

Abstract
Aims
Bindarit is an original compound with peculiar anti-inflammatory activity due to a selective inhibition of a subfamily of inflammatory chemokines, including the monocyte chemotactic proteins MCP-1/CCL2, MCP-3/CCL7, and MCP-2/CCL8. In this study, we investigated the effect of bindarit on neointima formation using two animal models of arterial injury: rat carotid artery balloon angioplasty and wire-induced carotid injury in apolipoprotein E-deficient (apoE−/−) mice.
Methods and results
Treatment of rats with bindarit (200 mg/kg/day) significantly reduced balloon injury-induced neointima formation by 39% at day 14 without affecting re-endothelialization and reduced the number of medial and neointimal proliferating cells at day 7 by 54 and 30%, respectively. These effects were associated with a significant reduction of MCP-1 levels both in sera and in injured carotid arteries of rats treated with bindarit. In addition, in vitro data showed that bindarit (10–300 µM) reduced rat vascular smooth muscle cell (VSMC) proliferation, migration, and invasion, processes contributing to the injury-induced neointima formation in vivo. Similar results were observed in hypercholesterolaemic apoE−/− mice in which bindarit administration resulted in a 42% reduction of the number of proliferating cells at day 7 after carotid injury and in a 47% inhibition of neointima formation at day 28. Analysis of the cellular composition in neointimal lesions of apoE−/− mice treated with bindarit showed that the relative content of macrophages and the number of VSMCs were reduced by 66 and 30%, respectively, compared with the control group.
Conclusion
This study demonstrates that bindarit is effective in reducing neointima formation in both non-hyperlipidaemic and hyperlipidaemic animal models of vascular injury by a direct effect on VSMC proliferation and migration and by reducing neointimal macrophage content. All of these data were associated with the inhibition of MCP-1 production.
Keywords: Bindarit, Neointima hyperplasia, Monocyte chemoattractant protein-1, Macrophages, Vascular smooth muscle cells
1. Introduction
Bindarit is an original indazolic derivative devoid of any immunosuppressive effects and with no activity on arachidonic acid metabolism that was shown to have anti-inflammatory activity in a number of experimental diseases including nephritis, arthritis, pancreatitis, and colitis.1–4 These pharmacological activities have been associated with its ability to interfere with monocyte recruitment, which has been ascribed to a selective inhibitory effect on the monocyte chemotactic protein (MCP) subfamily of CC inflammatory chemokines, including MCP-1/CCL2, MCP-3/CCL7, and MCP-2/CCL8.5 Phase II trials in patients with rheumatoid arthritis and lupus nephritis have shown that bindarit was well tolerated and significantly reduced urinary MCP-1 and albumin excretion in kidney disease.6,7
It is well known that chemokines have a crucial role in initiating and progressing neointima formation by controlling each step of the vascular remodelling in response to various noxious stimuli.8 Among pro-inflammatory CC chemokines, MCP-1 is receiving increasing attention. It has been demonstrated that eliminating the MCP-1 gene or blocking MCP-1 signalling decreases neointima hyperplasia after balloon- and stent-induced injury in several animal models;9–11 similarly, catheter-based adenovirus-mediated anti-monocyte chemoattractant gene therapy attenuates in-stent neointima formation in monkeys.12 Elevated circulating levels of MCP-1 were observed in patients with restenosis after coronary angioplasty.13
The induction of MCP-1 not only correlates with macrophage accumulation but there is strong evidence for an important role of MCP-1 in vascular smooth muscle cell (VSMC) proliferation and migration,14–16 processes that contribute substantially to neointima formation after arterial stenting and balloon angioplasty. In addition, it has been shown that MCP-1 has a pivotal role in vein graft thickening due to intima hyperplasia.17 These data suggest that an anti-inflammatory treatment based on the inhibition of MCP-1 may be an appropriate and reasonable approach for the prevention of neointima formation.
Here, we investigated the effect of bindarit on neointima formation in vivo using two well-known animal models of arterial injury: rat carotid artery balloon angioplasty and wire-induced carotid injury in apolipoprotein E-deficient (apoE−/−) mice. In addition, the effects of bindarit on VSMC proliferation and migration in vitro were also examined. The results provided in this study strongly support the beneficial effects of bindarit on the inflammatory/proliferative processes leading to neointima formation.
