

Abstract
Endometrial cancer is the most common invasive gynecologic malignancy, yet molecular mechanisms and signaling pathways underlying its etiology and pathophysiology remain poorly characterized. We sought to define a functional role for the protein kinase C (PKC) isoform, PKCα, in an established cell model of endometrial adenocarcinoma. Ishikawa cells depleted of PKCα protein grew slower, formed fewer colonies in anchorage-independent growth assays and exhibited impaired xenograft tumor formation in nude mice. Consistent with impaired growth, PKCα knockdown increased levels of the cyclin dependent kinase (CDK) inhibitors p21Cip1/WAF1 (p21) and p27Kip1 (p27). Despite the absence of functional phosphatase and tensin homologue (PTEN) protein in Ishikawa cells, PKCα knockdown reduced Akt phosphorylation at serine 473 and concomitantly inhibited phosphorylation of the Akt target, glycogen synthase kinase-3β (GSK-3β). PKCα knockdown also resulted in decreased basal ERK phosphorylation and attenuated ERK activation following EGF stimulation. p21 and p27 expression was not increased by treatment of Ishikawa cells with ERK and Akt inhibitors, suggesting PKCα regulates CDK expression independently of Akt and ERK. Immunohistochemical analysis of grade 1 endometrioid adenocarcinoma revealed aberrant PKCα expression, with foci of elevated PKCα staining, not observed in normal endometrium. These studies demonstrate a critical role for PKCα signaling in endometrial tumorigenesis by regulating expression of CDK inhibitors p21 and p27 and activation of Akt and ERK dependent proliferative pathways. Thus, targeting PKCα may provide novel therapeutic options in endometrial tumors.
Keywords: PKC, endometrial cancer, Akt, p21, p27
Introduction
The protein kinase C (PKC) family of serine-threonine signaling kinases regulate cellular processes critical to malignant transformation including proliferation, apoptosis, invasion, metastasis, and angiogenesis; and thus have gained recognition as potential therapeutic targets for the treatment of various malignancies 1, 2. In various types of cancer cells, PKCα is associated with increased proliferation 3, survival 4, invasion and metastasis 5, and tumorigenicity 6. However, PKCα can also exert growth inhibitory functions 7, underscoring the importance of defining PKCα function in specific tissues.
Endometrial cancer is the fourth most common malignancy in women in the United States, resulting in 7400 deaths per year 8. Despite these figures, signal transduction pathways that underlie endometrial carcinogenesis are poorly understood. PKC activity has been implicated in proliferation of normal and endometrial cancer cells, but few studies have addressed isoform specific effects 9. Others have suggested a role for PKCα in growth of both normal endometrium and endometrial tumors 10, and we have shown that PKCα regulates cell survival and apoptosis in endometrial cancer cells 11. In other cell types, PKCα has been implicated in regulation of the PI3 kinase/Akt pathway 12 13 14 which is frequently activated in endometrial tumors 15. However, the role of PKCα signaling in endometrial carcinogenesis had not been determined.
In this report, we show that PKCα regulates endometrial cancer cell proliferation, anchorage independent growth and xenograft tumorigenesis, indicating a fundamental role for PKCα signaling in endometrial carcinogenesis. Consistent with these observations, we demonstrate that PKCα knockdown in endometrial cancer cells increases expression of CDK inhibitor proteins p21 and p27, and suppresses activation of the Akt and extracellular signal-regulated kinase (ERK) growth pathways.
Materials and methods
Cell lines and reagents
Ishikawa human endometrial adenocarcinoma cells 16 were a generous gift from Dr. K. K. Leslie (University of New Mexico, Albuquerque). Unless stated otherwise, cells were cultured in DMEM supplemented with 12.5% horse serum, 2.5% FBS, 10 units/ml penicillin, 10 μg/ml streptomycin, and 200 μM L-glutamine (Mediatech Inc, Manassas, VA). The Akt inhibitor Akti-1/2 (Akt inhibitor VIII), and MEK1/MEK2 inhibitor U0126 were from Calbiochem (San Diego, CA). For PKCα knockdown, DNA oligomers matching a previously published shRNA sequence 5′-CAAGGCTTCCAGTGCCAAG-3′ 10 designed to target human PKCα were annealed and inserted into the pSuper retroviral vector (Oligoengine Inc., Seattle, WA). A vector generating shRNA's 5′-CGTACGCGGAATACTTCGA-3′ targeting the luciferase (Luc) protein was used as a control. To generate retrovirus, PT67 RetroPack cells (Clontech) were transiently transfected with a 3:1 ratio of pSuper to pCL-Ampho (Imgenex; San Diego, CA) using Polyfect (Qiagen). Cells were infected with retrovirus-containing media harvested twice at 48 and 56 h post-transfection, supplemented with 8 μg/mL polybrene, then filtered using a 0.45 μm syringe filter, (Sigma, St. Louis, MO), and immediately added to plates of dividing Ishikawa cells. Selection of stably transduced cells began 48 h following infection using 2 μg/mL puromycin, and mixed pools of stable cells remained under puromycin selection for at least two weeks before use or selection of clones. Clonal cell lines were generated by pipetting a single cell under a microscope and injecting the cell into a 96-well dish containing puromycin media. Of 96 clones isolated from the PKCα shRNA mixed population, 25 remained viable to be screened for their level of PKCα expression. From these 25 clones, the three clones with the lowest level of PKCα expression were selected. From the Luc shRNA mixed population, 20 cells were originally isolated, with eight remaining viable and three clones randomly chosen from this cohort.
Western blot analysis
Detailed protocols for cell harvest and western blotting have been published 11. Antibodies used for immunoblotting included PKCα (sc-208), PKC βI (sc-209), PKCβII (sc-210), PKCγ (sc-211), p53 (sc-126), p57 (sc-8298), Pol II (sc-900) and secondary anti-rabbit (sc-2004) or anti-mouse (sc-2005) IgG-HRP antibodies obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for p21 (#2946), p27 (#2552), phospho-Akt S473 (#4051), total Akt (#9272), phospho-GSK3β (#9336), total GSK-3β (#9315), phospho-ERK (#9101), and total ERK (#9102) were obtained from Cell Signaling Technology, Inc. (Beverly, MA). Loading controls were assessed using monoclonal anti-β-actin (A5316) or anti-β-tubulin (T8535) antibodies (Sigma) and molecular weight estimated using Rainbow markers (Amersham Biosciences, Piscataway, NJ). Quantitation of band intensity was performed by densitometry using the ChemDoc imaging system and Quantity One software (v. 4.5.1; Bio-Rad, Hercules, CA).
Cell proliferation and cell cycle analysis
Subconfluent cultures were maintained in either 60 or 100 mm tissue culture dishes with media containing serum. Cell number was determined using a Vi-Cell Coulter Counter (Beckman-Coulter Inc., Fullerton, CA). The proportion of cells in G1, S, and G2/M phases of the cell cycle was determined by staining cells with Krishan's stain (0.1% NP-40, 10 mM sodium acetate, 0.5% w/v Propidium Iodide) and analyzing DNA content by flow cytometry using a Beckman Coulter Cytomics FC 500 and Modfit analysis software (Verity, Topsham, ME).
