

Abstract
The formation and function of the mammalian cerebral cortex relies on the complex interplay of a variety of genetic and environmental factors through protracted periods of gestational and postnatal development. Biogenic amine systems are important neuromodulators, both in the adult nervous system, and during critical epochs of brain development. Abnormalities in developmental programming likely contribute to developmental delays and multiple neurological and psychiatric disorders, often with symptom onset much later than the actual induction of pathology. We review several genetic and pharmacological models of dopamine and serotonin modulation during development, each of which produces permanent changes in cerebral cortical structure and function. These models clearly illustrate the ability of these neurotransmitters to function beyond their classic roles and show their involvement in the development and modulation of fine brain circuitry that is sensitive to numerous effectors. Furthermore, these studies demonstrate the need to consider not only gene by environment interactions, but also gene by environment by developmental time interactions.
Keywords: dopamine, serotonin, cortex, cocaine, prenatal, postnatal
Introduction
Defects in the development of the cerebral cortex can have a profound impact on mature brain functions. Developmental anomalies in cortical development contribute to neuropsychiatric disorders such as schizophrenia (Lewis & Levitt, 2002; Weinberger, 1995), mental retardation, learning disabilities and autism (Charman, 1999; Levitt et al., 2004). The molecular and cellular bases that tie developmental defects to cortical dysfunction in these disorders remain unknown, but influences on cell-cell interactions that mediate specific developmental events are likely targets. This also seems to be true for non-genetic insults, such as prenatal exposure to toxicants, stress or drugs of abuse (Andersen, 2005; Rodier, 1995; Stanwood & Levitt, 2004; Trask & Kosofsky, 2000).
The biogenic amine neurotransmitters dopamine (DA), serotonin (5-HT) and norepinephrine (NE) are pleiotropic molecules (that is, each can produce multiple, diverse effects) that serve as regulators of distinct cellular functions at different times in neurodevelopment and adulthood. In this review, we will describe several key animal models which have examined the structural and functional consequences of altering these neurotransmitters in development. We hypothesize that genetic and environmental modulation of biogenic amines, during discrete sensitive periods of brain development (Knudsen, 2004; Rodier, 1994; Stanwood & Levitt, 2004), alter the formation and function of brain circuitry and modulate the development of modularity within the cerebral cortex.
Example 1: 5-HT and the Development of Anxiety Circuits and Behavior
5-HT is a well known modulator of behavioral, physiological and emotional functions, including learning and memory, appetite, temperature regulation, and mood (Albert & Lemonde, 2004; Hedlund & Sutcliffe, 2004; Holmes et al., 2005; Lucki, 1998; Meneses, 1999; D. L. Murphy & Lesch, 2008; Ressler & Nemeroff, 2000; Silverstone, 1992). Dysfunctions of serotonergic signaling may contribute to disorders including anxiety, aggression, depression, and autism (Burgess et al., 2006; Canli & Lesch, 2007; Leonardo & Hen, 2006; Mann et al., 2001; Popova, 2006; Sutcliffe et al., 2005). In the mammalian nervous system, 5-HT exerts its effects by binding to at least 15 distinct receptor subtypes, each with unique pharmacological properties, distribution, and activational profiles. The high affinity 5-HT transporter, by transporting 5-HT back into the presynaptic terminal, is the primary means of 5-HT removal from the synapse and extracellular fluid.
Developmentally, 5-HT signaling modulates cellular functioning even before the onset of neurogenesis, including events in the periphery such as craniofacial, gastrointestinal, and cardiovascular morphogenesis (Buznikov et al., 1996; Lauder, 1983; Whitaker-Azmitia, 1991; Whitaker-Azmitia et al., 1996). Maternal 5-HT has also been observed to influence left-right patterning in frog and chick embryos (Fukumoto et al., 2005); additional evidence also implicates maternal serotonin in mouse embryogenesis (Cote et al., 2007). Within the brain, 5-HT and its receptors are expressed early in prenatal development (Bonnin et al., 2006; Bonnin et al., 2007; Goldman-Rakic et al., 1990; Lambe et al., 2000; Whitaker-Azmitia et al., 1987), and can modify elements of dendritic and axonal differentiation (Bonnin et al., 2007; Persico et al., 2006) (see also Example 2, below). The discovery of alterations in thalamocortical development and somatosensory cortex cytoarchitecture in monoamine oxidase A knockout mice (this is the enzyme largely responsible for serotonin degradation in rodents) led to a recognition that different components of aminergic signaling might specifically influence discrete ontogenic events (Cases et al., 1996; Luo et al., 2003).
Genetic elimination of 5-HT1A receptors and the 5-HT transporter, along with known 5-HT pharmacology, implicated the 5-HT system in anxiety and depression (Holmes et al., 2003; Parks et al., 1998; Ramboz et al., 1998). To test for a possible developmental role of the 5-HT1A receptor in establishing anxiety circuitry, Hen and colleagues generated a conditional knockout mouse that allowed for temporally-restricted rescue of postsynaptic 5-HT1A receptors in the cerebral cortex and hippocampus (Gross et al., 2002). Using this strategy, they demonstrated that initiating expression of the receptor after postnatal day (P) 21 resulted in increased anxiety levels identical to constitutive 5-HT1A receptor knockout animals. Conversely, earlier expression of the 5-HT1A receptor, during the first three postnatal weeks, produced mice with anxiety levels that were indistinguishable from wild-type animals, even if the receptor was turned off in adulthood. These findings indicate that 5-HT1A receptors are essential to the establishment of normal anxiety-modulating circuits in the brain during early postnatal development in the rodent. In humans, this time frame would include the third trimester of pregnancy, and infancy (Clancy et al., 2007; Dobbing & Sands, 1979; Rodier, 1980).
Constitutive knockout of the 5-HT transporter also leads to increases in anxiety and alterations in cortical and subcortical information processing. This led to an intriguing study in which mice were administered the selective serotonin reuptake inhibitor (SSRI) fluoxetine from P4-P21, producing a pharmacological equivalent of a temporally restricted knockout (Ansorge et al., 2004). This treatment induced effects on anxiety behaviors that are quite similar to the 5-HT transporter gene knockout. Another group examined the effect of administration of the SSRI citalopram during P8–21 in rats, and observed long lasting effects on 5-HT metabolism and behavior (Maciag et al., 2006). Taken together, the 5-HT1A rescue experiments and early postnatal SSRI models suggest that 5-HT plays a critical role in the maturation of circuits relevant to anxiety prior to the third postnatal week. Intriguingly, a recent study examined the effects of foot-shock stress during P7–13 on adult responses and found no effects on anxiety in either wildtype or 5-HT transporter knockout mice (Carroll et al., 2007). These data suggest that the key sensitive period for altering adult anxiety responses may be between P14 and P21 in the rodent (Figure 1).
