

Abstract
Purpose
Tumors produce multiple pro-angiogenic factors, making it unlikely that agents targeting a single angiogenic pathway will be sufficient to treat the spectrum of tumors which occur clinically. PD-ECGF (platelet-derived endothelial cell growth factor) has angiogenic activity in vitro and in vivo and is overexpressed in most human cancers, where its expression has been correlated with increased microvessel density, more aggressive tumors, and poorer patient prognosis. PD-ECGF is identical to the enzyme thymidine phosphorylase (TP), and unlike other angiogenic factors, TP's pro-angiogenic actions are dependent upon its enzyme activity.
Experimental Design
A potent and specific small molecule inhibitor of the catalytic activity of TP, 6-(2-aminoethyl)amino-5-chlorouracil (AEAC), was tested for antiangiogenic and anti-tumor activity in human cancer xenografts in vivo.
Results
Oral administration of AEAC caused 40-50% reductions in the growth of A549 non-small cell lung cancer and PANC-1 pancreatic cancer xenografts, but it was not active against a second pancreatic tumor, BxPC-3. AEAC reduced the microvessel density in the tumors, providing evidence for an anti-angiogenic action. Equal or better activity was seen when the mice were treated with the VEGF-Trap, a soluble VEGF decoy receptor, and the combination of AEAC and the VEGF-Trap produced additive anti-tumor activity that was significantly greater than the VEGF-Trap alone. In the A549 tumors, the combination produced tumor regressions.
Conclusion
These studies show antitumor activity for a drug targeting TP, and suggest that inhibitors of TP could be used to augment the clinical efficacy of drugs targeting the VEGF pathway.
Keywords: Thymidine phosphorylase, VEGF-trap, angiogenesis
Introduction
The number of drugs that target tumor angiogenesis is growing, and emerging clinical data indicate these agents increase response rates and prolong survival in patients with advanced malignancies. Drugs targeting VEGF-mediated pathways are the furthest advanced and have provided proof-of-concept for this class of antitumor agents (1). Thus bevacizumab, a humanized variant of a mouse anti-VEGF-A antibody, increased response rates (in combination with cytotoxic drugs) and survival (or progression-free survival) in patients with metastatic colorectal cancer, non-small cell lung cancer, and metastatic breast cancer, and phase III studies of sunitinib and sorafenib, both small molecule inhibitors of the kinases of the VEGF and PDGF-β (and other) receptors, showed they had activity in renal cell carcinoma, one of the tumors most resistant to chemotherapy (1). These and other studies suggest that angiogenesis inhibitors are likely to have a broad spectrum of activity against solid tumors. Although these observations provide much promise, the percentage of patients showing significant responses in these trials is relatively small, and it is clear that the use of angiogenesis inhibitors which target a single angiogenic molecule or pathway will not be sufficient to treat the spectrum of heterogeneous tumors occurring clinically. The importance of targeting multiple angiogenesis pathways was recognized early in the consideration of anti-angiogenesis therapy, and was supported by early animal tumor experiments (2-3).
Our studies have focused on the angiogenic factor thymidine phosphorylase (TP), based on extensive evidence demonstrating its role in experimental and human cancer (4-5). TP was first identified as an enzyme involved in the cellular metabolism of thymidine, and recent studies have shown that it plays a critical role in maintaining steady state plasma thymidine levels in humans (6-7). A surprising observation was reported in 1992 when the sequencing of the cDNA of human TP revealed it to be identical to the factor PD-ECGF (platelet-derived endothelial cell growth factor) (8). PD-ECGF was first isolated from platelets and was subsequently found to stimulate the migration of endothelial cells (but not that of fibroblasts or smooth muscle cells) in vitro and angiogenesis in vivo (5-12). When transfected into 3T3, KB or MCF7 carcinoma cells, TP increased the vascularization and growth of tumor xenografts of these cells growing in nude mice; in contrast, it had no effect on the growth of the cells in vitro (13-16). TP also increased angiogenesis when transfected into the coronary arteries of dogs with myocardial ischemia due to coronary artery constriction, demonstrating that its angiogenic actions were operative in both neoplastic and non-neoplastic pathologies (17).
TP is distinct from most other angiogenic factors in that, unlike classical cytokines which bind to a cell surface receptor, it exerts its angiogenic actions through its catalytic activity (10, 12, 18). Thus site-directed mutagenesis experiments which utilized a series of TP proteins with single amino acid changes at residues thought to be critical for the binding of thymidine found that mutant TP proteins that lost enzymatic activity were inactive as angiogenic factors, while mutant proteins that retained partial enzyme activity had partial angiogenic activity (10, 12). Based on these observations, TP has been a widely-studied target for the development of anti-angiogenic compounds. We have synthesized a series of 5- and 6-amino-substituted uracil derivatives and evaluated them for their ability to inhibit TP activity (19). One of these compounds, 6-(2-aminoethyl)amino-5-chlorouracil (AEAC, figure 1), was found to be one of the most potent competitive inhibitors of TP (Ki of 165 nM) reported to date (19). AEAC was a selective inhibitor of TP, having no effect on the related enzymes uridine phosphorylase and purine nucleoside phosphorylase at concentrations up to 1 mM, and it was also found to block TP-mediated endothelial cell migration in vitro (19). In this report, we provide evidence that AEAC has anti-angiogenic and anti-tumor activity in mouse tumor xenograft models.
Figure 1.
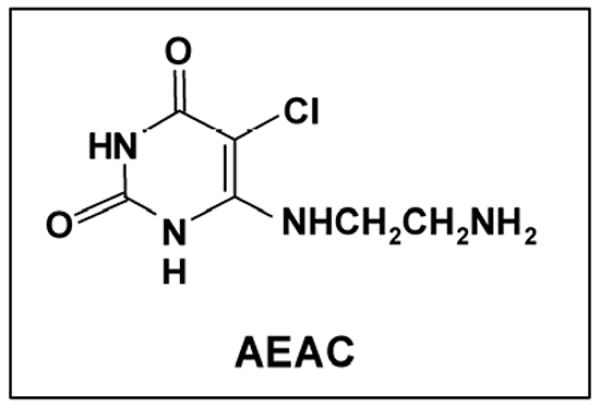
Structure of AEAC.