2. Methods
2.1. Treatments
Bindarit, 2-methyl-2-[[1-(phenylmethyl)-1H-indazol-3-yl]methoxy] propanoic acid (MW 324.38), was synthesized by Angelini (Angelini Research Center—ACRAF, Italy). Pharmacokinetic studies in rodents show that bindarit is well absorbed when administered by oral route, and it has a mean half-life of ∼9 h and, at dose regimen used in this study, reaches plasma levels in the range of 150–450 µM (Product data sheet, Angelini Research Center).
Animals were treated with bindarit, suspended in 0.5% methylcellulose aqueous solution, at the dose of 100 mg/kg given orally, by gastric gavage, twice a day.4 Rats were treated with bindarit from 2 days before angioplasty up to 14 days after, whereas apoE−/− mice were treated from 1 week before endothelial denudation up to 28 days after. In each experiment, control animals received an equal volume of methylcellulose (0.5 mL/100 g in rats; 0.1 mL/10 g in mice). The concentrations of bindarit used for in vitro experiments have been found previously to be effective at inhibiting MCP-1 synthesis in human monocytes and umbilical vein endothelial cells.5
2.2. Cell culture
Primary aortic VSMCs were isolated from the thoracic aorta of male Wistar rats or female apoE−/− mice as described previously16 and grown in Dulbecco's modified Eagle medium (DMEM; Cambrex Bio Science) supplemented with l-glutamine, 10% foetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin in a humidified incubator at 37°C in 5% CO2. Before initiation of assays, the VSMCs were switched into DMEM supplemented with 1% FBS for 48 h. Studies were performed with cells at passages 3–6.
2.3. Enzyme-linked immunosorbent assay for MCP-1 protein
Cells were used after the induction of quiescence in 24-well plastic culture plates at a density of 1.5 × 104 cells/well. The cells were stimulated with platelet derived growth factor-BB (PDGF-BB; 10 ng/mL; R&D Systems) in the presence or absence of bindarit (10–300 µM). After 6, 12, 24, and 48 h, media were collected, centrifuged at 2000 g for 15 min at 4°C, and supernatants were used for enzyme-linked immunosorbent assay (ELISA) (OptEIA™, Biosciences).
2.4. Proliferation assay
The cell proliferation assay was carried out using the MTT assay. VSMCs were plated on 24-well plastic culture plates at the density of 1.5 × 104 cells/well and then incubated with DMEM containing PDGF-BB (10 ng/mL) for 48 h in the presence or absence of bindarit (10–300 µM). The absorbance values were obtained with an ELISA assay reader (630 nm).
2.5. Chemotactic migration and invasion
VSMC migration was evaluated using a modified Boyden chamber (Corning 24 mm Transwell with 8.0 µm pore polycarbonate membrane insert) coated with rat-tail collagen I (Sigma-Aldrich). Biocoat Matrigel invasion chambers (with 8.0 µm pore) were used according to the manufacturer's instructions for invasion studies (Becton–Dickinson). Briefly, starved VSMCs were trypsinized and pre-treated or not with bindarit (10–300 µM) for 2 h. 5 × 105 cells were plated in the upper chamber in 150 µL of 1% FBS medium with or without bindarit (10–300 µM) and the lower chamber was filled with 600 µL of 1% FBS medium in the absence (untreated cells) or presence of PDGF-BB 10 ng/mL. After 6 h for migration assay or 48 h for invasion assay, the migrated cells were fixed and stained with haematoxylin. The number of migrated cells was counted in eight randomly chosen fields per insert.
2.6. Animals
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996), and Italian ministerial authorization (DL 116/92) was obtained to carry out the experimentation. Male Wistar rats (Harlan Laboratories) weighing 250 g and 8-week-old female apoE−/− mice (Charles River) were used for the present study. Animals were housed at the Department of Experimental Pharmacology, University of Naples Federico II.