Figure 1.
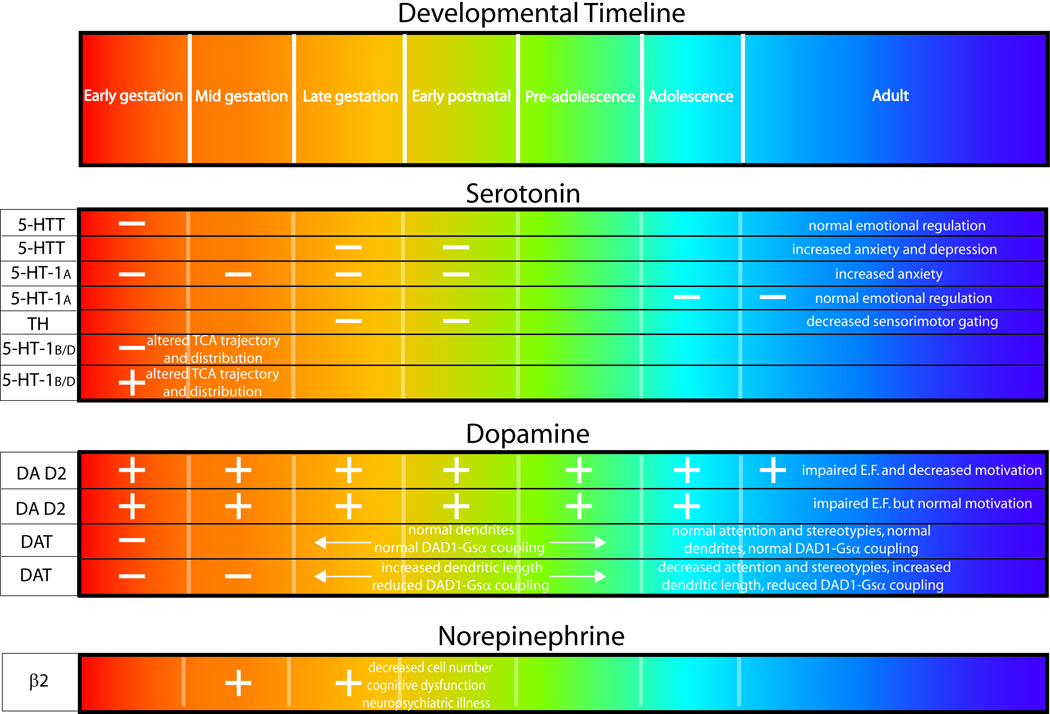
Different circuits and brain regions may have different sensitive ontogenic periods. Prenatal exposure to fluoxetine in rats (embryonic days (E)6-E20) was found not to significantly alter anxiety behavior in adult offspring (Bairy et al., 2007). Furthermore, recent work has implicated the activity of the 5-HT synthesis enzyme tryptophan hydroxylase in the proper maturation of sensorimotor gating between P21-P24 (Nakamura et al., 2006).
Consistent with the rodent models, data from human studies suggest that baseline anxiety levels are influenced early in life. By 2 years of age, most children have established cohesive patterns of response to novel environments, as measured by behavioral inhibition. These measures appear to be stable over many years (Hirshfeld et al., 1992; Rosenbaum et al., 1993; Schwartz et al., 1999) (although see also (Degnan & Fox, 2007)), and can predict one’s future risk of anxiety disorders (Kagan & Snidman, 1999; Kagan et al., 2007).
Not surprisingly, polymorphisms in 5-HT receptors, signaling partners, synthesis enzymes, and the 5-HT transporter have all been associated with anxiety- and depression-related symptoms and even autism (Albert & Lemonde, 2004; Dannlowski et al., 2008; Kim et al., 2006; D. L. Murphy & Lesch, 2008; Sutcliffe et al., 2005; Walderhaug et al., 2007). Moreover, many prominent psycho-therapeutics target the 5-HT system and are utilized for depression and anxiety among pregnant and nursing mothers. When behavioral therapy is not effective in treating depression in a pregnant woman, the potential risk of drug therapy must be weighed against the considerable risk for relapse of the disorder if pharmacological therapy is interrupted. Published literature to date suggests only modest alterations in neonatal outcome (Andrade et al., 2008; Maschi et al., 2008; Oberlander et al., 2008; Pearson et al., 2007), but given the likelihood of 5-HT modulation of brain circuitry raised by the animal data reviewed above, further study of the neurobehavioral consequences of antidepressant exposure on the developing fetus and infant are clearly warranted. Further, environmental toxicants such as organophosphate insecticides clearly alter developing serotonin systems with permanent consequences (Slotkin & Seidler, 2005); limiting exposures to such environmental chemicals and identifying strategies to ameliorate these effects are crucial.
Example 2: 5-HT1B/1D Receptor Activation Influences Thalamocortical Development
The next example illustrates the ability of the biogenic amines to directly influence cortical modularity. Specifically, we review data demonstrating cortical modulation is based on the effects of 5-HT signaling on the responsiveness of thalamocortical axons to guidance cues. In a detailed neuroanatomical study, Bonnin et al (Bonnin et al., 2006) demonstrated that expression patterns of select serotonin receptors overlapped with those of axon guidance receptors, including netrin receptors, in the embryonic mouse thalamus. In a follow-up study, the authors performed a series of in vitro explants studies to examine the response of thalamic axons to different guidance cues, and the role of serotonin in this response (Bonnin et al., 2007). It was found that the normal attraction of axons from posterior thalamic explants to netrin was reversed when the explants were simultaneously exposed to serotonin. Further experiments determined that this switch was due to activation of the 5-HT1B/1D receptors, as 5-HT1B/1D antagonists blocked the switching capacity of serotonin, and 5-HT1B/1D agonists directly mimicked the actions of serotonin. Elegant in utero electroporation studies then revealed that directly over-expressing or reducing 5-HT1B/1D receptor expression in vivo altered thalamocortical axon trajectory and distribution, demonstrating a direct neuromodulatory role for 5-HT in the developing mouse brain (Bonnin et al., 2007).
These experiments further illustrate the importance of normal 5-HT signaling during development. Altering 5-HT expression can disrupt normal development of brain circuitry, thereby increasing the risk of neurodevelopmental disorders.