Methods
Cell lines, drugs, and animals
Human umbilical vein endothelial cells (HUVEC; GlycoTech, Gaithersburg, MD by arrangement with the Developmental Therapeutics Program Angiogenesis Resource Center, National Cancer Institute) were grown in MCDB131, 2% fetal bovine serum (FBS), 10 ng/ml EGF, 12 μg/ml endothelial cell growth supplement, 1 μg/ml hydrocortisone, 10 units/ml heparin, 2 mM l-glutamine, penicillin G, and streptomycin sulfate. A549 human non-small cell lung cancer cells (American Type Culture Collection) were maintained in RPMI 1640 with 10% FBS; PANC-1 and BxPC-3 human pancreatic carcinoma cells (ATCC) were maintained in DMEM and RPMI 1640, respectively, with 10% FBS. Cells were maintained at 37°C with 5% CO2. AEAC, 6-(2-aminoethyl) amino-5-chlorouracil, was provided by the Developmental Therapeutics Program, NCI. The VEGF-Trap was from Regeneron Pharmaceuticals, Inc., and was provided by the Cancer Treatment Evaluation Program, NCI. Pathogen-free female NCR nude mice (6–8 weeks old, 19–24 g) were purchased from Taconic Laboratories (Germantown, NY). All mice were maintained in a pathogen-free animal facility for at least 1 week before each experiment. The animal use committees of the Albert Einstein College of Medicine and Montefiore Medical Center approved all animal study protocols described in this publication, and experiments were conducted in compliance with the Guide for the Care and Use of Laboratory Animals.
In vitro proliferation assays
A549, PANC-1 and BxPC-3 cells were plated at a density of 4,000 per well in 96-well plates in 100 μl of culture media supplemented with 10% FBS. After 24 h of incubation, 100 μl of complete media containing AEAC were added to each well. At 72 h time points, the number of viable cells was detected using the MTT method.
Xenograft tumor models and treatment
Nude mice were inoculated sc in their right flank with 2 × 106 (A549, PANC-1 and BxPC-3) or 12 × 106 (A549) cells in 100 μl PBS. When tumors became palpable (1-3 weeks), mice were randomly assigned into treatment and control groups. AEAC was dissolved in H2O containing 0.5% (w/v) methylcellulose, and was administered via oral gavage five days per week; mice in the control group received an equal volume of vehicle (0.5% methylcellulose). VEGF-Trap was administered via sc injection twice per week. Mice were weighed weekly, and caliper measurements of the tumors were done twice weekly and tumor volumes were calculated according to the formula of the volume of an ellipse: V = (l × w2) / 2, where l and w correspond to the longest and shortest diameters of the tumor, respectively. At the end of the experiments, animals were euthanized by CO2 overdose, and the tumors were removed, weighed, and portions were frozen or prepared for immunohistochemistry.
Western blotting
Total protein was extracted from tumor cell lines or tumor tissues using radioimmunoprecipitation assay (RIPA) buffer (Santa Cruz). Protein concentration was quantified with bicinchoninic acid reagent (Pierce). Protein extracts (20 μg) were subjected to Western immunoblot analysis with antibody for the detection of TP (Cell Signaling Technologies). An antibody to actin (Sigma) was used to evaluate protein loading in each lane. Immunoblots were developed with the Western Lightning Chemiluminescence Plus detection system (Perkin-Elmer) according to the manufacturer's protocol.
Immunohistochemical staining for blood vessels, macrophages, and TP
Tumor tissue was fixed in 4% buffered formalin, embedded in paraffin, sectioned, and stained with H&E using standard histological techniques. In addition, slides were immunostained for TP (Oncogene), CD34 (Cedarlane Labs), and macrophages (rat monoclonal antibody against isolated mouse macrophages; RM0029-11H3, Abcam). Antigen retrieval was performed by heating the slides at 95° to 97°C in 0.01M sodium citrate pH 6.0 for 10 minutes. Endogenous peroxidase activity was quenched by treatment with 3% hydrogen peroxide in methanol/water (1:1) for 10 minutes at room temperature (RT); sections were then incubated with 10% normal serum in PBS for 40 min at RT, followed by incubation with the primary antibody overnight at 4°C. Following incubation with the secondary antibody, the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) was used for detection with diaminobenzidine tetrahydrochloride (Vector) as the substrate. Slides were counterstained with hematoxylin. Negative control sections were incubated with pre-immune serum.
To determine the density of the vessels and macrophage in tumors, three “hot” spots of each section were photographed and the DAB pixel area in the lesion was quantified using the ImageJ program. Five mice per group were analyzed.
Statistical analyses
Statistical analysis was done with one way ANOVA, and pairwise comparison procedures including calculation of P values were done using the Tukey's multiple comparison test. P < 0.05 was considered significant.
Results
Both VEGF and TP inhibition are required to block carcinoma cell-mediated HUVEC migration in vitro
The migration of endothelial cells in vitro can be stimulated by purified angiogenic factors and by carcinoma cells that are producing these factors. HUVEC migration in a Boyden chamber was stimulated by VEGF, purified human TP, or by HT29 carcinoma cells which expressed both TP and VEGF (supplemental fig. 1). Under these conditions, neither a VEGF inhibitor nor a TP inhibitor, when used alone, was able to fully block HT29-mediated HUVEC migration, but the combination of the two inhibitors reduced HUVEC migration to that of control wells (no angiogenic factors or tumor cells) (supplemental fig. 1).
AEAC and the VEGF-Trap inhibited A549 NSCLC and PANC-1 pancreatic cancer xenograft growth in nude mice
The anti-tumor activity of AEAC was assessed in nude mice bearing xenografts of a human non-small cell lung cancer cell line, A549, or two human pancreatic cancer cell lines, PANC-1 and BxPC-3. AEAC was administered by oral gavage at 50 mg/kg once daily for 5 days each week, a dose and schedule which was chosen arbitrarily, since AEAC has not been previously tested in animals. In preliminary experiments, AEAC at doses up to 100 mg/kg had no effect on the body weight of mice with chronic administration, nor did it have any apparent acute toxic effects (data not shown). Since these cancer cells also express VEGF (20-22), we tested AEAC in combination with the VEGF-Trap. The VEGF-Trap binds to all forms of human and murine VEGF (including PIGF) with KDs < 1 pM, and has favorable pharmacokinetics that provide for long term VEGF blockade. When used at 10 mg/kg twice a week, it has been shown to have substantial anti-angiogenic and anti-tumor activity in preclinical models, including in pancreatic and lung cancers.