Colony formation assay
The growth area of a 35 mm tissue culture dish was coated with a 1% (w/v) mixture of agar and serum-containing medium. Once solidified, a second layer containing cells and 1% agar/media mixture was applied on top. The plates were incubated in a humidified chamber for 45 d before processing. To visualize colonies, plates were stained with 0.005% (w/v) crystal violet for 30 min, and then washed with distilled water to reduce background. Plates were photographed using a Gel-Doc imaging system (Bio-Rad) and colonies counted using the colony count feature in the Quantity-One Software package (v. 4.5.1; Bio-Rad).
Xenograft tumor formation assays
Intact, female athymic nu/nu mice between 5 and 6 weeks of age were obtained from Harlan Sprague-Dawley (Indianapolis, IN). Tumorigenicity of the three PKCα or Luc shRNA clones was assessed by subcutaneously injecting 10×106 cells bilaterally into five mice, so that a total n = 15 mice were used. Tumor measurement began one and a half weeks after injection and was conducted twice weekly with a digital calipers. Volume was calculated as ((1/2 length)(width2)). All procedures were approved by the University of Colorado Institutional Animal Care and Use Committee.
Cytoplasmic and nuclear fractionation
Reagents contained within the Nuclear Extract Kit (Active Motif, Carlsbad, CA) were used with minor modifications to the manufacturers protocol. Nuclei were pelleted by centrifugation at 1000 ×g for 1 min, and the cytoplasmic fraction was removed and centrifuged at 14,000 ×g for 15 min before removing supernatant to a fresh tube and adding Laemmli sample buffer (Bio-Rad). The pellets of intact nuclei were washed two additional times in hypotonic buffer to eliminate whole cells and cytoplasmic proteins. After the final wash, the provided detergent was added to the nuclei, followed by centrifugation at 14,000 ×g for 15 min. An aliquot of each fraction was retained for a protein assay using Bradford reagent (Bio-Rad) and an equal amount of protein from each fraction was resolved by SDS-PAGE.
Luciferase reporter assays
Creation of the p21-promoter luciferase (Luc) reporter construct has been described 17. The p27-Luc reporter plasmid, which contains -3568 to -12 bp of the p27 promoter 5′ fused to a Luc reporter, was a kind gift from T. Sakai (Kyoto Prefectual University of Medicine, Kyoto, Japan). Since clonal populations expressing our control vector (pSuper-Luc shRNA) would target the Luc reporter mRNA, we instead used Ishikawa clones stably transduced with pRev Tet-On vector (BD biosciences, San Jose, CA) that had been isolated under similar conditions. To compare p21 and p27 promoter activity in PKCα knockdown and pRev Tet-On clones, 2.0 × 105 cells per well were seeded into 12-well dishes and transiently transfected with 1.0 μg p21-Luc or p27-Luc reporter plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) per the manufacturers protocol. Transfection efficiency was determined by cotransfection of 0.5 μg plasmid encoding β-galactosidase under control of the CMV constitutive promoter. Luc and β-galactosidase assays were conducted as previously described 18.
Human endometrial specimens and immunohistochemistry
Twelve cases of grade 1, endometrioid type uterine endometrial adenocarcinoma and seven samples of normal cycling endometrium were obtained from the Department of Pathology at the University of Colorado Health Science Center under Colorado Multiple Institutional Review Board protocol number 00-1094. Tumor grade was determined according to the International Federation of Gynecology and Obstetrics (FIGO) criteria. Procedures for immunohistochemistry have been described 19, briefly, tissue was formalin-fixed, paraffin-embedded, sectioned into 5 μm slices, and blocked for endogenous peroxidase activity. Antigen retrieval was performed in citrate buffer (20 mmol/L, pH 6.0) for 10 minutes at 60°C. Sections were incubated with antibodies specific for PKCα (sc-8393; Santa Cruz) and stained using an indirect avidin biotin immunoperoxidase method on a Dako Autostainer. Negative controls (not shown) were run using an equivalent concentration of subclass-matched immunoglobulin G (BD Pharmingen, San Diego, CA) and specificity of staining was demonstrated by addition of PKCα blocking peptide (Santa Cruz). Sections were evaluated by blinded review by 4 independent observers.
Statistical analysis
Values shown in figures are given as the mean ± standard deviation or standard error of the mean. The data were analyzed using a paired Students T test. P values of <0.05 were considered significant.
Results
Knockdown of PKCα inhibits Ishikawa cell proliferation and colony formation
Ishikawa cells, derived from a well-differentiated endometrial adenocarcinoma 16, are considered one of the best characterized models of Type I, or sex steroid-dependent, endometrial cancers 20. To address the functional role of PKCα in endometrial cancer, we generated stable populations of Ishikawa cells with PKCα expression knocked down by short hairpin (sh) RNAs (Figure 1a). As shown in figure 1b, Ishikawa cells lacking PKCα protein grow slower than control cells expressing a control shRNA directed against luciferase (Luc). The reduced growth rate in the PKCα shRNA population was confirmed by flow cytometry, which indicated about a 10% increase in the percentage of non-dividing, or G0/G1 cells, in an unsynchronized population (Figure 1c). No significant differences in the fraction of viable cells were observed (data not shown).
Figure 1.
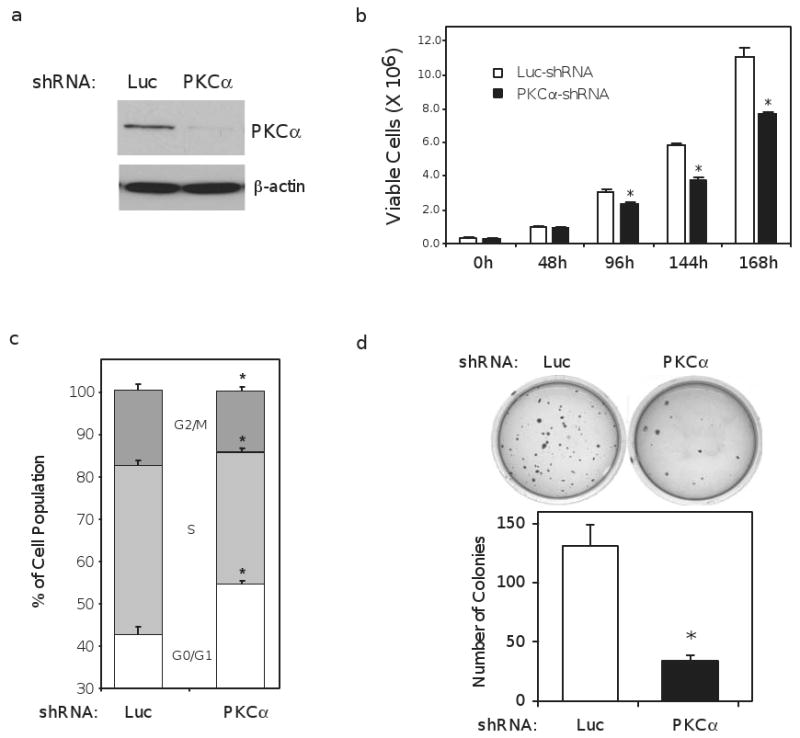
PKCα knockdown inhibits endometrial cancer cell proliferation and anchorage-independent growth. (a) Western blot depicting knockdown of PKCα expression in stable, mixed populations of Ishikawa cells expressing short hairpin (sh) RNA constructs targeting PKCα or luciferase (Luc). β-actin is included as loading control. (b) proliferation assay comparing the growth rate of Luc and PKCα shRNA cells. 50,000 cells were plated initially; with the first count (0h) conducted 24h later. The number of viable cells at each time point was determined using an automated Vi-Cell Coulter Counter (Beckman-Coulter Inc.). Growth profile is a single experiment and representative of three separate experiments. (c) Luc and PKCα shRNA cells were deprived of serum for 24h and the proportion of cells in the indicated phases of the cell cycle was determined by flow cytometry. Data represents an average calculated from three independent experiments. (d) anchorage-independent growth was assayed by plating 30,000 Ishikawa cells stably expressing a Luc or PKCα shRNA into soft agar and allowing growth for 45 days before staining with crystal violet and imaging. The number of colonies formed was determined using the colony counting feature in the Quantity One software package (BioRad). Bar graphs represent mean ± SEM; * denotes P < 0.01.