Example 3: Overexpression of Subcortical Dopamine D2 Receptors Alters Dopaminergic Responsiveness in the Prefrontal Cortex
Our next two examples are drawn from dopamine, and its effects on cortical modularity. An intriguing model system recently created within the dopaminergic system involves conditional transgenic mice over-expressing dopamine D2 receptors in the striatum (Kellendonk et al., 2006). Striata of the transgenic mice contain increased numbers of D2 receptor binding sites and increased effects on adenylyl cyclase, indicating that the transgenic receptors are functional. Adult mutant mice exhibit normal locomotor activity, sensorimotor gating, and anxiety behaviors. However, the mice display substantial defects in working memory tasks and behavioral flexibility typically associated with prefrontal cortical dysfunction. Consistent with this, the mutant mice display altered glucose metabolism, dopamine levels, and dopamine D1 receptor activation in the prefrontal cortex. An advantage of this mouse model is that over-expression of the transgenic receptor can be regulated temporally, such that administration of doxycycline restores dopamine D2 receptor to normal levels. Interestingly, doxycycline treatment in the adult does not reverse the cognitive defects, indicating that these deficits reflect striatal D2 receptor over-activity during fetal life. A recent follow-up study suggests that the effects of exogenous D2 receptor over-expression on interval timing in an operant task also are established developmentally, whereas motivational processes can be normalized by transgene elimination in the adult (Drew et al., 2007). Direct assessment of cortical architecture and connectivity in this model is still needed.
Example 4: Developmental Loss of Dopamine D1 Receptor Signaling Induces Alterations in Cortical Architecture and Function
This next example again demonstrates how monoamines can directly modify cortical development. Administration of cocaine in utero alters dopamine neurotransmission. The extent to which this prenatal exposure affects the development of the human fetus varies significantly, but it has been consistently established that there are long-term consequences, ranging from relatively mild to severe functional disruptions in cognition and attention (Dow-Edwards et al., 1999; Gingras & O'Donnell, 1998; Karmel & Gardner, 1996; Mayes et al., 1998; Richardson et al., 1996; Singer et al., 2004).
Different animal models, designed to mimic human drug use during gestation, confirm that prenatal cocaine exposure results in specific behavioral, cellular, and molecular changes that appear to be permanent (Harvey, 2004; Lidow, 2003; Mayes, 2002; Stanwood & Levitt, 2004). Our lab has developed a unique animal model of prenatal cocaine exposure to study the mechanisms underlying the complex, long-term adaptive changes and the functional outcomes of in utero cocaine exposure. This model utilizes a low-dose regimen of intravenous prenatal cocaine exposure in the rabbit, which was initially selected for ease of intravenous administration. Furthermore, the pharmacokinetic profile of intravenous cocaine in the rabbit (Parlaman et al., 2007) closely models what is seen when human users abuse cocaine (Evans et al., 1996; Jenkins et al., 2002), allowing for direct species comparisons and interpretations. A number of studies have established that the prenatal dosing is not teratologic, nor does it impact basic developmental parameters such as kit mortality, litter size, sex or growth rates (L. Jones et al., 1996; E. H. Murphy et al., 1997; X. H. Wang et al., 1995b; X. H. Wang et al., 1996). However, through control of length of drug exposure, age at drug exposure, and dosing, we have delineated a critical window of time (E 16–25, Figure 1) during which exposure to cocaine affects behavior, morphology, and cellular composition (Stanwood & Levitt, 2003; Stanwood et al., 2001a; Stanwood et al., 2001b; B. L. Thompson et al., 2005b). This window of time corresponds to the emergence of pre- and postsynaptic components of the DA system in the cerebral cortex (Stanwood et al., 2001a).
Our neuroanatomical and molecular analyses following prenatal cocaine exposure have delineated a number of highly specific changes in DA-rich cortical areas, including changes in GABA content, calcium binding protein expression, and a 40–50 percent increase in pyramidal neuron apical dendrite length within DA rich cortical areas (L. B. Jones et al., 2000; E. H. Murphy et al., 1997; Stanwood & Levitt, 2001; Stanwood & Levitt, 2007; Stanwood et al., 2006; Stanwood et al., 2001b). These cortical areas are involved in cognition and executive functioning tasks, including attention (Clark et al., 2004; Collette & Van der Linden, 2002; Elliott, 2003; Elston, 2003; Goldman-Rakic, 1996).
Consistent with the regional selectivity in the anatomical findings, extensive behavioral characterization of rabbits following in utero exposure to cocaine suggest that the behaviors disrupted appear to be limited to those mediated via select DA-rich cortical and sub-cortical regions (Stanwood & Levitt, 2003; B. L. Thompson et al., 2005b). For example, these animals exhibit decreases in spontaneous alternation as measured by the Y-maze following prenatal cocaine exposure. This decrease in attention is not accompanied by changes in open field behavior or two-object recognition. Additionally, offspring exposed to prenatal cocaine show a decreased number of head-bobs, a measure of stereotypy, following a single injection of amphetamine and display a blunted preference for cocaine in a conditioned place preference paradigm (Stanwood & Levitt, 2003; B. Thompson et al., 2005a).
Molecular analyses have determined that the dopamine D1 receptor exhibits permanent reduced coupling to its cognate G-protein, Gsα, following prenatal cocaine exposure (Friedman et al., 1996; L. B. Jones et al., 2000; H. Y. Wang et al., 1995a). This reduction in coupling is a result of dopamine D1 receptor remaining internalized and not trafficking properly to the cell membrane where it would then interact with Gsα (Stanwood & Levitt, 2007). Adult rabbits exposed to cocaine prenatally also exhibit greatly reduced psychostimulant-induced stereotypies, consistent with diminished D1 receptor signaling (Simansky & Kachelries, 1996; Stanwood & Levitt, 2003). Additional evidence to support a role for altered D1 receptor signaling in the cellular findings comes from our recent study of the D1 receptor knockout mouse, which exhibits similar cellular and morphological changes to the prenatal cocaine exposed rabbits (Stanwood et al., 2005). In contrast, D1 receptor overexpression has the opposite effects on dendritic architecture (Song et al., 2002).
There are two very important points to emphasize with respect to these findings. First, there is temporal requirement for all of the above results. Specifically, cocaine must be given while the dopaminergic system is simultaneously developing in the embryo. If cocaine is given before the DA system begins to develop, the neuroanatomical and behavioral changes are not observed. Second, there is spatial specificity to these data. The biochemical, morphological, and cellular alterations are only found within DA rich cortical and sub-cortical areas. Non-DA rich areas do not display similar disruptions following prenatal cocaine exposure.
Example 5: Developmental Exposure to a β Adrenergic Receptor Agonist Disrupts Cortical Development
Terbutaline is a β2-adrenergic receptor agonist that is wide used to abate preterm labor because of its ability to cause uterine muscle relaxation. Terbutaline crosses the placental and blood-brain barriers, suggesting that it might also activate fetal β adrenergic receptors on developing neurons and glia. In fact, alterations in glucose metabolism and tachycardia have been observed in neonatal offspring of women treated with terbutaline. Moreover, children exposed to terbutaline or related drugs prior to birth appear to show impaired school performance, cognitive dysfunction, and increased incidence of neuropsychiatric disease (Feenstra, 1992; Hadders-Algra et al., 1986; Pitzer et al., 2001). Although still preliminary, recent data suggest that functional polymorphisms in the β2 receptor may be associated with autism (Cheslack-Postava et al., 2007; Connors et al., 2005).