In the first set of experiments, nude mice were inoculated sc with 2 × 106 A549 cells, and tumors were allowed to grow until they reached an average size of 75 mm3, at which point they were divided into groups of 5 mice and therapy was begun. Mice were treated with vehicle only (control), AEAC, VEGF-Trap, or a combination of AEAC plus the VEGF-Trap. AEAC-alone significantly inhibited tumor growth by 39% ± 6.5 (p < 0.05) compared with control, and a larger antitumor effect was observed with the VEGF-Trap-alone (91% ± 0.9 inhibition) in these xenografts (figure 2A). A higher dose of AEAC (75 mg/kg) did not noticeably increase its anti-tumor efficacy. Even though the VEGF-Trap has substantial activity when used alone, a further significant reduction in tumor size was seen in the AEAC+VEGF-Trap group, when compared to either the AEAC- or the VEGF-Trap-alone groups (figure 2B). The combination notably also produced tumor regressions in 10 out of the 10 mice, versus in 3 out of 10 mice treated with the VEGF-Trap alone. The tumors in the combination group rapidly regressed with treatment to an average of 52% ± 6.3 of their original size, and tumor size remained stable for the first three weeks of drug therapy. At that point, tumor growth resumed and seemed to parallel an observed increase in growth rate in the VEGF-Trap-alone group. However, it cannot be ascertained from these experiments whether the renewed tumor growth resulted from a loss of sensitivity to AEAC, the VEGF-Trap, or both.
Figure 2. AEAC and the VEGF-Trap inhibit A549 NSCLC xenograft growth in nude mice.
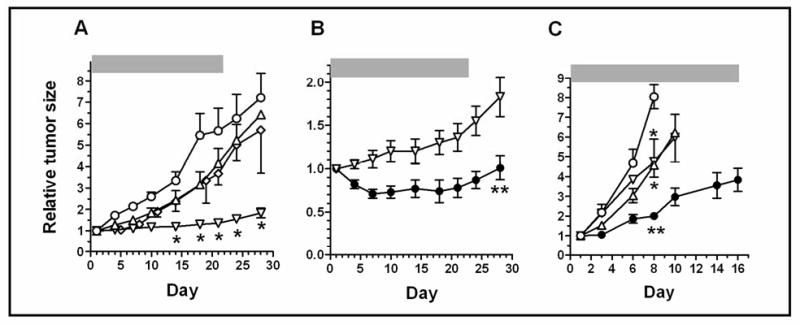
Mice were inoculated sc with 2 × 106 cells (panels A and B) or 12 × 106 cells (panel C) A549 cells. Tumors were allowed to grow until they reached an average size of approx. 75 mm3 (2 weeks; panels A and B) or 485 mm3 (1 week; panel C) at which point they were divided into groups of 5 mice, and therapy was begun. Mice were treated with vehicle (○), AEAC (oral gavage at 50 mg/kg (△) or 75 mg/kg (◊); once daily for 5 days each week); VEGF-Trap (10 mg/kg sc twice weekly) (▽); or the combination of AEAC (50 mg/kg) + VEGF-Trap (●), using the same schedule as when administered individually. Drug therapy was begun on day 1, and was continued for the time indicated by the horizontal bars. Tumor size was measured twice weekly. Tumor volumes were calculated as (l × w2) / 2, and are expressed relative to the tumor size at the start of therapy. The data in panels A and B are from the same experiments; note that the Y axis has been changed in panel B to better illustrate the difference between the VEGF-Trap alone (▽) and AEAC+VEGF-Trap (●) treated mice. Data in panels A & B are means ± SEM from 2 experiments (n=10), and in panel C from a single experiment (n=5).
* Indicates significantly different (p < 0.01) from control.
** Indicates significantly different from control, from AEAC alone, and from VEGF-Trap alone (p < 0.01; ANOVA with Tukey's multiple comparison test) (in panel B, all time points between days 7 and 28 were significantly different).
The drugs were also tested in mice bearing substantially larger A549 tumors, in which therapy was initiated seven days before the untreated mice had to be euthanized due to the size of their tumors and/or evidence of morbidity (figure 2C). Under these conditions, the VEGF-Trap was somewhat less effective, producing a 25% reduction in mean tumor growth relative to control, compared to a 50% reduction in the AEAC-treated mice (figure 2C and Table 1A). Evidence for an approximately additive effective of the combination was obtained in these mice as well, with an 86% reduction in mean tumor growth observed that was significantly different from either the AEAC- or the VEGF-Trap-alone groups. The efficacy of the combination was also reflected in an 85% reduction in the size of the A549 tumors from the combination group, when they were weighed at the end of the experiment (Table 1B).
Table 1.
| A. Tumor xenograft | Mean Tumor Growth Inhibition (% reduction compared to control) | |||
|---|---|---|---|---|
| AEAC | VEGF-Trap | AEAC + VEGF-Trap | ||
| PANC-1 (pancreatic cancer) | 38% ± 4.7* | 31% ± 8.1* | 80% ± 4.5** | |
| BXPC-3 (pancreatic cancer) | -4.4% ± 8.0 | 57% ± 1.8* | 79% ± 2.5** | |
| A549 (NSCLC) | 50% ± 5.4* | 25 ± 18* | 86% ± 8.2** | |
| B. Tumor xenograft | Tumor weight (mg) at conclusion of experiment | |||
| Control | AEAC | VEGF-Trap | AEAC + VEGF-Trap | |
| PANC-1 | 186 ± 20 | 85 ± 29* | 102 ± 16* | 68 ± 13* |
| BXPC-3 | 294 ± 40 | 304 ± 139 | 79 ± 15 | 57 ± 20* |
| A549 | 527 ± 77 | 329 ± 24* | 114 ± 14* | 45 ± 24** |
A. SC tumors were measured twice weekly, and tumor volumes calculated as (length × width2)/2. Mean tumor growth inhibition was calculated from tumor volume measurements determined at time points during drug treatment, using the following formula (50): % inhibition = [1- (VTx / VCx) / 1- VCx] × 100, where VTx equals the tumor volume in drug-treated mice on day × expressed as ratios of the tumor volumes of the same mice on day 0, and VCx equals the tumor volumes in control mice on day × expressed as ratios of the tumor volumes of the same mice on day 0.
B. Tumors were excised at the completion of the experiments, dissected from surrounding connective tissue, and weighed.
Values are means ± SEM, n=5.
Indicates significantly different from control, p< 0.05;
Indicates significantly different from control and from single-agent AEAC or VEGF-Trap, p< 0.05 (ANOVA with Tukey's multiple comparisons test).
Experiments next utilized two human pancreatic cancer xenografts, PANC-1 and BxPC-3. Drug therapy was the same as for the A549 experiments, beginning when tumors reached an average size of 75-100 mm3, and continuing for an additional 4 weeks. AEAC and the VEGF-Trap had equivalent effects on PANC-1 tumors, inhibiting tumor growth by 38% and 31%, respectively (figure 3A and Table 1A). Similar to the A549 tumors, the drug combination produced an approximately additive tumor growth inhibition of 80%, which was significantly different from the AEAC- and the VEGF-Trap-alone groups. In contrast to the other two tumor models, AEAC did not affect BxPC-3 tumor growth, although interestingly, it did significantly enhance the anti-tumor effect of the VEGF-Trap (79% growth inhibition for the combination versus 57% for the VEGF-Trap alone; figure 3B and Table 1). In all cases, the inhibitory effects of AEAC and VEGF-Trap were confirmed when the individual tumors were weighed at the completion of the experiments (Table 1B).