Anchorage-independent growth is characteristic of the transformed cell 21. To test if PKCα modulates anchorage-independent growth, PKCα and Luc shRNA cells were plated into soft agar colony formation assays. As shown in figure 1d, Ishikawa cells with the Luc shRNA generated approximately 4 times as many colonies as the PKCα shRNA cells, indicating a requirement for PKCα in anchorage-independent growth.
PKCα regulates tumor formation in nude mice
We next examined the effects of reduced PKCα expression on Ishikawa cell growth and tumorigenesis in vivo. For these studies, three Luc control and three PKCα shRNA clonal populations were established and their level of PKCα protein expression is compared in figure 2a. We also confirmed targeting specificity of the shRNA constructs by examining expression of other conventional PKC isoforms- PKCβI, PKCβII and PKCγ. As shown in figure 2a, expression of these closely related PKC isoforms was not decreased. A modest increase in PKCβII and PKCγ expression may reflect compensatory changes in response to knockdown of PKCα. We also conducted a cell cycle analysis to confirm that the clones reflected the growth phenotype observed in the mixed population, and indeed, the PKCα shRNA clones exhibited an increase of approximately 12% in the fraction of non-dividing cells in G0/G1 (Figure 2b), consistent with changes observed in the mixed population (Figure 1c).
Figure 2.
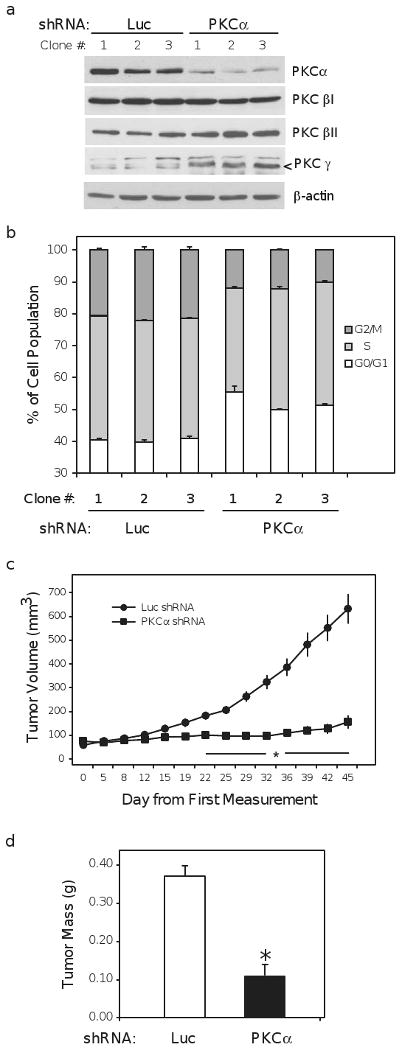
PKCα knockdown impairs tumorigenesis in vivo. (a) Ishikawa cell clones expressing luciferase (Luc) or PKCα shRNA constructs were isolated as described in materials and methods. Lysate from clonal populations was harvested after 24h of serum deprivation and probed for PKCα PKCβI, PKCβII, and PKCγ. β-actin is included as loading control and arrow (<) indicates the correct molecular weight band for PKCγ. (b) Luc and PKCα shRNA clones were deprived of serum for 24h and the proportion of cells in the indicated phases of the cell cycle was determined by flow cytometry. Data represent an average derived from two independent experiments. (c) Volume of xenograft tumors resulting from the subcutaneous injection of 10 million cells from Luc and PKCα shRNA clonal cell lines into athymic, nude mice. Each PKCα knockdown clone was injected contra-laterally to a Luc control in a total of 5 mice, thus data represent the combined average from n = 15 animals. (d) Average final weight of excised xenograft tumors assessed after the final dimension measurement. Bar graphs represent mean ± SEM; * denotes P < 0.01.
Tumorigenicity of the Luc and PKCα shRNA clonal populations was compared by injecting 10 ×106 cells from each clone subcutaneously on contra-lateral sides of five nude mice, for a total of (n = 15) mice. All Luc shRNA clones established a vigorous tumor in every recipient mouse. Conversely, PKCα shRNA clones formed significantly smaller tumors (Figure 2c) with some regressing entirely (n = 4) or showing no increase in size relative to their initial measurement (n = 4). Consistent with a reduction in tumor volume, the final mass of the tumors was also significantly reduced by PKCα knockdown (Figure 2d). This large disparity in tumor forming capacity between the Luc and PKCα shRNA clones clearly indicates that PKCα signaling pathways are important for tumor formation in vivo by endometrial cancer cells.
PKCα dependent expression of CDK inhibitors
Considering the reduced rate of proliferation and increased proportion of cells in G0/G1 observed in vitro (Figures 1b and 1c), we next examined expression of proteins inhibitory to cell cycle progression, the CDK inhibitors of the INK4 (p15, p16, p18, and p19) and CIP/KIP (p21, p27, and p57) families.
Western blotting for the various CDK inhibitors revealed a robust increase in expression of p21 and p27 protein in the PKCα shRNA clones (Figure 3a). Conversely, there was no change in expression of the other CIP/KIP family member, p57Kip2, or the INK4 protein, p15INK4B (Figure 3a). p16INK4A expression was undetectable (data not shown). Inhibition of cell cycle progression by p21 and p27 requires their localization to the nucleus 22. Therefore, we examined the sub-cellular distribution of p21 and p27 in both the PKCα and Luc shRNA clones. In Luc controls, all detectable p27 was localized to the nuclear fraction, and p21 was present in both nuclear and cytoplasmic fractions (Figure 3b). This pattern is consistent with a low level of nuclear-localized p21 and p27 being required to promote cyclin D and CDK 4 and 6 complex formation in G1-phase 22. In the PKCα knockdown clones, despite the significant increase in levels of p21 and p27, the relative proportions in nuclear and cytoplasmic fractions were equivalent to control. Thus, reduced PKCα did not alter p21 and p27 localization; however, abundant p21 and p27 expression leads to their accumulation in the nucleus, which may underlie the reduction in growth rate and tumorigenicity in these cells.
Figure 3.