Neurochemical and neuroanatomical findings in animal models suggest that during particular times of gestation terbutaline can enter the fetal brain and alter patterns of cellular differentiation and synaptogenesis, perhaps via increases in cAMP (Slotkin et al., 2003). In particular, loss of cerebellar Purkinje cells, cerebellar thinning, a reduction in the proportion of pyramidal neurons in the somatosensory cortex with a concomitant increase in smaller nonpyramidal cells, and sex-dependent neuroglial activation have all been reported (Rhodes et al., 2004; Zerrate et al., 2007). In the rat, early postnatal development (P2-P5) appears to be a very vulnerable period (see Figure 1), corresponding to early third trimester in human (Rhodes et al., 2004), when the drug is utilized clinically. Providing an intriguing example of how multiple environmental exposures can influence one another, neonatal terbutaline exposure (P2–5) augments the long-lasting and deleterious effects of later exposure to the insecticide chlorpyrifos (P11–14) (Meyer et al., 2005).
Summary
In recent years, diverse roles for classical neurotransmitters, in particular DA, NE and 5-HT, have been described in the developing brain. Disruption of these signaling pathways during development can lead to permanent alterations in neuronal signaling, brain architecture and behavioral outcome, depending on the time at which disruption occurs. Thus, altering DA, NE or 5-HT neurotransmission during discrete prenatal and early postnatal periods disrupts the developmental processes that establish essential functional circuits required for typical adult function.
Insights into the developmental role of these neurotransmitters primarily have come from animal models. These animal models provide the opportunity to manipulate directly key developmental variables and measure their functional outcome. Through such manipulations, the scientific community is beginning to identify gene by environment interactions that alter developmental trajectory, bringing us closer to understanding the etiology of developmental disorders. However, as the studies highlighted in this paper illustrate, there can be different outcomes based on the developmental state during which the genetic or environmental insult occurs. The delineation of additional “sensitive” and “critical” periods of neurodevelopment is a crucial next step in understanding both normal biology and pathophysiology. As the incidence of neurodevelopmental disorders rises, we emphasize that the relevant variable of developmental time must be included when considering the role of gene by environment interactions in the etiology of neuropsychiatric and neurological disease.
Acknowledgements
Drs. Thompson and Stanwood currently receive support from NICHD core grant P30HD15052 (GDS), F32DA020981 (BLT) and the Vanderbilt Kennedy Center (GDS & BLT). We thank Dr. Pat Levitt for useful discussions, insightful comments, and partial financial support. We also thank Dr. Kathie Eagleson and Dr. Daniel Campbell for critical reading of this manuscript.
Contributor Information
Barbara L. Thompson, Department of Pharmacology; 8114 MRBIII, Nashville, TN 37232, USA; (615) 936-3865 (phone); (615) 936-3747 (fax); barbara.thompson@vanderbilt.edu
Gregg D. Stanwood, Department of Pharmacology, Vanderbilt Kennedy Center for Research on Human Development, & Center for Molecular Neuroscience; 8405 MRBIV, Nashville, TN 37232, USA; (615) 936-3861 (phone); (615) 936-2202 (fax); gregg.stanwood@vanderbilt.edu
References
- Albert PR, Lemonde S. 5-ht1a receptors, gene repression, and depression: Guilt by association. Neuroscientist. 2004;10(6):575–593. doi: 10.1177/1073858404267382. [DOI] [PubMed] [Google Scholar]
- Andersen SL. Stimulants and the developing brain. Trends Pharmacol Sci. 2005;26(5):237–243. doi: 10.1016/j.tips.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Andrade SE, Raebel MA, Brown J, Lane K, Livingston J, Boudreau D, et al. Use of antidepressant medications during pregnancy: A multisite study. Am J Obstet Gynecol. 2008;198(2) doi: 10.1016/j.ajog.2007.07.036. 194 e191-195. [DOI] [PubMed] [Google Scholar]
- Ansorge MS, Zhou M, Lira A, Hen R, Gingrich JA. Early-life blockade of the 5-ht transporter alters emotional behavior in adult mice. Science. 2004;306(5697):879–881. doi: 10.1126/science.1101678. [DOI] [PubMed] [Google Scholar]
- Bairy KL, Madhyastha S, Ashok KP, Bairy I, Malini S. Developmental and behavioral consequences of prenatal fluoxetine. Pharmacology. 2007;79(1):1–11. doi: 10.1159/000096645. [DOI] [PubMed] [Google Scholar]
- Bonnin A, Peng W, Hewlitt W, Levitt P. Expression mapping of 5-ht1 serotonin receptor subtypes during fetal and early postnatal mouse forebrain development. Neuroscience. 2006 doi: 10.1016/j.neuroscience.2006.04.036. in press. [DOI] [PubMed] [Google Scholar]
- Bonnin A, Torii M, Wang L, Rakic P, Levitt P. Serotonin modulates the response of embryonic thalamocortical axons to netrin-1. Nat Neurosci. 2007;10(5):588–597. doi: 10.1038/nn1896. [DOI] [PubMed] [Google Scholar]
- Burgess NK, Sweeten TL, McMahon WM, Fujinami RS. Hyperserotoninemia and altered immunity in autism. J Autism Dev Disord. 2006;36(5):697–704. doi: 10.1007/s10803-006-0100-7. [DOI] [PubMed] [Google Scholar]
- Buznikov GA, Shmukler YB, Lauder JM. From oocyte to neuron: Do neurotransmitters function in the same way throughout development? Cellular & Molecular Neurobiology. 1996;16(5):537–559. doi: 10.1007/BF02152056. [DOI] [PubMed] [Google Scholar]
- Canli T, Lesch KP. Long story short: The serotonin transporter in emotion regulation and social cognition. Nat Neurosci. 2007;10(9):1103–1109. doi: 10.1038/nn1964. [DOI] [PubMed] [Google Scholar]
- Carroll JC, Boyce-Rustay JM, Millstein R, Yang R, Wiedholz LM, Murphy DL, et al. Effects of mild early life stress on abnormal emotion-related behaviors in 5-htt knockout mice. Behav Genet. 2007;37(1):214–222. doi: 10.1007/s10519-006-9129-9. [DOI] [PubMed] [Google Scholar]