Figure 3. AEAC and the VEGF-Trap inhibit pancreatic cancer xenograft growth in nude mice.

Mice were inoculated sc with 2 × 106 PANC-1 (panel A) or BxPC-3 (panel B) pancreatic cancer cells. Tumors were allowed to grow until they reached an average size of approx. 75-100 mm3 at which point they were divided into groups of 5 mice, and therapy was begun. Mice were treated with vehicle (○); AEAC, oral gavage at 50 mg/kg (△) once daily for 5 days each week; VEGF-Trap, 10 mg/kg sc twice weekly (▽); or the combination of AEAC + VEGF-Trap (●). Drug therapy was begun on day 1, and was continued for the duration of the experiment. Tumor size was measured twice weekly. Tumor volumes were calculated as (l × w2) / 2, and are expressed relative to the tumor size at the start of therapy.
* Indicates significantly different from control, p< 0.05.
** Indicates significantly different from control and from single-agent AEAC or VEGF-Trap, p< 0.05 (ANOVA with Tukey's multiple comparisons test).
Collectively, these in vivo data suggest that AEAC inhibited selected xenograft tumor growth, and importantly, it also had an additive inhibitory effect on these models when coadministered with the VEGF-Trap.
AEAC had no effect on tumor cell growth in vitro
To evaluate the possibility that the antitumor activity of AEAC could be due to a direct effect on the tumor cells, the growth of A549, PANC-1 and BxPC-3 cells was assessed in vitro after drug treatment for 72 hrs. AEAC had no effect on cell proliferation; at the highest concentration tested, 100 μM, inhibition was less than 10% (data not shown).
Changes in TP expression in human tumor xenografts
TP expression was examined in the excised tumors by immunohistochemistry and by Western blotting (figure 4). TP was most prominently expressed in the tumor cells, with expression also occasionally noted in stromal cells (figure 4A). Shown in figure 4B are whole cell protein extracts from the cancer cells grown in tissue culture (lane 1), or from individual tumors (labeled 1, 2 or 3) from mice treated as in figure 3 with vehicle, AEAC, VEGF-Trap, or the combination of AEAC+VEGF-Trap. TP expression was equal to or higher in all the in vivo tumors compared to the same tumor cells in tissue culture. This may have been due to the upregulation of TP expression that we and others have shown to occur in response to a variety of cytokines, including TNFα, interferon, and IL-8 (reviewed in 5). However, there was a large degree of heterogeneity observed in TP expression among the individual tumors, preventing any conclusions from being drawn regarding the relationship between the level of TP expression and response to therapy.
Fig. 4. Expression of TP in human tumor xenografts.
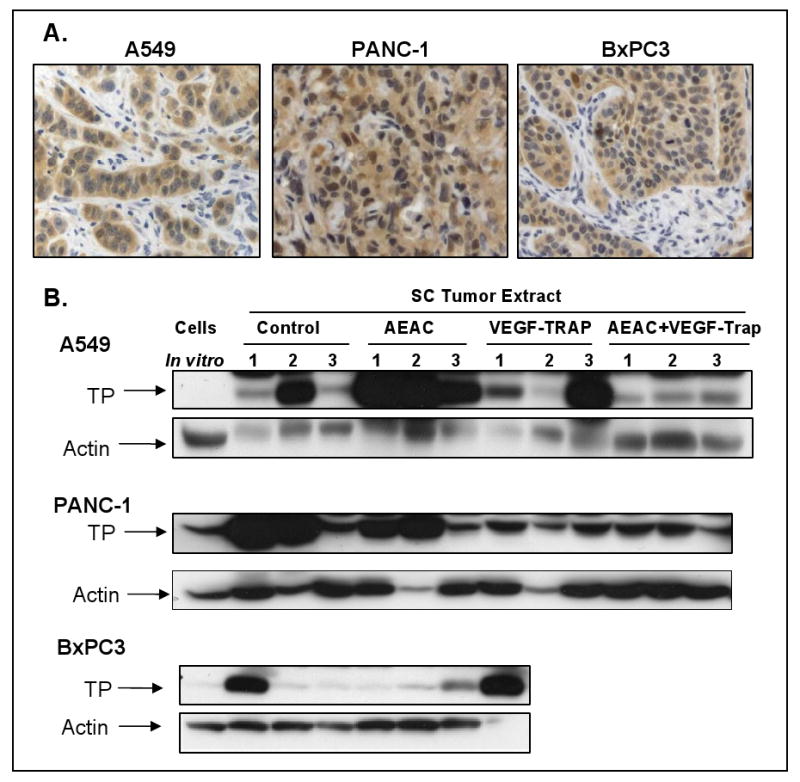
Tumors were removed at the end of the experiments shown in figure 3, and analyzed for TP expression by IHC (4A) or Western blots (4B). TP expression from the tissue culture cells used to inoculate the mice are shown in the first lane of 4B, and expression in individual tumors, obtained from mice treated as indicated, are shown in the remaining lanes.
Using an in vitro assay, AEAC was found to inhibit TP activity in extracts from the A549 tumors (supplemental figure 2).
AEAC and VEGF-Trap reduced tumor microvessel density
The effect of AEAC and the VEGF-Trap on microvessel density was examined in the tumors, as a measure of their anti-angiogenic actions (figure 5A). Blood vessels in 10 μm-thick histological sections were immunohistochemically stained with CD-34, an endothelial cell marker commonly used for microvessel quantification, and blood vessel density was quantitated by analysis with Image J (supplemental figure 3). Both AEAC and the VEGF-Trap had anti-angiogenic activity, which correlated with their anti-tumor actions (figure 4 and supplemental figure 3). Thus AEAC at 50 mg/kg significantly reduced microvessel density in the A549 and PANC-1 tumors but not in the BxPC-3 tumors, while VEGF-trap was active in all three xenografts. More importantly, the combination therapy of AEAC and VEGF-Trap significantly reduced the tumor MVD compared with control in all three models, and there was also a significant difference for the combination when compared to the VEGF-Trap alone for the PANC-1 tumors (figure 5A).
Fig. 5. AEAC and the VEGF-Trap reduce microvessel density and macrophage infiltration in tumors.