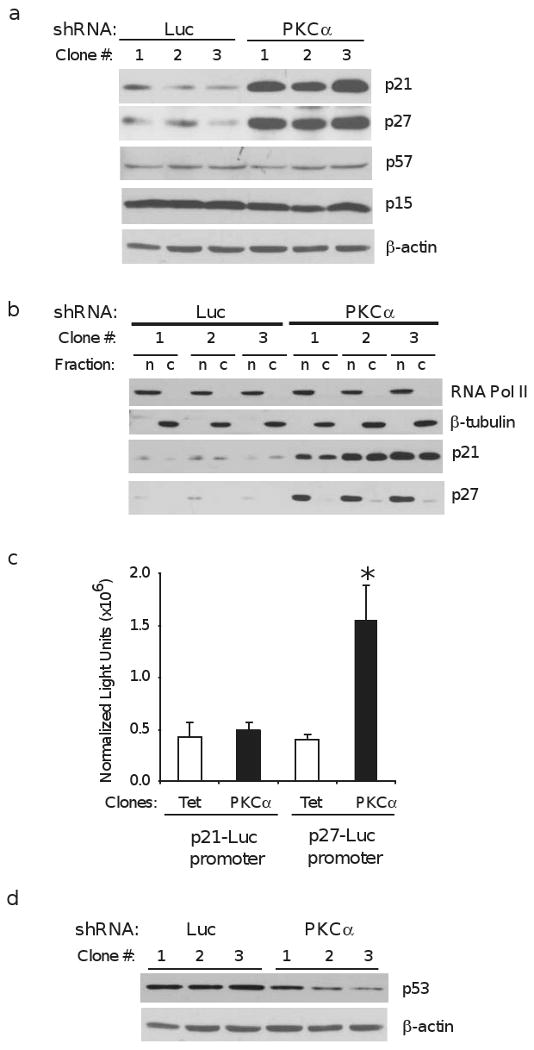
PKCα knockdown increases CDK inhibitors p21 and p27. (a) Western blot analysis of CDK inhibitor proteins p21, p27, p57, and p15 in Ishikawa cell clones expressing shRNA's directed against luciferase (Luc) or PKCα. Clones were deprived of serum growth factors for 24h before harvest; β-actin is included as loading control. (b) Luc and PKCα clones were deprived of serum for 24h before isolating nuclear and cytoplasmic fractions as described in materials and methods. RNA polymerase II (RNA Pol II) and β-tubulin are included as markers of nuclear (n) and cytoplasmic (c) fractions, respectively. (c) basal transcriptional activity of the p21 and p27 promoters was determined in PKCα shRNA clones and Ishikawa clones expressing the Tet-On transcription factor. Data are presented as luciferase-derived light units normalized to β-galactosidase activity and are mean ± SEM of three experiments conducted in triplicate, * - P < 0.05. (d) Western blots of Luc and PKCα shRNA clones probed for p53, a transcriptional activator of the p21 promoter. β-actin is loading control.
To determine the relative basal activity of the p27 and p21 promoters, the PKCα shRNA clones were compared with clones isolated in a similar fashion, but instead expressing the Tet-On transcription factor, since the Luc shRNA would target the luciferase reporters. As shown in figure 3c, no significant changes in activity of the p21 promoter were detected in response to PKCα knockdown. In contrast, p27 promoter activity was increased approximately 3-fold in the PKCα knockdown clones, suggesting that increased p27 transcription contributes to its increased expression. We also examined expression of the tumor suppressor protein, p53, an activator of the p21 promoter 23. Levels of p53 protein were decreased in the PKCα knockdown clones (Figure 3d), suggesting that increased p21 protein in the PKCα knockdown clones are not attributable to increased p53-dependent transcriptional activation.
PKCα modulates activation of ERK and AKT pathways
To further investigate a potential underlying mechanism that would explain the reduced proliferation and impaired tumorigenicity in the PKCα knockdown cells, we chose to examine the activation status of the Akt and ERK growth pathways, which are frequently dysregulated in endometrial and other cancers. As in a majority of low-grade endometrial adenocarcinoma's 24, 25, Ishikawa cells have an inactivating mutation in the phosphatase and tensin homologue (PTEN) tumor suppressor gene. Thus, the lipid kinase activity of phosphatidylinositol-3-OH kinase (PI3K), which is wild-type in these cells 25, is unopposed and leads to activation of the Akt oncogenic pathway. Accordingly, even under serum-deprived conditions, Luc clones showed high basal Akt phosphorylation at serine 473 (S473), the primary regulatory site targeted by the mTORC2 complex 26 (Figure 4a and b). Intriguingly, the PKCα knockdown clones showed a reduction in the level of basal Akt activation, as S473 phosphorylation was virtually undetectable in two clones. This reduction in Akt phosphorylation occurred despite an apparent increase in total Akt expression in the PKCα knockdown clones (Figure 4a). To substantiate diminished Akt activation, the same lysates were probed for glycogen synthase kinase-3β (GSK-3β) phosphorylated on serine 9 (S9), an Akt target site 27. Consistent with reduced Akt activity, the relative level of GSK-3β phosphorylation was proportionately decreased in the PKCα knockdown cells (Figure. 4a).
Figure 4.

PKCα knockdown inhibits Akt and ERK activation. (a) Ishikawa clones expressing shRNA constructs directed against luciferase (Luc) or PKCα were deprived of serum for 24h before cell harvest. Western blots were probed with phospho-specific antibodies to residues S473 of Akt (P-Akt (S473)) and S9 of GSK-3β (P-GSK-3β (S9)), and antibodies to total Akt and total GSK-3β as indicated. (b) Quantitation of P-AKT/total AKT, results are mean ± s.d. N=6,**p<0.002. (c) Luc and PKCα shRNA clones were cultured in 1% FBS for 24h before cell harvest. Western blots of cell lysates were probed with phospho-specific antibodies to ERK (P-ERK) and antibodies directed against total ERK. (d) Bands were quantitated and expressed as mean ± s.d. P-ERK/total ERK. N=6, **p <0.002 (e) Representative Western blots of P-ERK and total ERK in a mixed, non-clonal population of Ishikawa cells stably expressing Luc or PKCα shRNA's. Following 24h of serum deprivation, the cells were treated with 10 ng/mL epidermal growth factor (EGF) and harvested at the times indicated. (f) Quantitation of P-ERK/total ERK results are mean ± s.d. of 4 independent experiments, *p<0.05, **p<0.002.
Similar to Akt, the ERK signaling pathway is frequently upregulated in cancer 28, and PKCα has been shown in other systems to be an upstream activator of ERK 29. Western blotting for activated phosphorylated ERK (P-ERK) and total ERK protein in the clonal populations revealed that PKCα knockdown caused a modest decrease in levels of P-ERK and concomitant increase in the amount of total ERK, reflected in the decreased fraction of active kinase (P-ERK/total ERK) (Figure 4c and d). Since others have suggested that the ERK pathway is highly resilient to suppression and can quickly adapt to inhibitory pressures 30, the clonal isolates could have potentially undergone compensatory changes in ERK expression and activity during selection. To address this, we used early passage, mixed populations of Luc and PKCα knockdown cells to reexamine ERK activation under basal and stimulated conditions. As shown in figure 4e and f, PKCα knockdown reduced basal P-ERK in serum-deprived cells (0 min time point), and ERK phosphorylation following stimulation with epidermal growth factor (EGF) was substantially diminished. These results implicate PKCα as a modulator of growth factor dependent ERK activity in Ishikawa cells.