- Cases O, Vitalis T, Seif I, De Maeyer E, Sotelo C, Gaspar P. Lack of barrels in the somatosensory cortex of monoamine oxidase a-deficient mice: Role of a serotonin excess during the critical period. Neuron. 1996;16(2):297–307. doi: 10.1016/s0896-6273(00)80048-3. [DOI] [PubMed] [Google Scholar]
- Charman T. Autism and the pervasive developmental disorders. Current Opinion in Neurology. 1999;12:155–159. doi: 10.1097/00019052-199904000-00005. [DOI] [PubMed] [Google Scholar]
- Cheslack-Postava K, Fallin MD, Avramopoulos D, Connors SL, Zimmerman AW, Eberhart CG, et al. Beta2-adrenergic receptor gene variants and risk for autism in the agre cohort. Mol Psychiatry. 2007;12(3):283–291. doi: 10.1038/sj.mp.4001940. [DOI] [PubMed] [Google Scholar]
- Clancy B, Kersh B, Hyde J, Darlington RB, Anand KJ, Finlay BL. Web-based method for translating neurodevelopment from laboratory species to humans. Neuroinformatics. 2007;5(1):79–94. doi: 10.1385/ni:5:1:79. [DOI] [PubMed] [Google Scholar]
- Clark L, Cools R, Robbins TW. The neuropsychology of ventral prefrontal cortex: Decision-making and reversal learning. Brain Cogn. 2004;55(1):41–53. doi: 10.1016/S0278-2626(03)00284-7. [DOI] [PubMed] [Google Scholar]
- Collette F, Van der Linden M. Brain imaging of the central executive component of working memory. Neurosci Biobehav Rev. 2002;26(2):105–125. doi: 10.1016/s0149-7634(01)00063-x. [DOI] [PubMed] [Google Scholar]
- Connors SL, Crowell DE, Eberhart CG, Copeland J, Newschaffer CJ, Spence SJ, et al. Beta2-adrenergic receptor activation and genetic polymorphisms in autism: Data from dizygotic twins. J Child Neurol. 2005;20(11):876–884. doi: 10.1177/08830738050200110401. [DOI] [PubMed] [Google Scholar]
- Cote F, Fligny C, Bayard E, Launay JM, Gershon MD, Mallet J, et al. Maternal serotonin is crucial for murine embryonic development. Proc Natl Acad Sci U S A. 2007;101(1):329–334. doi: 10.1073/pnas.0606722104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannlowski U, Ohrmann P, Bauer J, Deckert J, Hohoff C, Kugel H, et al. 5-httlpr biases amygdala activity in response to masked facial expressions in major depression. Neuropsychopharmacology. 2008;33(2):418–424. doi: 10.1038/sj.npp.1301411. [DOI] [PubMed] [Google Scholar]
- Degnan KA, Fox NA. Behavioral inhibition and anxiety disorders: Multiple levels of a resilience process. Dev Psychopathol. 2007;19(3):729–746. doi: 10.1017/S0954579407000363. [DOI] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3(1):79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- Dow-Edwards D, Mayes L, Spear L, Hurd Y. Cocaine and development: Clinical, behavioral, and neurobiological perspectives--a symposium report. Neurotoxicol Teratol. 1999;21(5):481–490. doi: 10.1016/s0892-0362(99)00008-2. [DOI] [PubMed] [Google Scholar]
- Drew MR, Simpson EH, Kellendonk C, Herzberg WG, Lipatova O, Fairhurst S, et al. Transient overexpression of striatal d2 receptors impairs operant motivation and interval timing. J Neurosci. 2007;27(29):7731–7739. doi: 10.1523/JNEUROSCI.1736-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott R. Executive functions and their disorders. Br Med Bull. 2003;65:49–59. doi: 10.1093/bmb/65.1.49. [DOI] [PubMed] [Google Scholar]
- Elston GN. Cortex, cognition and the cell: New insights into the pyramidal neuron and prefrontal function. Cereb Cortex. 2003;13(11):1124–1138. doi: 10.1093/cercor/bhg093. [DOI] [PubMed] [Google Scholar]
- Evans SM, Cone EJ, Henningfield JE. Arterial and venous cocaine plasma concentrations in humans: Relationship to route of administration, cardiovascular effects and subjective effects. J Pharmacol Exp Ther. 1996;279(3):1345–1356. [PubMed] [Google Scholar]
- Feenstra MG. Functional neuroteratology of drugs acting on adrenergic receptors. Neurotoxicology. 1992;13(1):55–63. [PubMed] [Google Scholar]
- Friedman E, Yadin E, Wang HY. Effect of prenatal cocaine on dopamine receptor-g protein coupling in mesocortical regions of the rabbit brain. Neuroscience. 1996;70(3):739–747. doi: 10.1016/s0306-4522(96)83011-9. [DOI] [PubMed] [Google Scholar]
- Fukumoto T, Kema IP, Levin M. Serotonin signaling is a very early step in patterning of the left-right axis in chick and frog embryos. Curr Biol. 2005;15(9):794–803. doi: 10.1016/j.cub.2005.03.044. [DOI] [PubMed] [Google Scholar]
- Gingras JL, O'Donnell KJ. State control in the substance-exposed fetus. I. The fetal neurobehavioral profile: An assessment of fetal state, arousal, and regulation competency. Annals of the New York Academy of Sciences. 1998;846:262–276. [PubMed] [Google Scholar]
- Goldman-Rakic PS. The prefrontal landscape: Implications of functional architecture for understanding human mentation and the central executive. Philosophical Transactions of the Royal Society of London - Series B: Biological Sciences. 1996;351(1346):1445–1453. doi: 10.1098/rstb.1996.0129. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Lidow MS, Gallager DW. Overlap of dopaminergic, adrenergic, and serotoninergic receptors and complementarity of their subtypes in primate prefrontal cortex. Journal of Neuroscience. 1990;10(7):2125–2138. doi: 10.1523/JNEUROSCI.10-07-02125.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Zhuang X, Stark K, Ramboz S, Oosting R, Kirby L, et al. Serotonin1a receptor acts during development to establish normal anxiety-like behaviour in the adult. Nature. 2002;416(6879):396–400. doi: 10.1038/416396a. [DOI] [PubMed] [Google Scholar]
- Hadders-Algra M, Touwen BC, Huisjes HJ. Long-term follow-up of children prenatally exposed to ritodrine. Br J Obstet Gynaecol. 1986;93(2):156–161. doi: 10.1111/j.1471-0528.1986.tb07880.x. [DOI] [PubMed] [Google Scholar]