2.7. Rat carotid balloon angioplasty
Rats were anaesthetized with an intraperitoneal injection of ketamine (100 mg/kg) (Gellini International) and xylazine (5 mg/kg) (Sigma). Endothelial denudation of the left carotid artery was performed with a balloon embolectomy catheter (2 F, Fogarty, Edwards Lifesciences) according to the procedure well validated in our laboratories.18 Some animals were subjected to anaesthesia and surgical procedure, without balloon injury (sham-operated group). Rats were euthanized 1, 7, and 14 days after angioplasty. Blood and carotid arteries were collected and processed as described below.
2.8. Atherogenic murine model of vascular injury
ApoE−/− mice were fed an atherogenic diet (21% fat, 0.15% cholesterol, 19.5% casein, wt/wt; TD88137, Mucedola) from 1 week before until 4 weeks after carotid injury performed as described previously,19 with minor modification. Briefly, mice were anaesthetized as described above, and endothelial injury of the left common carotid artery was performed with a 0.35 mm diameter flexible nylon wire introduced through the left external carotid artery and advanced to the aortic arch. The endothelium was damaged by passing the wire through the lumen of the artery three times. Blood and carotid arteries were collected 7 and 28 days after wire injury and processed as described below.
2.9. Evaluation of neointima formation
Carotid arteries from rats or apoE−/− mice were fixed by perfusion with phosphate-buffered saline (PBS; pH 7.2) followed by PBS containing 4% formaldehyde through a cannula placed in the left ventricle. Paraffin-embedded sections were cut (6 µm thick) from the approximate middle portion of the artery and stained with haematoxylin and eosin to demarcate cell types. Ten sections from each carotid artery were reviewed and scored under blind conditions. The cross-sectional areas of media and neointima were determined by a computerized analysis system (LAS, Leica).
2.10. Proliferating cell nuclear antigen analysis in injured rat carotid artery
Proliferating cell nuclear antigen (PCNA) analysis was used to quantify the proliferative activity of cells at the injury sites and was performed using monoclonal mouse anti-PCNA antibody (1:250, PC10, Sigma) and biotinylated anti-mouse secondary antibody (1:400, DakoCytomation). Slides were treated with streptavidin–HRP (DakoCytomation) and exposed to diaminobenzidine chromogen (DakoCytomation) with haematoxylin counterstain. The proliferating cell number in the rat carotid arteries was scored in 10 fields for each section, six sections from each carotid artery, and expressed as the percentage of total medial and neointimal cells positive for PCNA 7 days after angioplasty. The proliferating cell number in the mouse apoE−/− carotid arteries was scored in 10 sections from each carotid artery and expressed as the percentage of positive cells 7 days after wire injury.
2.11. MCP-1 immunohistochemistry
Rat carotid arteries (1, 7, and 14 days after angioplasty, or naive) were snap-frozen in liquid nitrogen in OCT embedding medium (Tissue Tek, Sakura Finetek). Ten cross-sections were cut (6 µm) from the approximate middle portion of the artery and used for MCP-1 detection. Sections were incubated with polyclonal goat anti-MCP-1 antibody (1:50, R-17, Santa Cruz) diluted in blocking buffer/0.3% Triton X-100 (MP Biomedicals) in PBS overnight before being washed in TNT wash buffer (Tris–HCl, pH 7.5, 0.15 M NaCl, and 0.05% Tween 20; Sigma). Sections incubated with goat non-immune serum were used as negative controls. Subsequently, sections were incubated with biotinylated anti-goat secondary antibody (1:400, DakoCytomation) diluted in blocking buffer/0.3% Triton X-100, washed in TNT wash buffer, treated with streptavidin–HRP, and exposed to diaminobenzidine chromogen with haematoxylin counterstain. The sections were photographed and the images were stored in the image analysis system (LAS, Leica).
2.12. Enzyme-linked immunosorbent assay
Rat carotid artery was crushed into powder and resuspended in 100 µL of lysis buffer (20 mM HEPES, 0.4 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, and 20% glycerol) with protease inhibitors (1 mM DTT, 0.5 mM PMSF, 15 µg/mL Try-inhibitor, 3 µg/mL pepstatin-A, 2 µg/mL leupeptin, and 40 µM benzamidine). After centrifugation at 13 000 g at 4°C for 30 min, MCP-1 in the supernatant was quantified using an ELISA kit (OptEIA™, Biosciences). All measurements were performed in duplicate. The values were corrected by protein concentrations measured by the Bio-Rad protein assay kit (Bio-Rad).