Expression, stability, and localization of p21 and p27 protein may be regulated by both Akt 31, 32 and ERK 30 pathways in other cell types. To address the possibility that increased p21 and p27 associated with PKCα knockdown is a consequence of reduced Akt and ERK activity, cells were treated with inhibitors of Akt (Akti-1/2), or the MEK 1/2 inhibitor (U0126), to block activation of ERK. Treatment of Ishikawa cells with these inhibitors specifically reduced levels of activated P-Akt and P-ERK without altering total kinase (Figure 5), demonstrating the efficacy of the inhibitors. Contrary to expectations, inhibition of Akt or ERK activity resulted in a decrease in p21 protein, while p27 was relatively unchanged (Figure 5). These results suggest that enhanced CDK inhibitor expression in PKCα knockdown cells is not directly attributable to reduced Akt and ERK activation and that PKCα regulation of p21 and p27 expression is mediated through Akt and ERK independent signaling pathways.
Figure 5.
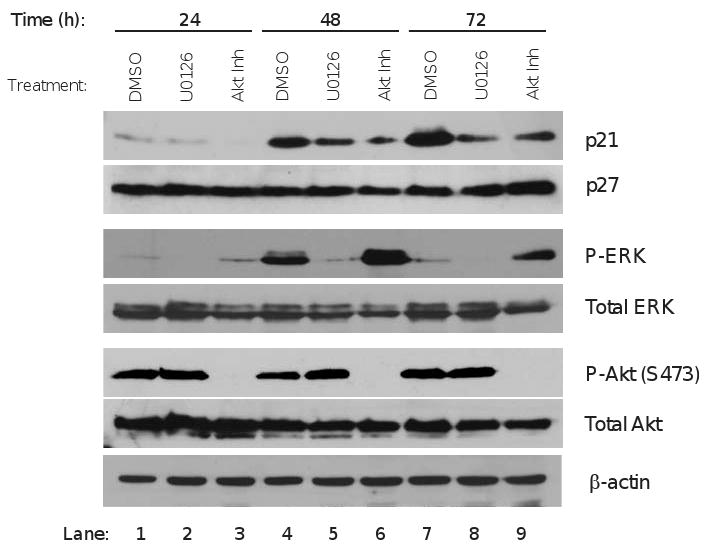
Effects of Akt and ERK on p21 and p27 expression. Ishikawa cells were serum starved for 8h before treatment with vehicle (DMSO); Akt inhibitor Akti-1/2 (5μM); or the MEK1/MEK2 inhibitor U0126 (5 nM) to block ERK activation. Cells were treated in medium containing 0.5% serum and vehicle or inhibitors were added fresh at 24 and 48h time points. Treated cells were harvested at 24, 48 and 72h and probed using antibodies to CDK inhibitors p21 and p27; phospho-ERK (P-ERK), Akt phosphorylated on S473 (P-Akt (S473)), total Akt and total ERK. β-actin is loading control. Blots shown are representative of two separate experiments.
Immunohistochemical analysis of PKCα expression in endometrial tumors
Given the observed role of PKCα in the regulation of growth of Ishikawa cells, we examined expression of PKCα in samples of normal endometrium and low grade tumors. Immunohistochemical staining of PKCα was performed on paraffin sections of normal endometrial tissue acquired during the proliferative (n = 4) or secretory (n = 3) phases of the menstrual cycle and grade 1 endometrioid adenocarcinomas (n = 12). In normal proliferative (Figure 6a) and secretory (Figure 6b) endometrium, PKCα was detected in both stroma and glandular epithelium, with epithelial staining typically concentrated at the lumen (Figure 6a). There was no consistent difference in PKCα expression between proliferative and secretory endometrial epithelium, whereas stromal staining was generally more pronounced in secretory endometrium (Figure 6b). PKCα staining was detected in all grade 1 endometrial adenocarcinomas; however, the pattern and intensity was highly variable within tumors and between different patients (Figure 6c). All tumors exhibited regions where PKCα expression was low or undetectable, despite staining in adjacent stroma (Figure 6c), and a subset of tumors showed uniform, high intensity staining throughout a majority of the section (Figure 6d). The majority of tumors exhibited staining intensities comparable to, or greater than normal tissue, with higher intensity PKCα staining most frequently observed in multiple discrete foci within continuous gland-like structures (Figure 6e and 6f). Considerable intratumoral variation in stromal staining was also observed, but maximal levels of stromal PKCα in tumors did not exceed those in normal stroma. Overall, in comparison to normal tissue, endometrial adenocarcinoma exhibited aberrant PKCα staining, consistent with the functional role of PKCα in Ishikawa cell proliferation and tumorigenesis.
Figure 6.
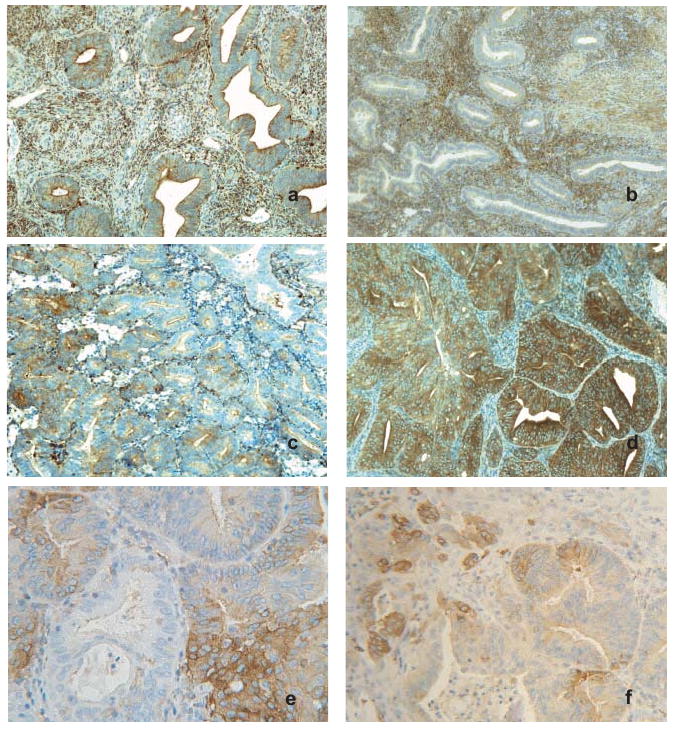
PKCα is abnormally expressed in endometrial tumors. Representative images of immunohistochemical staining for PKCα in normal proliferative (a) and secretory (b) endometrium. Staining in normal glandular epithelial cells was typically uniform and of low intensity; whereas endometrial stroma showed variable expression and was generally higher in stroma from secretory endometrium. Original magnification, ×100. (c) and (d), PKCα expression in representative grade 1 endometrioid adenocarcinomas. Original magnification, ×100. (c) typical PKCα staining in tumors was highly variable, with more intense staining frequently observed in discrete foci. (d) PKCα staining observed in a subset of tumors where high intensity expression occurred throughout the majority of the tissue section. (e) and (f), images from gland-like structures in endometrial tumors displaying focal variation in PKCα staining. Original magnification, ×250 and ×160, respectively.