- Harvey JA. Cocaine effects on the developing brain: Current status. Neurosci Biobehav Rev. 2004;27(8):751–764. doi: 10.1016/j.neubiorev.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Hedlund PB, Sutcliffe JG. Functional, molecular and pharmacological advances in 5-ht7 receptor research. Trends Pharmacol Sci. 2004;25(9):481–486. doi: 10.1016/j.tips.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Hirshfeld DR, Rosenbaum JF, Biederman J, Bolduc EA, Faraone SV, Snidman N, et al. Stable behavioral inhibition and its association with anxiety disorder. J Am Acad Child Adolesc Psychiatry. 1992;31(1):103–111. doi: 10.1097/00004583-199201000-00016. [DOI] [PubMed] [Google Scholar]
- Holmes A, le Guisquet AM, Vogel E, Millstein RA, Leman S, Belzung C. Early life genetic, epigenetic and environmental factors shaping emotionality in rodents. Neurosci Biobehav Rev. 2005;29(8):1335–1346. doi: 10.1016/j.neubiorev.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Holmes A, Lit Q, Murphy DL, Gold E, Crawley JN. Abnormal anxiety-related behavior in serotonin transporter null mutant mice: The influence of genetic background. Genes Brain Behav. 2003;2(6):365–380. doi: 10.1046/j.1601-1848.2003.00050.x. [DOI] [PubMed] [Google Scholar]
- Jenkins AJ, Keenan RM, Henningfield JE, Cone EJ. Correlation between pharmacological effects and plasma cocaine concentrations after smoked administration. J Anal Toxicol. 2002;26(7):382–392. doi: 10.1093/jat/26.7.382. [DOI] [PubMed] [Google Scholar]
- Jones L, Fischer I, Levitt P. Nonuniform alteration of dendritic development in the cerebral cortex following prenatal cocaine exposure. Cerebral Cortex. 1996;6(3):431–445. doi: 10.1093/cercor/6.3.431. [DOI] [PubMed] [Google Scholar]
- Jones LB, Stanwood GD, Reinoso BS, Washington RA, Wang HY, Friedman E, et al. In utero cocaine-induced dysfunction of dopamine d1 receptor signaling and abnormal differentiation of cerebral cortical neurons. Journal of Neuroscience. 2000;20(12):4606–4614. doi: 10.1523/JNEUROSCI.20-12-04606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan J, Snidman N. Early childhood predictors of adult anxiety disorders. Biol Psychiatry. 1999;46(11):1536–1541. doi: 10.1016/s0006-3223(99)00137-7. [DOI] [PubMed] [Google Scholar]
- Kagan J, Snidman N, Kahn V, Towsley S. The preservation of two infant temperaments into adolescence. Monogr Soc Res Child Dev. 2007;72(2):1–75. doi: 10.1111/j.1540-5834.2007.00436.x. vii; discussion 76-91. [DOI] [PubMed] [Google Scholar]
- Karmel BZ, Gardner JM. Prenatal cocaine exposure effects on arousal-modulated attention during the neonatal period. Developmental Psychobiology. 1996;29(5):463–480. doi: 10.1002/(SICI)1098-2302(199607)29:5<463::AID-DEV5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Kellendonk C, Simpson EH, Polan HJ, Malleret G, Vronskaya S, Winiger V, et al. Transient and selective overexpression of dopamine d2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron. 2006;49(4):603–615. doi: 10.1016/j.neuron.2006.01.023. [DOI] [PubMed] [Google Scholar]
- Kim H, Lim SW, Kim S, Kim JW, Chang YH, Carroll BJ, et al. Monoamine transporter gene polymorphisms and antidepressant response in koreans with late-life depression. Jama. 2006;296(13):1609–1618. doi: 10.1001/jama.296.13.1609. [DOI] [PubMed] [Google Scholar]
- Knudsen EI. Sensitive periods in the development of the brain and behavior. J Cogn Neurosci. 2004;16(8):1412–1425. doi: 10.1162/0898929042304796. [DOI] [PubMed] [Google Scholar]
- Lambe EK, Krimer LS, Goldman-Rakic PS. Differential postnatal development of catecholamine and serotonin inputs to identified neurons in prefrontal cortex of rhesus monkey. J Neurosci. 2000;20(23):8780–8787. doi: 10.1523/JNEUROSCI.20-23-08780.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauder JM. Hormonal and humoral influences on brain development. Psychoneuroendocrinology. 1983;8(2):121–155. doi: 10.1016/0306-4530(83)90053-7. [DOI] [PubMed] [Google Scholar]
- Leonardo ED, Hen R. Genetics of affective and anxiety disorders. Annu Rev Psychol. 2006;57:117–137. doi: 10.1146/annurev.psych.57.102904.190118. [DOI] [PubMed] [Google Scholar]
- Levitt P, Eagleson KL, Powell EM. Regulation of neocortical interneuron development and the implications for neurodevelopmental disorders. Trends Neurosci. 2004;27(7):400–406. doi: 10.1016/j.tins.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- Lidow MS. Consequences of prenatal cocaine exposure in nonhuman primates. Brain Res Dev Brain Res. 2003;147(1–2):23–36. doi: 10.1016/j.devbrainres.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Lucki I. The spectrum of behaviors influenced by serotonin. Biol Psychiatry. 1998;44(3):151–162. doi: 10.1016/s0006-3223(98)00139-5. [DOI] [PubMed] [Google Scholar]
- Luo X, Persico AM, Lauder JM. Serotonergic regulation of somatosensory cortical development: Lessons from genetic mouse models. Dev Neurosci. 2003;25(2–4):173–183. doi: 10.1159/000072266. [DOI] [PubMed] [Google Scholar]
- Maciag D, Simpson KL, Coppinger D, Lu Y, Wang Y, Lin RC, et al. Neonatal antidepressant exposure has lasting effects on behavior and serotonin circuitry. Neuropsychopharmacology. 2006;31(1):47–57. doi: 10.1038/sj.npp.1300823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann JJ, Brent DA, Arango V. The neurobiology and genetics of suicide and attempted suicide: A focus on the serotonergic system. Neuropsychopharmacology. 2001;24(5):467–477. doi: 10.1016/S0893-133X(00)00228-1. [DOI] [PubMed] [Google Scholar]
- Maschi S, Clavenna A, Campi R, Schiavetti B, Bernat M, Bonati M. Neonatal outcome following pregnancy exposure to antidepressants: A prospective controlled cohort study. Bjog. 2008;115(2):283–289. doi: 10.1111/j.1471-0528.2007.01518.x. [DOI] [PubMed] [Google Scholar]
- Mayes LC. A behavioral teratogenic model of the impact of prenatal cocaine exposure on arousal regulatory systems. Neurotoxicol Teratol. 2002;24(3):385–395. doi: 10.1016/s0892-0362(02)00200-3. [DOI] [PubMed] [Google Scholar]
- Mayes LC, Grillon C, Granger R, Schottenfeld R. Regulation of arousal and attention in preschool children exposed to cocaine prenatally. Annals of the New York Academy of Sciences. 1998;846:126–143. [PubMed] [Google Scholar]