Serum MCP-1 levels (1, 7, and 14 days after angioplasty) were also measured in the same animals used above by ELISA (OptEIA™, Biosciences). The results are expressed as nanograms per millilitre.
2.13. Western blot analysis
The levels of CD68 were evaluated in total extracts prepared from two pooled rat carotid arteries. The extraction procedure was performed as described above. Protein concentration was determined by the Bio-Rad protein assay kit (Bio-Rad). Equivalent amounts of protein (50 µg) from each sample were electrophoresed in an 8% discontinuous polyacrylamide minigel. The proteins were transferred onto nitrocellulose membranes according to the manufacturer's instructions (Bio-Rad). The membranes were saturated by incubation with 10% non-fat dry milk in PBS/0.1% Triton X-100 for 3 h at room temperature and then incubated with anti-CD68 mouse antibody (1:1000; Serotec) or anti-β-actin (1:5000; Sigma) mouse antibody overnight at 4°C. The membranes were washed three times with 0.1% Tween 20 in PBS and then incubated with anti-mouse immunoglobulins coupled to peroxidase (1:1000; DakoCytomation) for 1 h at room temperature. The immune complexes were visualized by enhanced chemiluminescence (Amersham). Subsequently, the relative intensities of the bands were quantified by densitometric scanning of the X-ray films with a GS-800 Imaging Densitometer (Bio-Rad) and the computer program ‘Quantity One’ (Bio-Rad). Results are expressed as arbitrary units of CD68 protein levels, normalized to protein levels of the housekeeping protein β-actin.
2.14. Evaluation of re-endothelialization in injured rat carotid artery
Re-endothelialization was assessed 2 weeks after balloon injury by staining with Evans Blue dye (0.5 mL of 0.5% Evans Blue dye iv; Sigma) as described previously.20 Planimetric analysis with an image analysis program (LAS, Leica) was performed. Re-endothelialization was expressed as the percentage of re-endothelialized area vs. the total denuded area. To verify that the Evans Blue stain accurately depicted the presence or absence of endothelium, sections of completely or partially re-endothelialized carotid arteries (based on Evans Blue appearance) were stained with antibody to von Willebrand factor (vWF, Cytomation Dako) as described previously.21
2.15. Immunohistochemisty analysis in injured apoE−/− mouse carotid artery
Carotid arteries were snap-frozen in liquid nitrogen in OCT embedding medium. Fifteen cross-sections were cut (6 µm) from the approximate middle portion of the artery and used for MCP-1, α-smooth muscle actin (α-SMA), and macrophage detection by immunofluorescence. For staining, the sections were processed as described above and incubated with polyclonal goat anti-mouse MCP-1 antibody (1:50, M-18, Santa Cruz) or rat anti-F4/80 monoclonal antibody (1:50, clone BM8, Abcam) diluted in blocking buffer/0.3% Triton X-100 (MP Biomedicals) in PBS overnight before being washed in TNT wash buffer. Sections incubated with non-immune goat serum or an isotype-matched control antibody were used as negative controls. Subsequently, the sections were incubated with 1:75 Texas Red-donkey anti-goat IgG (Jackson ImmunoResearch Laboratories) or with 1:200 biotinylated anti-rat secondary antibody (DakoCytomation), amplified with Tyramide Signal Amplification Systems (PerkinElmer), and revealed with streptavidin–FITC (1:50, DakoCytomation). Monoclonal anti-α-SMA FITC (1:250, clone 1A4, Sigma) was added in blocking buffer for 1 h before washing as described above. DAPI was used to identify nuclei. Images were taken using an AxioCam HRc video-camera (Zeiss) connected to an Axioplan fluorescence microscope (Zeiss) using the AxioVision 3.1 software.
The neointimal areas stained for F4/80 were determined in digitized images (five sections per mouse), and positive areas for specific immunostaining were quantified (NIH Imaging; http://rsb.info.nih.gov/ij). Data are expressed as the percentage of the immunostained area per total neointimal area.
Serial carotid paraffin-embedded sections were cut (5 µm) and used for detection of calponin (1:100, clone hCP, Sigma). Ten sections from each carotid artery were reviewed and scored under blind conditions. The number of calponin-positive cells in neointima was counted.