Histological sections in tumors from mice treated as described in figures 2 and 3 were immunohistochemically stained for endothelial cells (A) or macrophages (B). For each tumor, images in 3 high-powered microscope fields were analyzed for total vessel area (pixels/hpf) using Image J. Data are means ± SEM for 5 mice per group.
* Indicates significantly different from control, ANOVA with Tukey's Multiple Comparison test or Dunnett's test, p < 0.05.
** Indicates significantly different from both control and from VEGF-Trap alone, p < 0.05.
The VEGF-Trap reduced macrophage infiltration into xenograft tumors
TP expression appeared to be lower in tumors from VEGF-Trap-treated mice (i.e. both the VEGF-Trap alone and the AEAC+VEGF-Trap groups), when compared to the control and AEAC-alone groups (figure 4B). Since TP is highly expressed in macrophages as well as in tumor epithelial cells, a reduction in tumor-associated macrophages could have contributed to a reduction in tumor TP. As shown in supplemental figure 4 and figure 5B, for the A549 and PANC-1 tumors, both the VEGF-Trap alone and combination of AEAC and VEGF-Trap significantly inhibited macrophage infiltration into the tumor tissues. Interestingly, the VEGF-Trap did not cause a reduction in macrophages in the BxPC-3 tumors. While tumors from AEAC-alone treated mice had a reduction in macrophage infiltration, it was not statistically significant, and in fact, a significant increase in macrophage infiltration was seen in the BxPC-3 tumors.
Discussion
The angiogenic factor TP is overexpressed in a wide range of human solid tumors, often at levels 10-fold higher than in adjacent uninvolved tissue (5, 23). The degree to which TP expression is elevated varies widely, however, even among those tumors of the same histological type; thus overexpression of TP is not an inevitable characteristic of solid tumor formation. Rather most studies have found a correlation between a high level of TP expression and increased tumor microvessel density, increased tumor invasion and metastasis, and shorter patient survival time (5). TP, VEGF and bFGF expression were examined in the same specimens in some of these studies. In a study of 96 patients with colorectal cancer, TP was expressed in 83% of the patients' tumors, and the level of expression was significantly correlated with vessel counts, but not with the level of VEGF expression (24). In a study of 104 human non-small cell lung cancer patients, high TP expression was seen in 32% of the patient tumors and was associated with increased vascular density (25). VEGF was highly expressed in 47% of the NSCLC patient tumors and was likewise correlated with vessel counts. Linear regression analysis revealed only a weak positive correlation between TP and VEGF expression in the lung cancers. TP was overexpressed in 67% of 384 pancreatic cancer specimens, when compared either to adjacent uninvolved pancreatic tissue or to tissue from benign pancreatic diseases. Overexpression of VEGF and bFGF were observed in 54% and 58%, respectively, of the pancreatic cancer specimens, and there was a significant correlation reported for the co-expression of TP and VEGF (26-29). In addition to elevated expression in the pancreatic tumor cells, 40-50% of patient specimens had elevated TP expression in adjacent stromal cells (macrophages, lymphocytes and/or fibroblasts) (26, 28, 30). Overall the conclusion from clinical studies in which both TP and VEGF have been evaluated was that TP is an independent prognostic marker of tumor aggressiveness, and may be the predominant angiogenic factor in tumors that express low or no detectable VEGF.
TP-expressing cells mediate endothelial cell migration via the intracellular metabolism of thymidine to thymine and dR-1-P, the intracellular conversion of dR-1-P to 2dR, and the subsequent extracellular release of 2dR, which then forms a concentration and chemotactic gradient (31). We have previously shown that 2dR has direct effects on cell signaling pathways and cell migration in endothelial cells (32). State-of-the-art proteomics also recently identified TP as a key regulator of the angiogenic potential of endothelial progenitor cells in vitro (33). These critical studies provided the basis for the hypothesis that small molecule inhibitors directed at the catalytic site of TP would have anti-angiogenic and antitumor therapeutic activities.
A large number of TP inhibitors have been evaluated in vitro since the first compounds were synthesized over 40 years ago. A majority of these were analogs of uracil, deoxyuridine, thymidine, or related acyclonucleosides (34). Few, however, have been found to inhibit TP activity with IC50s less than 1 μM, the exceptions being an aminoimidazolyl-methyluracil analog, an iminopyrrolidinyl-methyluracil derivative called TPI, and AEAC (19, 35, 36). Using a different strategy, a bicyclic pyrimidine nucleoside multisubstrate inhibitor (i.e. binds to both the nucleoside and phosphate binding sites on the enzyme) has also been reported to inhibit human TP with nanomolar activity (37). Thus there are only a small number of TP inhibitors with sufficient potency to be considered for use in vivo, and to date, only TPI has been reported to have anti-tumor activity in preclinical models (38, 39). In those studies, TPI was shown to decrease the growth rate, increase the apoptotic index, reduce the microvessel density and suppress liver metastases of KB (HeLa) cells in mouse xenografts. Interestingly, activity was seen only with tumors from KB cells that had been transfected with a TP cDNA and which overexpressed TP protein by 100-fold. Thus our studies are the first to demonstrate activity of a TP inhibitor in tumors from cells that have not been manipulated to overexpress TP, and also in tumors which are representative of human cancers that are often refractory to other chemotherapeutic agents.
Our experiments combined AEAC with the VEGF-Trap, a novel, soluble VEGF decoy receptor protein, which is comprised of fragments of the VEGF receptors VEGFR-1 (flt1) and VEGFR-2 (flk1, KDR) (40-41). The VEGF-Trap has shown substantial anti-angiogenic and anti-tumor activity in preclinical models (including in pancreatic cancer), and is currently in Phase III clinical trials for patients with NSCLC, pancreatic, prostate and colorectal cancers (40-41). In our studies, we found that AEAC had antitumor activity in pancreatic cancer and NSCLC xenografts, and more importantly, it significantly increased the anti-tumor activity of the VEGF-Trap in two different pancreatic cancers and in a lung cancer xenograft. For these studies, AEAC was administered at 50 mg/kg/day. In other experiments, daily administration of AEAC at doses up to 100 mg/kg had no effect on body weight nor did it produce any apparent acute toxic effects on mice (data not shown). Since VEGF inhibitors are now approved for use in some human solid tumors and are being widely studied in others, it is likely that there will be future opportunities to conduct clinical trials which add a relatively non-toxic, orally available inhibitor of a second angiogenic pathway. While the present studies do not directly demonstrate that AEAC is acting via an effect on TP, we have previously shown that AEAC did not inhibit VEGF-induced endothelial cell migration at concentrations that completely inhibited TP-induced endothelial cell migration (19). Since it is possible that AEAC could have additional actions in vivo, mechanisms of AEAC action that are not directly due to TP inhibition cannot be ruled out by the current studies.