Discussion
The functions of specific PKC isoforms are clearly dependent on cell type and context. Thus, PKCα has been shown to mediate either cell survival and proliferation 3 or growth inhibition and apoptosis 7. PKCα dependent signaling has been implicated in the pathogenesis of endometrial tumors 9, though the functional role of PKCα in endometrial cancers had not been investigated. Defining PKCα dependent responses is critical to provide a rational basis for potential therapeutic intervention using isoform specific inhibitors 1.
Our experiments clearly demonstrate a requirement for PKCα in xenograft tumor formation by Ishikawa endometrial adenocarcinoma cells (Figures 2c and d), consistent with inhibition of xenograft tumor growth by PKCα knockdown in stomach, lung, bladder, and colon cancer cell lines 6, 33, 34, and increased tumor size from MCF-7 breast cancer cells overexpressing PKCα 35. We had previously shown that PKCα is important for endometrial cancer cell survival 11, and these studies show PKCα signaling is important in endometrial cancer cell proliferation (Figure 1b and c) and anchorage-independent growth (Figure 1d). Our data are consistent with other studies demonstrating a small reduction in Ishikawa cell growth in response to transient knockdown of PKCα10; the larger responses we observed may reflect stable knockdown of the kinase. Collectively, these data indicate a functional requirement for PKCα in endometrial tumorigenesis.
Analysis of cell cycle regulators suggests that PKCα regulates endometrial cell proliferation through expression of the CDK inhibitor proteins, p21 and p27. Low-grade human endometrial tumors have dramatically reduced expression of p27 relative to normal endometrium 36, 37, suggesting an important role for this CDK inhibitor in not only regulating normal proliferation, but also as a barrier to dysregulated growth in endometrial hyperplasia and carcinoma. In addition, physiologic inhibitors of normal endometrial cell growth like progestins and transforming-growth factor β (TGF-β) increase p27 38, 39, whereas PTEN mutations are correlated with reduced p27 expression 40. Our data indicate that transcriptional regulation of the p27 promoter is partially responsible for the observed increase in expression associated with PKCα knockdown (Figure 3c). Akt has also been shown to reduce p27 protein stability 41, another potential mechanism by which PKCα knockdown could lead to p27 accumulation. However, since pharmacologic inhibition of Akt had little effect on p27 expression (Figure 5), it appears the predominant effect of PKCα on p27 is independent of Akt in these cells.
Akt has also been shown to directly phosphorylate p21, increasing its stability 42 and promoting translocation out of the nucleus 31. Hence, the reduction in p21 following pharmacological inhibition of Akt in Ishikawa cells (Figure 5) is consistent with a role for Akt in p21 protein stability. However, despite a similar reduction in Akt activation with PKCα knockdown (Figure 4a and b), which would predict a decrease in p21 stability and translocation to the cytoplasm, p21 protein was elevated (Figure 3a) and no shift in subcellular distribution was observed (Figure 3b). Thus, the disparity between Akt activity and p21 expression suggests that PKCα also regulates p21 independent of Akt.
Whilst our data cannot exclude a transcriptional basis for increased p21 protein (Figure 3a), PKCα knockdown cells showed no enhancement of the p21 promoter (Figure 3c) or expression of the transactivator p53 (Figure 3d). Studies examining p21 in endometrial cancer cell lines are in agreement with our observation that increased p21 protein is associated with growth inhibition 43, 44. Moreover, p21 expression has been reported to be reduced in endometrial tumors 45, with lower levels correlating with disease recurrence 46.
Unregulated activation of the PI3K/Akt pathway is arguably one of the most critical steps in endometrial carcinogenesis 15. It has been estimated that 50% of endometrial tumors have mutations in the lipid phosphatase PTEN 47, and female PTEN heterozygous mice develop endometrial carcinoma with 100% penetrance 48. PKCα knockdown caused a reduction in Akt phosphorylation and activity in Ishikawa cells (Figure 4a), which also lack functional PTEN 20. PKCα has been implicated in activation of Akt in myeloid progenitor cells 12, monocytes 13 and endothelial cells 14. To our knowledge, this is the first report implicating PKCα in the regulation of Akt in carcinoma cells. Interestingly, recent reports have shown that PIK3CA and PTEN gene mutations coexist at a high frequency in endometrial carcinomas 24, 25, thus it was proposed that additional inputs are required to fully activate Akt, despite PTEN mutation, in endometrial carcinoma cells 25. Our observation that PKCα is required in Ishikawa cells to elicit and maintain full Akt activation suggests that PKCα may provide such a stimulus.
Our data indicate PKCα signals upstream of the ERK MAPK signal transduction pathway. ERK has been implicated in the proliferation of endometrial cancer cells in response to estrogen and growth factors 9, 49. Ishikawa cells deficient in PKCα show reduced basal ERK activity (Figure 4c and d), and an attenuation of ERK activation in response to EGF (Figure 4e and f), suggesting that PKCα is required for optimal activity of this proliferative signaling pathway.
Finally, immunohistochemical analysis of PKCα expression in endometrial adenocarcinomas demonstrated considerable variability in staining of both stromal and epithelial derived components, with many tumors exhibiting focal regions of higher PKCα expression that were not observed in normal endometrium. Estrogen has been shown to regulate PKCα expression in the endometrium 10, suggesting a possible link to estrogen receptor-α (ERα) status of the tumor cells. Conversely, another study of endometrial tumors correlated increased PKCα with a loss in ERα expression and poorer patient prognosis 50; however, this latter study used western blot analysis of tumor extracts, which would not discriminate between PKCα expressed in the tumor or tumor-associated stroma.
In conclusion, these studies demonstrate a requirement for PKCα in xenograft tumor formation by Ishikawa endometrial carcinoma cells. Impaired tumor growth resulting from PKCα knockdown was associated with a dramatic increase in expression and nuclear accumulation of the CDK inhibitor proteins p21 and p27, suggesting that PKCα dependent signaling may underlie the observed decreases in p21 and p27 expression in endometrial tumors 36, 37, 45. PKCα knockdown also reduced activation of Akt and ERK, implicating, PKCα as an upstream regulator of these critical growth and survival signaling pathways. Thus, PKCα inhibitors may provide a basis for simultaneous therapeutic targeting of both Akt and ERK dependent signaling in endometrial tumors. PKCα inhibitors may provide a basis for simultaneous therapeutic targeting of both Akt and ERK dependent signaling in endometrial tumors.
Acknowledgments
The authors would like to thank Dr.'s Wood, Gutierrez-Hartmann, Moralize, and Jackson for critical reading of the manuscript and Dr. Kenneth Shroyer for advice and assistance with immunohistochemical analyses. We also acknowledge Jaime Fornetti for technical contributions, and Drs Robert Sloane and Rajesh Agawam for providing the p21 and p27 reporter constructs.
Funding: Funded by NCI CA 104875 to APB. Assistance provided by the University of Colorado Cancer Center's Cytogenesis and Flow Cytometry Core.