- Meneses A. 5-ht system and cognition. Neurosci Biobehav Rev. 1999;23(8):1111–1125. doi: 10.1016/s0149-7634(99)00067-6. [DOI] [PubMed] [Google Scholar]
- Meyer A, Seidler FJ, Aldridge JE, Slotkin TA. Developmental exposure to terbutaline alters cell signaling in mature rat brain regions and augments the effects of subsequent neonatal exposure to the organophosphorus insecticide chlorpyrifos. Toxicol Appl Pharmacol. 2005;203(2):154–166. doi: 10.1016/j.taap.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Murphy DL, Lesch KP. Targeting the murine serotonin transporter: Insights into human neurobiology. Nat Rev Neurosci. 2008;9(2):85–96. doi: 10.1038/nrn2284. [DOI] [PubMed] [Google Scholar]
- Murphy EH, Fischer I, Friedman E, Grayson D, Jones L, Levitt P, et al. Cocaine administration in pregnant rabbits alters cortical structure and function in their progeny in the absence of maternal seizures. Experimental Brain Research. 1997;114(3):443–441. doi: 10.1007/pl00005652. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Koyama Y, Takahashi K, Tsurui H, Xiu Y, Ohtsuji M, et al. Requirement of tryptophan hydroxylase during development for maturation of sensorimotor gating. J Mol Biol. 2006;363(2):345–354. doi: 10.1016/j.jmb.2006.08.051. [DOI] [PubMed] [Google Scholar]
- Oberlander TF, Bonaguro RJ, Misri S, Papsdorf M, Ross CJ, Simpson EM. Infant serotonin transporter (slc6a4) promoter genotype is associated with adverse neonatal outcomes after prenatal exposure to serotonin reuptake inhibitor medications. Mol Psychiatry. 2008;13(1):65–73. doi: 10.1038/sj.mp.4002007. [DOI] [PubMed] [Google Scholar]
- Parks CL, Robinson PS, Sibille E, Shenk T, Toth M. Increased anxiety of mice lacking the serotonin1a receptor. Proc Natl Acad Sci U S A. 1998;95(18):10734–10739. doi: 10.1073/pnas.95.18.10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlaman JP, Thompson BL, Levitt P, Stanwood GD. Pharmacokinetic profile of cocaine following intravenous administration in the female rabbit. Eur J Pharmacol. 2007;563(1–3):124–129. doi: 10.1016/j.ejphar.2007.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson KH, Nonacs RM, Viguera AC, Heller VL, Petrillo LF, Brandes M, et al. Birth outcomes following prenatal exposure to antidepressants. J Clin Psychiatry. 2007;68(8):1284–1289. doi: 10.4088/jcp.v68n0817. [DOI] [PubMed] [Google Scholar]
- Persico AM, Di Pino G, Levitt P. Multiple receptors mediate the trophic effects of serotonin on ventroposterior thalamic neurons in vitro. Brain Res. 2006;1095(1):17–25. doi: 10.1016/j.brainres.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Pitzer M, Schmidt MH, Esser G, Laucht M. Child development after maternal tocolysis with beta-sympathomimetic drugs. Child Psychiatry Hum Dev. 2001;31(3):165–182. doi: 10.1023/a:1026419720410. [DOI] [PubMed] [Google Scholar]
- Popova NK. From genes to aggressive behavior: The role of serotonergic system. Bioessays. 2006;28(5):495–503. doi: 10.1002/bies.20412. [DOI] [PubMed] [Google Scholar]
- Ramboz S, Oosting R, Amara DA, Kung HF, Blier P, Mendelsohn M, et al. Serotonin receptor 1a knockout: An animal model of anxiety-related disorder. Proc Natl Acad Sci U S A. 1998;95(24):14476–14481. doi: 10.1073/pnas.95.24.14476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler KJ, Nemeroff CB. Role of serotonergic and noradrenergic systems in the pathophysiology of depression and anxiety disorders. Depress Anxiety. 2000;12 Suppl 1:2–19. doi: 10.1002/1520-6394(2000)12:1+<2::AID-DA2>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Rhodes MC, Seidler FJ, Abdel-Rahman A, Tate CA, Nyska A, Rincavage HL, et al. Terbutaline is a developmental neurotoxicant: Effects on neuroproteins and morphology in cerebellum, hippocampus, and somatosensory cortex. J Pharmacol Exp Ther. 2004;308(2):529–537. doi: 10.1124/jpet.103.060095. [DOI] [PubMed] [Google Scholar]
- Richardson GA, Conroy ML, Day NL. Prenatal cocaine exposure: Effects on the development of school-age children. Neurotoxicology & Teratology. 1996;18(6):627–634. doi: 10.1016/s0892-0362(96)00121-3. [DOI] [PubMed] [Google Scholar]
- Rodier PM. Chronology of neuron development: Animal studies and their clinical implications. Dev Med Child Neurol. 1980;22(4):525–545. doi: 10.1111/j.1469-8749.1980.tb04363.x. [DOI] [PubMed] [Google Scholar]
- Rodier PM. Vulnerable periods and processes during central nervous system development. Environ Health Perspect. 1994;102 Suppl 2:121–124. doi: 10.1289/ehp.94102121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier PM. Developing brain as a target of toxicity. Environ Health Perspect. 1995;103 Suppl 6:73–76. doi: 10.1289/ehp.95103s673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum JF, Biederman J, Bolduc-Murphy EA, Faraone SV, Chaloff J, Hirshfeld DR, et al. Behavioral inhibition in childhood: A risk factor for anxiety disorders. Harv Rev Psychiatry. 1993;1(1):2–16. doi: 10.3109/10673229309017052. [DOI] [PubMed] [Google Scholar]
- Schwartz CE, Snidman N, Kagan J. Adolescent social anxiety as an outcome of inhibited temperament in childhood. J Am Acad Child Adolesc Psychiatry. 1999;38(8):1008–1015. doi: 10.1097/00004583-199908000-00017. [DOI] [PubMed] [Google Scholar]
- Silverstone T. Appetite suppressants. A review. Drugs. 1992;43(6):820–836. doi: 10.2165/00003495-199243060-00003. [DOI] [PubMed] [Google Scholar]
- Simansky KJ, Kachelries WJ. Prenatal exposure to cocaine selectively disrupts motor responding to d-amphetamine in young and mature rabbits. Neuropharmacology. 1996;35(1):71–78. doi: 10.1016/0028-3908(95)00151-4. [DOI] [PubMed] [Google Scholar]