In human colon and other GI tumors, TP overexpression occurred more often in tumor-associated macrophages and other stromal cells, when compared to expression in the colon cancer epithelial cells (24). High levels of expression of TP in tumor-associated macrophages have also been observed in human breast, prostate, lung and brain tumors (42-46). These findings suggest that TP in tumor-associated macrophages may play a more direct role in tumor angiogenesis, and it has been hypothesized that tumors cells can amplify their own angiogenic activity by recruiting or activating macrophages, which then express high levels of angiogenic factors (47). The possibility that overexpression of TP in certain malignancies is a consequence of a host response suggests that TP-directed therapy may have a broad applicability. Thus anti-TP therapy could be useful even in instances where the tumor epithelial cells themselves are not over-expressing TP. Interestingly, we found that macrophage infiltration was substantially reduced in tumors from mice treated with the VEGF-Trap. This confirms a recent report which showed that tumor-associated macrophages express the VEGF receptor-2, and that selective inhibition of VEGFR-2 inhibits recruitment of macrophages into pancreatic tumors in vivo (48). The reduced number of macrophages in the tumors from the VEGF-Trap treated mice could also have contributed to the reduced levels of TP expression observed in these tumors.
In addition to contributing to angiogenesis in tumors, TP may play a pro-angiogenic role in inflammatory and other non-neoplastic pathologies, as well as in normal physiology (5). Studies have also shown that carcinoma cells that were transfected with and overexpressed TP were more invasive and metastatic in vitro and in vivo, than were control-transfected cells (16, 49-50). While our studies with AEAC in sc tumor xenografts were not designed to detect drug effects on tumor invasion and metastasis, data showing that this compound also had direct effects on cancer cells in vivo would support the continuing evaluation of this novel drug.
Supplementary Material
Acknowledgments
Grant support: NCI/NIH grants RO1 CA98456 and RO1 CA89352 (E.L. Schwartz).
Footnotes
Translational Relevance: Although angiogenesis inhibitors that target the VEGF pathway have a broad spectrum of activity against human solid tumors, the percentage of patients showing significant responses to these drugs in clinical trials has been relatively small. It is likely that angiogenesis inhibitors which target a single angiogenic molecule or pathway will not be sufficient to treat the spectrum of heterogeneous tumors occurring clinically. The angiogenic factor TP is overexpressed in a wide range of human solid tumors, often at levels 10-fold higher than in adjacent uninvolved tissue, and high levels of TP have been correlated with increased tumor microvessel density, increased tumor invasion and metastasis, and shorter patient survival time. TP stimulates endothelial cell migration and is a key regulator of endothelial progenitor cells in culture. In this report, we demonstrate anti-angiogenic and antitumor activities for a small molecule inhibitor of TP in vivo, and suggest that it or related compounds could be used clinically to complement the therapeutic activity of VEGF inhibitors.
References
- 1.Kowanetz M, Ferrara N. Vascular endothelial growth factor signaling pathways: therapeutic perspective. Clin Cancer Res. 2006;12:5018–22. doi: 10.1158/1078-0432.CCR-06-1520. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931–4. [PubMed] [Google Scholar]
- 3.Parangi S, O'Reilly M, Christofori G, Holmgren L, Grosfeld J, Folkman J, Hanahan D. Antiangiogenic therapy of transgenic mice impairs de novo tumor growth. Proc Natl Acad Sci USA. 1996;93:2002–7. doi: 10.1073/pnas.93.5.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Folkman J. What is the role of thymidine phosphorylase in tumor angiogenesis? J Natl Cancer Inst. 1996;88:1091–2. doi: 10.1093/jnci/88.16.1091. [DOI] [PubMed] [Google Scholar]
- 5.Liekens S, Bronckaers A, Pérez-Pérez MJ, Balzarini J. Targeting platelet-derived endothelial cell growth factor/ thymidine phosphorylase for cancer therapy. Biochem Pharmacol. 2007;74:1555–67. doi: 10.1016/j.bcp.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 6.Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–92. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- 7.Spinazzola A, Marti R, Nishino I, Andreu AL, Naini A, Tadesse S, Pela I, Zammarchi E, Donati MA, Oliver JA, Hirano M. Altered thymidine metabolism due to defects of thymidine phosphorylase. J Biol Chem. 2002;277:4128–33. doi: 10.1074/jbc.M111028200. [DOI] [PubMed] [Google Scholar]
- 8.Furukawa T, Yoshimura A, Sumizawa T, Haraguchi M, Akiyama SI, Fukui K, Ishizawa M, Yamada Y. Angiogenic factor. Nature. 1992;356:668. doi: 10.1038/356668a0. [DOI] [PubMed] [Google Scholar]
- 9.Ishikawa F, Miyazono K, Hellman U, Drexler H, Wernstedt C, Usuki K, Takaku F, Risau W, Heldin CH. Identification of angiogenic activity and the cloning and expression of platelet-derived endothelial cell growth factor. Nature. 1989;338:557–62. doi: 10.1038/338557a0. [DOI] [PubMed] [Google Scholar]
- 10.Moghaddam A, Bicknell R. Expression of PD-ECGF factor in E. coli and confirmation of its thymidine phosphorylase activity. Biochemistry. 1992;31:12141–6. doi: 10.1021/bi00163a024. [DOI] [PubMed] [Google Scholar]
- 11.Sumizawa T, Furukawa T, Haraguchi M, Yoshimura A, Takeysu A, Ishizawa M, Yamada Y, Akiyama SI. Thymidine phosphorylase activity associated with platelet-derived endothelial cell growth factor. J Biochem. 1993;114:9–14. doi: 10.1093/oxfordjournals.jbchem.a124146. [DOI] [PubMed] [Google Scholar]
