] [PubMed] [Google Scholar]
- 16.Sougioultzis S, Simeonidis S, Bhaskar KR, Chen X, Anton PM, Keates S, Pothoulakis C, Kelly CP. Saccharomyces boulardii produces a soluble anti-inflammatory factor that inhibits NF-kappaB-mediated IL-8 gene expression. Biochem Biophys Res Commun. 2006;343:69–76. doi: 10.1016/j.bbrc.2006.02.080. [DOI] [PubMed] [Google Scholar]
- 17.Jiang D, Yang H, Willson JK, Liang J, Humphrey LE, Zborowska E, Wang D, Foster J, Fan R, Brattain MG. Autocrine transforming growth factor alpha provides a growth advantage to malignant cells by facilitating re-entry into the cell cycle from suboptimal growth states. J Biol Chem. 1998;273:31471–9. doi: 10.1074/jbc.273.47.31471. [DOI] [PubMed] [Google Scholar]
- 18.Huang SM, Harari PM. Epidermal growth factor receptor inhibition in cancer therapy: biology, rationale and preliminary clinical results. Invest New Drugs. 1999;17:259–69. doi: 10.1023/a:1006384521198. [DOI] [PubMed] [Google Scholar]
- 19.Nautiyal J, Rishi AK, Majumdar AP. Emerging therapies in gastrointestinal cancers. World J Gastroenterol. 2006;12:7440–50. doi: 10.3748/wjg.v12.i46.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts RB, Min L, Washington MK, Olsen SJ, Settle SH, Coffey RJ, Threadgill DW. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc Natl Acad Sci U S A. 2002;99:1521–6. doi: 10.1073/pnas.032678499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moran AE, Hunt DH, Javid SH, Redston M, Carothers AM, Bertagnolli MM. Apc deficiency is associated with increased Egfr activity in the intestinal enterocytes and adenomas of C57BL/6J-Min/+ mice. J Biol Chem. 2004;279:43261–72. doi: 10.1074/jbc.M404276200. [DOI] [PubMed] [Google Scholar]
- 22.Stern DF. ERBB3/HER3 and ERBB2/HER2 duet in mammary development and breast cancer. J Mammary Gland Biol Neoplasia. 2008;13:215–23. doi: 10.1007/s10911-008-9083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–28. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 24.LeRoith D, Roberts CT., Jr The insulin-like growth factor system and cancer. Cancer Lett. 2003;195:127–37. doi: 10.1016/s0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- 25.Le Roy C, Wrana JL. Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat Rev Mol Cell Biol. 2005;6:112–26. doi: 10.1038/nrm1571. [DOI] [PubMed] [Google Scholar]
- 26.Miljan EA, Bremer EG. Regulation of growth factor receptors by gangliosides. Sci STKE. 2002;2002:RE15. doi: 10.1126/stke.2002.160.re15. [DOI] [PubMed] [Google Scholar]
- 27.Thiel KW, Carpenter G. ErbB-4 and TNF-alpha converting enzyme localization to membrane microdomains. Biochem Biophys Res Commun. 2006;350:629–33. doi: 10.1016/j.bbrc.2006.09.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirayama K, Rafter J. The role of probiotic bacteria in cancer prevention. Microbes Infect. 2000;2:681–6. doi: 10.1016/s1286-4579(00)00357-9. [DOI] [PubMed] [Google Scholar]
- 29.Rafter J. Probiotics and colon cancer. Best Practice & Research Clinical Gastroenterology. 2003;17:849–859. doi: 10.1016/s1521-6918(03)00056-8. [DOI] [PubMed] [Google Scholar]
- 30.Brady LJ, Gallaher DD, Busta FF. The Role of Probiotic Cultures in the Prevention of Colon Cancer. J Nutr. 2000;130:410S–414S. doi: 10.1093/jn/130.2.410S. [DOI] [PubMed] [Google Scholar]
- 31.Chen X, Kelly C. Saccharomyces spp. In: Versalovic J, Wilson M, editors. Therapeutic Micriobiology: Probiotics and Related Strategies. 1st. Washington, DC: ASM Press; 2008. pp. 51–60. [Google Scholar]
- 32.Pothoulakis C, Kelly CP, Joshi MA, Gao N, O'Keane CJ, Castagliuolo I, Lamont JT. Saccharomyces boulardii inhibits Clostridium difficile toxin A binding and enterotoxicity in rat ileum. Gastroenterology. 1993;104:1108–15. doi: 10.1016/0016-5085(93)90280-p. [DOI] [PubMed] [Google Scholar]
- 33.Gedek BR. Adherence of Escherichia coli serogroup O 157 and the Salmonella typhimurium mutant DT 104 to the surface of Saccharomyces boulardii. Mycoses. 1999;42:261–4. doi: 10.1046/j.1439-0507.1999.00449.x. [DOI] [PubMed] [Google Scholar]
- 34.Philpott DJ, McKay DM, Sherman PM, Perdue MH. Infection of T84 cells with enteropathogenic Escherichia coli alters barrier and transport functions. Am J Physiol. 1996;270:G634–45. doi: 10.1152/ajpgi.1996.270.4.G634. [DOI] [PubMed] [Google Scholar]
- 35.Buts JP, Bernasconi P, Vaerman JP, Dive C. Stimulation of secretory IgA and secretory component of immunoglobulins in small intestine of rats treated with Saccharomyces boulardii. Digestive Diseases And Sciences. 1990;35:251–256. doi: 10.1007/BF01536771. [DOI] [PubMed] [Google Scholar]
- 36.Qamar A, Aboudola S, Warny M, Michetti P, Pothoulakis C, LaMont JT, Kelly CP. Saccharomyces boulardii stimulates intestinal immunoglobulin A immune response to Clostridium difficile toxin A in mice. Infection And Immunity. 2001;69:2762–2765. doi: 10.1128/IAI.69.4.2762-2765.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dalmasso G, Cottrez F, Imbert V, Lagadec P, Peyron JF, Rampal P, Czerucka D, Groux H, Foussat A, Brun V. Saccharomyces boulardii inhibits inflammatory bowel disease by trapping T cells in mesenteric lymph nodes. Gastroenterology. 2006;131:1812–25. doi: 10.1053/j.gastro.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Sun X, LaMont JT, Pardee AB, Li CJ. Selective killing of cancer cells by beta -lapachone: Direct checkpoint activation as a strategy against cancer. PNAS. 2003;100:2674–2678. doi: 10.1073/pnas.0538044100. [DOI] [PMC free article] [PubMed] [Google Scholar]