- Singer LT, Minnes S, Short E, Arendt R, Farkas K, Lewis B, et al. Cognitive outcomes of preschool children with prenatal cocaine exposure. Jama. 2004;291(20):2448–2456. doi: 10.1001/jama.291.20.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin TA, Auman JT, Seidler FJ. Ontogenesis of beta-adrenoceptor signaling: Implications for perinatal physiology and for fetal effects of tocolytic drugs. J Pharmacol Exp Ther. 2003;306(1):1–7. doi: 10.1124/jpet.102.048421. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Seidler FJ. The alterations in cns serotonergic mechanisms caused by neonatal chlorpyrifos exposure are permanent. Brain Res Dev Brain Res. 2005;158(1–2):115–119. doi: 10.1016/j.devbrainres.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Song ZM, Undie AS, Koh PO, Fang YY, Zhang L, Dracheva S, et al. D1 dopamine receptor regulation of microtubule-associated protein-2 phosphorylation in developing cerebral cortical neurons. J Neurosci. 2002;22(14):6092–6105. doi: 10.1523/JNEUROSCI.22-14-06092.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanwood GD, Levitt P. The effects of cocaine on the developing nervous system. In: Nelson CA, Luciana M, editors. Handbook of developmental cognitive neuroscience. MIT Press; 2001. pp. 519–536. [Google Scholar]
- Stanwood GD, Levitt P. Repeated i.V. Cocaine exposure produces long-lasting behavioral sensitization in pregnant adults, but behavioral tolerance in their offspring. Neuroscience. 2003;122(3):579–583. doi: 10.1016/j.neuroscience.2003.08.029. [DOI] [PubMed] [Google Scholar]
- Stanwood GD, Levitt P. Drug exposure early in life: Functional repercussions of changing neuropharmacology during sensitive periods of brain development. Current Opinion in Pharmacology. 2004;4:65–71. doi: 10.1016/j.coph.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Stanwood GD, Levitt P. Prenatal exposure to cocaine produces unique developmental and long-term adaptive changes in dopamine d1 receptor activity and subcellular distribution. J Neurosci. 2007;27(1):152–157. doi: 10.1523/JNEUROSCI.4591-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanwood GD, Parlaman JP, Levitt P. Anatomical abnormalities in dopaminoceptive regions of the cerebral cortex of dopamine d(1) receptor mutant mice. J Comp Neurol. 2005;487(3):270–282. doi: 10.1002/cne.20548. [DOI] [PubMed] [Google Scholar]
- Stanwood GD, Parlaman JP, Levitt P. Genetic or pharmacological inactivation of the dopamine d1 receptor differentially alters the expression of regulator of g-protein signalling (rgs) transcripts. Eur J Neurosci. 2006;24(3):806–818. doi: 10.1111/j.1460-9568.2006.04970.x. [DOI] [PubMed] [Google Scholar]
- Stanwood GD, Washington RA, Levitt P. Identification of a sensitive period of prenatal cocaine exposure that alters the development of the anterior cingulate cortex. Cerebral Cortex. 2001a;11:430–440. doi: 10.1093/cercor/11.5.430. [DOI] [PubMed] [Google Scholar]
- Stanwood GD, Washington RA, Shumsky JS, Levitt P. Prenatal cocaine exposure produces consistent developmental alterations in dopamine-rich regions of the cerebral cortex. Neuroscience. 2001b;106(1):5–14. doi: 10.1016/s0306-4522(01)00256-1. [DOI] [PubMed] [Google Scholar]
- Sutcliffe JS, Delahanty RJ, Prasad HC, McCauley JL, Han Q, Jiang L, et al. Allelic heterogeneity at the serotonin transporter locus (slc6a4) confers susceptibility to autism and rigid-compulsive behaviors. Am J Hum Genet. 2005;77(2):265–279. doi: 10.1086/432648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson B, Stanwood G, Levitt P. Society For Neuroscience. Washington, DC: 2005a. Double dissociation of the reinforcing properties of cocaine. [Google Scholar]
- Thompson BL, Levitt P, Stanwood GD. Prenatal cocaine exposure specifically alters spontaneous alternation behavior. Behav Brain Res. 2005b;164(1):107–116. doi: 10.1016/j.bbr.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Trask CL, Kosofsky BE. Developmental considerations of neurotoxic exposures. Neurol Clin. 2000;18(3):541–562. doi: 10.1016/s0733-8619(05)70210-3. [DOI] [PubMed] [Google Scholar]
- Walderhaug E, Magnusson A, Neumeister A, Lappalainen J, Lunde H, Refsum H, et al. Interactive effects of sex and 5-httlpr on mood and impulsivity during tryptophan depletion in healthy people. Biol Psychiatry. 2007;62(6):593–599. doi: 10.1016/j.biopsych.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Wang HY, Runyan S, Yadin E, Friedman E. Prenatal exposure to cocaine selectively reduces d1 dopamine receptor-mediated activation of striatal gs proteins. Journal of Pharmacology & Experimental Therapeutics. 1995a;273(1):492–498. [PubMed] [Google Scholar]
- Wang XH, Levitt P, Grayson DR, Murphy EH. Intrauterine cocaine exposure of rabbits: Persistent elevation of gaba-immunoreactive neurons in anterior cingulate cortex but not visual cortex. Brain Research. 1995b;689(1):32–46. doi: 10.1016/0006-8993(95)00528-x. [DOI] [PubMed] [Google Scholar]
- Wang XH, Levitt P, O'Brien Jenkins A, Murphy EH. Normal development of tyrosine hydroxylase and serotonin immunoreactive fibers innervating anterior cingulate cortex and visual cortex in rabbits exposed prenatally to cocaine. Brain Research. 1996;715(1–2):221–224. doi: 10.1016/0006-8993(96)00012-1. [DOI] [PubMed] [Google Scholar]
- Weinberger DR. From neuropathology to neurodevelopment. Lancet. 1995;36(8974):552–557. doi: 10.1016/s0140-6736(95)91386-6. [DOI] [PubMed] [Google Scholar]
- Whitaker-Azmitia PM. Role of serotonin and other neurotransmitter receptors in brain development: Basis for developmental pharmacology. Pharmacol Rev. 1991;43(4):553–561. [PubMed] [Google Scholar]
- Whitaker-Azmitia PM, Druse M, Walker P, Lauder JM. Serotonin as a developmental signal. Behavioural Brain Research. 1996;73(1–2):19–29. doi: 10.1016/0166-4328(96)00071-x. [DOI] [PubMed] [Google Scholar]
- Whitaker-Azmitia PM, Lauder JM, Shemmer A, Azmitia EC. Postnatal changes in serotonin receptors following prenatal alterations in serotonin levels: Further evidence for functional fetal serotonin receptors. Brain Res. 1987;430(2):285–289. doi: 10.1016/0165-3806(87)90161-1. [DOI] [PubMed] [Google Scholar]
- Zerrate MC, Pletnikov M, Connors SL, Vargas DL, Seidler FJ, Zimmerman AW, et al. Neuroinflammation and behavioral abnormalities after neonatal terbutaline treatment in rats: Implications for autism. J Pharmacol Exp Ther. 2007;322(1):16–22. doi: 10.1124/jpet.107.121483. [DOI] [PubMed] [Google Scholar]