- 12.Finnis C, Dodsworth N, Pollitt CE, Carr G, Sleep D. Thymidine phosphorylase activity of platelet-derived endothelial cell growth factor is responsible for endothelial cell mitogenicity. Eur J Biochem. 1993;212:201–10. doi: 10.1111/j.1432-1033.1993.tb17651.x. [DOI] [PubMed] [Google Scholar]
- 13.Moghaddam A, Zhang HT, Fan TPD, Hu DE, Lees VC, Turley H, Fox SB, Gatter KC, Harris AL, Bicknell R. Thymidine phosphorylase is angiogenic and promotes tumor growth. Proc Nat Acad Sci USA. 1995;92:998–1002. doi: 10.1073/pnas.92.4.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyadera K, Sumizawa T, Haraguchi M, Yoshida H, Konstanty W, Yamada Y, Akiyama S. Role of thymidine phosphorylase activity in the angiogenic effect of platelet-derived endothelial cell growth factor/thymidine phosphorylase. Cancer Res. 1995;55:1687–90. [PubMed] [Google Scholar]
- 15.Matsushita S, Nitanda T, Furukawa T, Sumizawa T, Tani A, Nishimoto K, Akiba S, Miyadera K, Fukushima M, Yamada Y, Yoshida H, Kanzaki T, Akiyama S. The effect of a thymidine phosphorylase inhibitor on angiogenesis and apoptosis in tumors. Cancer Res. 1999;59:1911–6. [PubMed] [Google Scholar]
- 16.Nakajima Y, Gotanda T, Uchimiya H, Furukawa T, Haraguchi M, Ikeda R, Sumizawa T, Yoshida H, Akiyama S. Inhibition of metastasis of tumor cells overexpressing thymidine phosphorylase by 2-deoxy-L-ribose. Cancer Res. 2004;64:1794–801. doi: 10.1158/0008-5472.can-03-2597. [DOI] [PubMed] [Google Scholar]
- 17.Li W, Tanaka K, Ihaya A, Fujibayashi Y, Takamatsu S, Morioka K, Sasaki M, Uesaka T, Kimura T, Yamada N, Tsuda T, Chiba Y. Gene therapy for chronic myocardial ischemia using platelet-derived endothelial cell growth factor in dogs. Am J Physiol Heart Circ Physiol. 2005:H408–415. doi: 10.1152/ajpheart.00176.2004. [DOI] [PubMed] [Google Scholar]
- 18.Haraguchi M, Miyadera K, Uemura K, Sumizawa T, Furukawa T, Yamada K, Akiyama SI, Yamada Y. Angiogenic activity of enzymes. Nature. 1992;368:198–9. doi: 10.1038/368198a0. [DOI] [PubMed] [Google Scholar]
- 19.Klein RS, Lim T, Lenzi M, Hotchkiss KA, Schwartz EL. Novel 6-substituted uracil analogs as inhibitors of the angiogenic actions of thymidine phosphorylase. Biochem Pharm. 2001;62:1257–63. doi: 10.1016/s0006-2952(01)00783-3. [DOI] [PubMed] [Google Scholar]
- 20.Boussat S, Eddahibi S, Coste A, Fataccioli V, Gouge M, Housset B, Adnot S, Maitre B. Expression and regulation of vascular endothelial growth factor in human pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2000;279:L371–8. doi: 10.1152/ajplung.2000.279.2.L371. [DOI] [PubMed] [Google Scholar]
- 21.Itakura J, Ishiwata T, Friess H, Fujii H, Matsumoto Y, Büchler MW, Korc M. Enhanced expression of vascular endothelial growth factor in human pancreatic cancer correlates with local disease progression. Clin Cancer Res. 1997;3:1309–16. [PubMed] [Google Scholar]
- 22.Keyes KA, Mann L, Teicher B, Alvarez E. Site-dependent angiogenic cytokine production in human tumor xenografts. Cytokine. 2003;21:98–104. doi: 10.1016/s1043-4666(03)00015-2. [DOI] [PubMed] [Google Scholar]
- 23.Takebayashi Y, Yamada K, Miyadera K, Sumizawa T, Furukawa T, Kinoshita F, Aoki D, Okumura H, Yamada Y, Akiyama S, Aikou T. The activity and expression of thymidine phosphorylase in human solid tumors. Eur J Cancer. 1996;32A:1227–32. doi: 10.1016/0959-8049(96)00061-5. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi Y, Bucana CD, Liu W, Yoneda J, Kitadai Y, Cleary KR, Ellis LM. Platelet-derived endothelial cell growth factor in human colon cancer angiogenesis: role of infiltrating cells. J Natl Cancer Inst. 1996;88:1146–51. doi: 10.1093/jnci/88.16.1146. [DOI] [PubMed] [Google Scholar]
- 25.O'Byrne KJ, Koukourakis MI, Giatromanolaki A, Cox G, Turley H, Steward WP, Gatter K, Harris AL. Vascular endothelial growth factor, platelet-derived endothelial cell growth factor and angiogenesis in non-small-cell lung cancer. Br J Cancer. 2000;82:1427–32. doi: 10.1054/bjoc.1999.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujimoto K, Hosotani R, Wada M, Lee JU, Koshiba T, Miyamoto Y, Tsuji S, Nakajima S, Doi R, Imamura M. Expression of two angiogenic factors, vascular endothelial growth factor and platelet-derived endothelial cell growth factor in human pancreatic cancer, and its relationship to angiogenesis. Eur J Cancer. 1998;34:1439–47. doi: 10.1016/s0959-8049(98)00069-0. [DOI] [PubMed] [Google Scholar]
- 27.Ikeda N, Adachi M, Taki T, Huang C, Hashida H, Takabayashi A, Sho M, Nakajima Y, Kanehiro H, Hisanaga M, Nakano H, Miyake M. Prognostic significance of angiogenesis in human pancreatic cancer. Br J Cancer. 1999;79:1553–63. doi: 10.1038/sj.bjc.6690248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujioka S, Yoshida K, Yanagisawa S, Kawakami M, Aoki T, Yamazaki Y. Angiogenesis in pancreatic carcinoma. Thymidine phosphorylase expression in stromal cells and intratumoral microvessel density as independent predictors of overall and relapse-free survival. Cancer. 2001;92:1788–97. doi: 10.1002/1097-0142(20011001)92:7<1788::aid-cncr1695>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 29.Kuwahara K, Sasaki T, Kuwada Y, Murakami M, Yamasaki S, Chayama K. Expressions of angiogenic factors in pancreatic ductal carcinoma: a correlative study with clinicopathological parameters and patient survival. Pancreas. 2003;26:344–9. doi: 10.1097/00006676-200305000-00006. [DOI] [PubMed] [Google Scholar]
- 30.Miyake K, Imura S, Yoshizumi T, Ikemoto T, Morine Y, Shimada M. Role of thymidine phosphorylase and orotate phosphoribosyltransferase mRNA expression and its ratio to dihydropyrimidine dehydrogenase in the prognosis and clinicopathological features of patients with pancreatic cancer. Int J Clin Oncol. 2007;12:111–9. doi: 10.1007/s10147-006-0634-x. [DOI] [PubMed] [Google Scholar]