Abbreviations
- CDK
Cyclin dependent kinase
- DMSO
dimethyl sulfoxide
- Luc
luciferase
- MAPK
Mitogen activated protein kinase
- PBS
phosphate-buffered saline
- PKC
protein kinase C
Footnotes
The authors have no conflicts of interest
References
- 1.Mackay HJ, Twelves CJ. Targeting the protein kinase C family: are we there yet? Nat Rev Cancer. 2007;7:554–62. doi: 10.1038/nrc2168. [DOI] [PubMed] [Google Scholar]
- 2.Koivunen J, Aaltonen V, Peltonen J. Protein kinase C (PKC) family in cancer progression. Cancer Lett. 2005 doi: 10.1016/j.canlet.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 3.Mandil R, Ashkenazi E, Blass M, Kronfeld I, Kazimirsky G, Rosenthal G, Umansky F, Lorenzo PS, Blumberg PM, Brodie C. Protein kinase Calpha and protein kinase Cdelta play opposite roles in the proliferation and apoptosis of glioma cells. Cancer Res. 2001;61:4612–9. [PubMed] [Google Scholar]
- 4.Jiffar T, Kurinna S, Suck G, Carlson-Bremer D, Ricciardi MR, Konopleva M, Andreeff M, Ruvolo PP. PKC alpha mediates chemoresistance in acute lymphoblastic leukemia through effects on Bcl2 phosphorylation. Leukemia. 2004;18:505–12. doi: 10.1038/sj.leu.2403275. [DOI] [PubMed] [Google Scholar]
- 5.Sliva D. Signaling pathways responsible for cancer cell invasion as targets for cancer therapy. Curr Cancer Drug Targets. 2004;4:327–36. doi: 10.2174/1568009043332961. [DOI] [PubMed] [Google Scholar]
- 6.Dean N, McKay R, Miraglia L, Howard R, Cooper S, Giddings J, Nicklin P, Meister L, Ziel R, Geiger T, Muller M, Fabbro D. Inhibition of growth of human tumor cell lines in nude mice by an antisense of oligonucleotide inhibitor of protein kinase C-alpha expression. Cancer Res. 1996;56:3499–507. [PubMed] [Google Scholar]
- 7.Frey MR, Clark JA, Leontieva O, Uronis JM, Black AR, Black JD. Protein kinase C signaling mediates a program of cell cycle withdrawal in the intestinal epithelium. J Cell Biol. 2000;151:763–78. doi: 10.1083/jcb.151.4.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 9.Bamberger AM, Bamberger CM, Schulte HM. Molecular mechanisms of proliferation in endometrial tumour cells. Hum Reprod Update. 1998;4:526–31. doi: 10.1093/humupd/4.5.526. [DOI] [PubMed] [Google Scholar]
- 10.Wu H, Chen Y, Liang J, Shi B, Wu G, Zhang Y, Wang D, Li R, Yi X, Zhang H, Sun L, Shang Y. Hypomethylation-linked activation of PAX2 mediates tamoxifen-stimulated endometrial carcinogenesis. Nature. 2005;438:981–7. doi: 10.1038/nature04225. [DOI] [PubMed] [Google Scholar]
- 11.Haughian JM, Jackson TA, Koterwas DM, Bradford AP. Endometrial cancer cell survival and apoptosis is regulated by protein kinase C alpha and delta. Endocr Relat Cancer. 2006;13:1251–67. doi: 10.1677/erc.1.01278. [DOI] [PubMed] [Google Scholar]
- 12.Li W, Zhang J, Flechner L, Hyun T, Yam A, Franke TF, Pierce JH. Protein kinase C-alpha overexpression stimulates Akt activity and suppresses apoptosis induced by interleukin 3 withdrawal. Oncogene. 1999;18:6564–72. doi: 10.1038/sj.onc.1203065. [DOI] [PubMed] [Google Scholar]
- 13.Preiss S, Namgaladze D, Brune B. Critical role for classical PKC in activating Akt by phospholipase A2-modified LDL in monocytic cells. Cardiovasc Res. 2007;73:833–40. doi: 10.1016/j.cardiores.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 14.Partovian C, Simons M. Regulation of protein kinase B/Akt activity and Ser473 phosphorylation by protein kinase Calpha in endothelial cells. Cell Signal. 2004;16:951–7. doi: 10.1016/j.cellsig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 15.Hecht JL, Mutter GL. Molecular and pathologic aspects of endometrial carcinogenesis. J Clin Oncol. 2006;24:4783–91. doi: 10.1200/JCO.2006.06.7173. [DOI] [PubMed] [Google Scholar]
- 16.Nishida M, Kasahara K, Kaneko M, Iwasaki H, Hayashi K. Establishment of a new human endometrial adenocarcinoma cell line, Ishikawa cells, containing estrogen and progesterone receptors. Nippon Sanka Fujinka Gakkai Zasshi. 1985;37:1103–11. [PubMed] [Google Scholar]
- 17.Xiao H, Hasegawa T, Isobe K. Both Sp1 and Sp3 are responsible for p21waf1 promoter activity induced by histone deacetylase inhibitor in NIH3T3 cells. J Cell Biochem. 1999;73:291–302. [PubMed] [Google Scholar]
- 18.Jackson TA, Schweppe RE, Koterwas DM, Bradford AP. Fibroblast growth factor activation of the rat PRL promoter is mediated by PKCdelta. Mol Endocrinol. 2001;15:1517–28. doi: 10.1210/mend.15.9.0683. [DOI] [PubMed] [Google Scholar]
- 19.Reno EM, Haughian JM, Dimitrova IK, Jackson TA, Shroyer KR, Bradford AP. Analysis of protein kinase C (delta) expression in endometrial tumors. Hum Pathol. 2007 doi: 10.1016/j.humpath.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Albitar L, Pickett G, Morgan M, Davies S, Leslie KK. Models representing type I and type II human endometrial cancers: Ishikawa H and Hec50co cells. Gynecol Oncol. 2007;106:52–64. doi: 10.1016/j.ygyno.2007.02.033. [DOI] [PubMed] [Google Scholar]
- 21.Wang LH. Molecular signaling regulating anchorage-independent growth of cancer cells. Mt Sinai J Med. 2004;71:361–7. [PubMed] [Google Scholar]
- 22.LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–62. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Prives C. Increased and altered DNA binding of human p53 by S and G2/M but not G1 cyclin-dependent kinases. Nature. 1995;376:88–91. doi: 10.1038/376088a0. [DOI] [PubMed] [Google Scholar]
- 24.Hayes MP, Wang H, Espinal-Witter R, Douglas W, Solomon GJ, Baker SJ, Ellenson LH. PIK3CA and PTEN mutations in uterine endometrioid carcinoma and complex atypical hyperplasia. Clin Cancer Res. 2006;12:5932–5. doi: 10.1158/1078-0432.CCR-06-1375. [DOI] [PubMed] [Google Scholar]
- 25.Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005;65:10669–73. doi: 10.1158/0008-5472.CAN-05-2620. [DOI] [PubMed] [Google Scholar]