2.16. Evaluation of MCP-1, total cholesterol, and triglyceride serum levels in apoE−/− mice
The concentrations of serum MCP-1, triglycerides, and cholesterol were determined using enzymatic immunoassays according to the manufacturer's instructions (OptEIA™, Biosciences; Serum Triglyceride Kit, Sigma; Cholesterol Assay Kit, Cayman Chemical).
2.17. Statistical analysis
Results are expressed as mean ± SEM of n animals for in vivo experiments and mean ± SEM of multiple experiments for in vitro assays. Student's t-test was used to compare two groups and ANOVA (two-tailed P-value) was used with the Dunnett post hoc test for multiple groups using Graph Pad Instat 3 software (San Diego, CA, USA). The level of statistical significance was 0.05 per test.
3. Results
3.1. Effect of bindarit on rat VSMC proliferation and migration
Initiation and maintenance of VSMCs proliferation is a critical event in the pathogenesis of neointima formation. As shown in Figure 1A, bindarit at 100 and 300 µM significantly inhibited PDGF-BB-induced rat VSMCs proliferation by 27% (P < 0.05, n = 3) and 42% (P < 0.01, n = 3), respectively. Similar results were obtained with apoE−/− mice VSMC (data not shown). Another key mechanism of neointima formation is mitogen-mediated migration of VSMCs. Therefore, we evaluated the effects of bindarit on PDGF-BB-induced rat VSMCs chemotaxis. Bindarit inhibited significantly (P < 0.01, n = 3) chemotactic migration at 100 and 300 µM by 45 and 50%, respectively (Figure 1B). Moreover, bindarit (100 µM) also significantly reduced rat VSMCs invasion (by 30%, P < 0.01, n = 3, Figure 1C) through the Matrigel barrier which mimics extracellular matrix. Analysis of cell viability (>95%) demonstrated that it was not affected by bindarit at the concentrations used in this study (data not shown).
Figure 1.

(A) Effect of bindarit (10–300 µM) on rat VSMC proliferation, (B) migration, and (C) invasion performed as described in Section 2. Results are expressed as mean ± SEM from three separate experiments. *P < 0.05, **P < 0.01 vs. platelet derived growth factor-BB (PDGF-BB); °°P < 0.01, °°°P < 0.001 vs. unstimulated cells.
3.2. Effect of bindarit on MCP-1 production
To determine whether the anti-proliferative and anti-migratory effects of bindarit were associated with MCP-1 inhibition, protein concentration of MCP-1 in the supernatant of cultured rat VSMCs was determined by ELISA. As shown in Table 1, stimulation of VSMCs with PDGF-BB (10 ng/mL) caused a time-dependent increased release of MCP-1 compared with that observed in unstimulated cells. When rat VSMCs were stimulated with PDGF-BB in the presence of bindarit (10–300 µM), a concentration-related inhibition of MCP-1 production was observed.
Table 1.
Effect of bindarit on MCP-1 production by platelet derived growth factor-BB (PDGF-BB)-stimulated VSMCs
| MCP-1 (ng/mL) |
||||
|---|---|---|---|---|
| 6 h | 12 h | 24 h | 48 h | |
| Unstimulated cells | 0.32 ± 0.02 | 3.2 ± 0.21 | 18.5 ± 0.9 | 34.1 ± 1.4 |
| PDGF-BB 10 ng/mL | 3.1 ± 0.1°° | 20.8 ± 1.58°° | 59.6 ± 0.4°° | 142.6 ± 2.8°° |
| PDGF-BB 10 ng/mL + bindarit 10 µM | 3.0 ± 0.2 | 18.3 ± 1.2 | 50.9 ± 1.2** | 128.0 ± 3.7** |
| PDGF-BB 10 ng/mL + bindarit 30 µM | 1.5 ± 0.1** | 13.7 ± 0.5** | 42.8 ± 0.6** | 87.3 ± 1.7** |
| PDGF-BB 10 ng/mL + bindarit 100 µM | 1.3 ± 0.2** | 11.9 ± 0.4** | 34.0 ± 1.0** | 80.1 ± 1.8** |
| PDGF-BB 10 ng/mL + bindarit 300 µM | 0.8 ± 0.1** | 4.8 ± 0.5** | 29.3 ± 1.1** | 72.1 ± 1.3** |
Results are expressed as mean ± SEM of three separate experiments performed in triplicate.