- 31.Hotchkiss KA, Ashton AW, Klein RS, Lenzi ML, Zhu GH, Schwartz EL. Mechanisms by which tumor cells and monocytes expressing the angiogenic factor thymidine phosphorylase mediate human endothelial cell migration. Cancer Res. 2003;63:527–33. [PubMed] [Google Scholar]
- 32.Hotchkiss KA, Ashton AW, Schwartz EL. The angiogenic factors thymidine phosphorylase and 2-deoxyribose stimulate human endothelial cell migration by activation of integrins α5β1 and αVβ3. J Biol Chemistry. 2003;278:19272–9. doi: 10.1074/jbc.M212670200. [DOI] [PubMed] [Google Scholar]
- 33.Pula G, Mayr U, Evans C, Prokopi M, Vara DS, Yin X, Astroulakis Z, Xiao Q, Hill J, Xu Q, Mayr M. Proteomics identifies thymidine phosphorylase as a key regulator of the angiogenic potential of colony-forming units and endothelial progenitor cell cultures. Circulation Res. 2009;104:32–40. doi: 10.1161/CIRCRESAHA.108.182261. [DOI] [PubMed] [Google Scholar]
- 34.Baker BR, Kelley JL. Irreversible enzyme inhibitors. 188. Inhibition of mammalian thymidine phosphorylase. J Med Chem. 1971;14:812–6. doi: 10.1021/jm00291a009. [DOI] [PubMed] [Google Scholar]
- 35.Reigan P, Edwards PN, Gbaj A, Cole C, Barry ST, Page KM, Ashton SE, Luke RW, Douglas KT, Stratford IJ, Jaffar M, Bryce RA, Freeman S. Aminoimidazolylmethyluracil analogues as potent inhibitors of thymidine phosphorylase and their bioreductive nitroimidazolyl prodrugs. J Med Chem. 2005;48:392–402. doi: 10.1021/jm049494r. [DOI] [PubMed] [Google Scholar]
- 36.Fukushima M, Suzuki N, Emura T, Yano S, Kazuno H, Tada Y, Yamada Y, Asao T. Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2′-deoxribonucleosides. Biochem Pharmacol. 2000;59:1227–36. doi: 10.1016/s0006-2952(00)00253-7. [DOI] [PubMed] [Google Scholar]
- 37.Allan AL, Gladstone PL, Price ML, Hopkins SA, Juarez JC, Doñate F, Ternansky RJ, Shaw DE, Ganem B, Li Y, Wang W, Ealick S. Synthesis and evaluation of multisubstrate bicyclic pyrimidine nucleoside inhibitors of human thymidine phosphorylase. J Med Chem. 2006;49:7807–15. doi: 10.1021/jm060428u. [DOI] [PubMed] [Google Scholar]
- 38.Matsushita S, Nitanda T, Furukawa T, Sumizawa T, Tani A, Nishimoto K, Akiba S, Miyadera K, Fukushima M, Yamada Y, Yoshida H, Kanzaki T, Akiyama S. The effect of a thymidine phosphorylase inhibitor on angiogenesis and apoptosis in tumors. Cancer Res. 1999;59:1911–16. [PubMed] [Google Scholar]
- 39.Takao S, Akiyama SI, Nakajo A, Yoh H, Kitazono M, Natsugoe S, Miyadera K, Fukushima M, Yamada Y, Aikou T. Suppression of metastasis by thymidine phosphorylase inhibitor. Cancer Res. 2000;60:5345–8. [PubMed] [Google Scholar]
- 40.Holash J, Davis S, Papadopoulos N, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci USA. 2002;99:11393–8. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fukasawa M, Korc M. Vascular endothelial growth factor-trap suppresses tumorigenicity of multiple pancreatic cancer cell lines. Clin Cancer Res. 2004;10:3327–32. doi: 10.1158/1078-0432.CCR-03-0820. [DOI] [PubMed] [Google Scholar]
- 42.Sivridis E, Giatromanolaki A, Papadopoulos I, Gatter KC, Harris AL, Koukourakis MI. Thymidine phosphorylase expression in normal, hyperplastic and neoplastic prostates: correlation with tumour associated macrophages, infiltrating lymphocytes, and angiogenesis. Br J Cancer. 2002;86:1465–71. doi: 10.1038/sj.bjc.6600281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toi M, Ueno T, Matsumoto H, Saji H, Funata N, Koike M, Tominaga T. Significance of thymidine phosphorylase as a marker of protumor monocytes in breast cancer. Clin Cancer Res. 1999;5:1131–7. [PubMed] [Google Scholar]
- 44.Koukourakis MI, Giatromanolaki A, Kakolyris S, O'Byrn KJ, Apostolikas N, Skarlatos J, Gatter KC, Harris AL. Different patterns of stromal and cancer cell thymidine phosphorylase reactivity in non-small cell lung cancer: Impact on tumor neoangiogenesis and survival. Br J Cancer. 1998;77:1696–1703. doi: 10.1038/bjc.1998.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee AHS, Dublin EA, Bobrow LG. Angiogenesis and expression of thymidine phosphorylase by inflammatory cells in ductal carcinoma in situ of the breast. J Pathol. 1999;187:285–90. doi: 10.1002/(SICI)1096-9896(199902)187:3<285::AID-PATH238>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 46.Yao Y, Kubota T, Sato K, Kitai R. Macrophage infiltration-associated thymidine phosphorylase expression correlates with increased microvessel density and poor prognosis in astrocytic tumors. Clin Cancer Res. 2001;7:4021–6. [PubMed] [Google Scholar]
- 47.Polverini PJ, Leibovich SJ. Induction of neovascularization in vivo and endothelial proliferation in vitro by tumor-associated macrophages. Lab Invest. 1984;51:635–642. [PubMed] [Google Scholar]
- 48.Dineen SP, Lynn KD, Holloway SE, Miller AF, Sullivan JP, Shames DS, Beck AW, Barnett CC, Fleming JB, Brekken RA. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008;68:4340–6. doi: 10.1158/0008-5472.CAN-07-6705. [DOI] [PubMed] [Google Scholar]
- 49.Jones A, Fujiyama C, Turner K, Cranston D, Williams K, Stratford I, Bicknell R, Harris AL. Role of thymidine phosphorylase in an in vitro model of human bladder cancer invasion. J Urol. 2002;167:1482–6. [PubMed] [Google Scholar]
- 50.Yu EJ, Lee Y, Rha SY, Kim TS, Chung HC, Oh BK, Yang WI, Noh SH, Jeung HC. Angiogenic factor thymidine phosphorylase increases cancer cell invasion activity in patients with gastric adenocarcinoma. Mol Cancer Res. 2008;6:1554–66. doi: 10.1158/1541-7786.MCR-08-0166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.