- 26.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 27.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 28.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–90. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 29.Schonwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol Cell Biol. 1998;18:790–8. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chambard JC, Lefloch R, Pouyssegur J, Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta. 2007;1773:1299–310. doi: 10.1016/j.bbamcr.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 31.Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3:245–52. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 32.Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, Franssen E, Slingerland JM. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–60. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 33.Jiang XH, Tu SP, Cui JT, Lin MC, Xia HH, Wong WM, Chan AO, Yuen MF, Jiang SH, Lam SK, Kung HF, Soh JW, et al. Antisense targeting protein kinase C alpha and beta1 inhibits gastric carcinogenesis. Cancer Res. 2004;64:5787–94. doi: 10.1158/0008-5472.CAN-03-1172. [DOI] [PubMed] [Google Scholar]
- 34.Wang XY, Repasky E, Liu HT. Antisense inhibition of protein kinase Calpha reverses the transformed phenotype in human lung carcinoma cells. Exp Cell Res. 1999;250:253–63. doi: 10.1006/excr.1999.4529. [DOI] [PubMed] [Google Scholar]
- 35.Ways DK, Kukoly CA, deVente J, Hooker JL, Bryant WO, Posekany KJ, Fletcher DJ, Cook PP, Parker PJ. MCF-7 breast cancer cells transfected with protein kinase C-alpha exhibit altered expression of other protein kinase C isoforms and display a more aggressive neoplastic phenotype. J Clin Invest. 1995;95:1906–15. doi: 10.1172/JCI117872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lahav-Baratz S, Ben-Izhak O, Sabo E, Ben-Eliezer S, Lavie O, Ishai D, Ciechanover A, Dirnfeld M. Decreased level of the cell cycle regulator p27 and increased level of its ubiquitin ligase Skp2 in endometrial carcinoma but not in normal secretory or in hyperstimulated endometrium. Mol Hum Reprod. 2004;10:567–72. doi: 10.1093/molehr/gah084. [DOI] [PubMed] [Google Scholar]
- 37.Masciullo V, Susini T, Zamparelli A, Bovicelli A, Minimo C, Massi D, Taddei G, Maggiano N, De Iaco P, Ceccaroni M, Bovicelli L, Amunni G, et al. Frequent loss of expression of the cyclin-dependent kinase inhibitor p27(Kip1) in estrogen-related Endometrial adenocarcinomas. Clin Cancer Res. 2003;9:5332–8. [PubMed] [Google Scholar]
- 38.Lecanda J, Parekh TV, Gama P, Lin K, Liarski V, Uretsky S, Mittal K, Gold LI. Transforming growth factor-beta, estrogen, and progesterone converge on the regulation of p27Kip1 in the normal and malignant endometrium. Cancer Res. 2007;67:1007–18. doi: 10.1158/0008-5472.CAN-06-0235. [DOI] [PubMed] [Google Scholar]
- 39.Shiozawa T, Horiuchi A, Kato K, Obinata M, Konishi I, Fujii S, Nikaido T. Up-regulation of p27Kip1 by progestins is involved in the growth suppression of the normal and malignant human endometrial glandular cells. Endocrinology. 2001;142:4182–8. doi: 10.1210/endo.142.10.8455. [DOI] [PubMed] [Google Scholar]
- 40.An HJ, Lee YH, Cho NH, Shim JY, Kim JY, Lee C, Kim SJ. Alteration of PTEN expression in endometrial carcinoma is associated with down-regulation of cyclin-dependent kinase inhibitor, p27. Histopathology. 2002;41:437–45. doi: 10.1046/j.1365-2559.2002.01455.x. [DOI] [PubMed] [Google Scholar]
- 41.Gesbert F, Sellers WR, Signoretti S, Loda M, Griffin JD. BCR/ABL regulates expression of the cyclin-dependent kinase inhibitor p27Kip1 through the phosphatidylinositol 3-Kinase/AKT pathway. J Biol Chem. 2000;275:39223–30. doi: 10.1074/jbc.M007291200. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Dowbenko D, Lasky LA. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J Biol Chem. 2002;277:11352–61. doi: 10.1074/jbc.M109062200. [DOI] [PubMed] [Google Scholar]
- 43.Watanabe J, Kamata Y, Seo N, Okayasu I, Kuramoto H. Stimulatory effect of estrogen on the growth of endometrial cancer cells is regulated by cell-cycle regulators. J Steroid Biochem Mol Biol. 2007;107:163–71. doi: 10.1016/j.jsbmb.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 44.Kawaguchi M, Watanabe J, Hamano M, Kamata Y, Arai T, Nishimura Y, Obokata A, Jobo T, Kuramoto H. Medroxyprogesterone acetate stimulates cdk inhibitors, p21 and p27, in endometrial carcinoma cells transfected with progesterone receptor-B cDNA. Eur J Gynaecol Oncol. 2006;27:33–8. [PubMed] [Google Scholar]
- 45.Palazzo JP, Mercer WE, Kovatich AJ, McHugh M. Immunohistochemical localization of p21(WAF1/CIP1) in normal, hyperplastic, and neoplastic uterine tissues. Hum Pathol. 1997;28:60–6. doi: 10.1016/s0046-8177(97)90280-x. [DOI] [PubMed] [Google Scholar]
- 46.Pijnenborg JM, van de Broek L, Dam de Veen GC, Roemen GM, de Haan J, van Engeland M, Voncken JW, Groothuis PG. TP53 overexpression in recurrent endometrial carcinoma. Gynecol Oncol. 2006;100:397–404. doi: 10.1016/j.ygyno.2005.09.056. [DOI] [PubMed] [Google Scholar]
- 47.Kong D, Suzuki A, Zou TT, Sakurada A, Kemp LW, Wakatsuki S, Yokoyama T, Yamakawa H, Furukawa T, Sato M, Ohuchi N, Sato S, et al. PTEN1 is frequently mutated in primary endometrial carcinomas. Nat Genet. 1997;17:143–4. doi: 10.1038/ng1097-143. [DOI] [PubMed] [Google Scholar]
- 48.Vilgelm A, Lian Z, Wang H, Beauparlant SL, Klein-Szanto A, Ellenson LH, Di Cristofano A. Akt-mediated phosphorylation and activation of estrogen receptor alpha is required for endometrial neoplastic transformation in Pten+/- mice. Cancer Res. 2006;66:3375–80. doi: 10.1158/0008-5472.CAN-05-4019. [DOI] [PubMed] [Google Scholar]
- 49.Suga S, Kato K, Ohgami T, Yamayoshi A, Adachi S, Asanoma K, Yamaguchi S, Arima T, Kinoshita K, Wake N. An inhibitory effect on cell proliferation by blockage of the MAPK/estrogen receptor/MDM2 signal pathway in gynecologic cancer. Gynecol Oncol. 2007;105:341–50. doi: 10.1016/j.ygyno.2006.12.030. [DOI] [PubMed] [Google Scholar]
- 50.Fournier DB, Chisamore M, Lurain JR, Rademaker AW, Jordan VC, Tonetti DA. Protein kinase C alpha expression is inversely related to ER status in endometrial carcinoma: possible role in AP-1-mediated proliferation of ER-negative endometrial cancer. Gynecol Oncol. 2001;81:366–72. doi: 10.1006/gyno.2001.6164. [DOI] [PubMed] [Google Scholar]