°°P < 0.01 vs. unstimulated cells.
**P < 0.01 vs. PDGF-BB.
3.3. Effect of bindarit on neointima formation in rat carotid arteries
To determine the efficacy of a systemic treatment with bindarit for the limitation of neointimal hyperplasia, a rat carotid arterial injury model was used. A remarkable increase in the number of PCNA-positive cells was demonstrated in both the media and neointima 7 days after injury in control rats, which was significantly reduced (P < 0.001, n = 9) in the bindarit-treated group (200 mg/kg/day) by 54 and 30%, respectively (Figure 2A). Bindarit caused a significant inhibition of neointima formation by 39% (P < 0.01, n = 19) at day 14 compared with the control animals (Figure 2B). Medial area (0.124 ± 0.003 mm2 in the sham group) was not affected by both vascular injury and bindarit.
Figure 2.

(A) Photomicrographs showing the effect of bindarit (200 mg/kg/day) on PCNA-positive cells in rat carotid arteries at day 7 after injury (magnification ×400). Results are expressed as mean ± SEM of the percentage of total medial and neointimal cells positive for PCNA as described in Section 2, where n = 9 rats for each group. ***P < 0.001 vs. control group. (B) Photomicrographs showing the effect of bindarit (200 mg/kg/day) on neointima formation in rat carotid arteries 14 days after balloon injury (magnification ×100). Results are expressed as mean ± SEM, where n = 15–19 rats. **P < 0.01 vs. control group. (C) Effect of bindarit (200 mg/kg/day) on monocyte/macrophage marker CD68 protein expression in carotid arteries 14 days after angioplasty. Equal loading was confirmed by β-acting staining. Results are expressed as mean ± SEM of three separate experiments. **P < 0.01 vs. control group. (D) Effect of bindarit (200 mg/kg/day) on MCP-1 expression in rat carotid arteries 1, 7, and 14 days after balloon injury evaluated by ELISA. Results are expressed as mean ± SEM of MCP-1 levels normalized with protein concentrations, where n = 5 (see Section 2). *P < 0.05; ***P < 0.001 vs. control group; °°°P < 0.001 vs. sham-operated animals.
3.4. Effect of bindarit on monocytes/macrophages infiltration in rat carotid arteries
Western blot analysis was performed to examine the effect of bindarit on the carotid monocyte/macrophage content. Monocyte/macrophage marker CD68 was highly expressed in carotid arteries 14 days after angioplasty when compared with that of sham-operated animals (Figure 2C). Bindarit significantly reduced CD68 levels as shown by relative densitometric analysis.
3.5. Effect of bindarit on MCP-1 production in rat carotid arteries
We observed a significant time-dependent increase in MCP-1 production in the injured arteries at 1, 7, and 14 days after angioplasty when compared with that of sham-operated animals (Figure 2D). Bindarit was able to inhibit the MCP-1 protein expression throughout the time course considered, by 31% (P < 0.05; n = 5), 38% (P < 0.05; n = 5), and 49% (P < 0.001; n = 5) at 1, 7, and 14 days after injury, respectively. In the contralateral carotid artery, no significant changes were observed throughout the observation period compared with those measured in sham-operated animals. In carotid arteries from naive animals, the levels of MCP-1 measured were 0.74 ± 0.34 ng/mg (n = 5).
3.6. Effect of bindarit on MCP-1 localization in rat carotid arteries
Non-injured carotid arteries lacked immunoreactivity for MCP-1, whereas injured arteries stained strongly for MCP-1 (Figure 3). Negative controls showed no signal. MCP-1-positive staining was detectable in media of injured vessel from days 1 up to 14 and in neointimal cells at days 7 and 14. MCP-1 localization was not modified by bindarit, whereas the drug treatment resulted in a lower MCP-1 expression in both media and neointima (Figure 3).
Figure 3.

Immohistochemical localization of MCP-1 expression in rat carotid arteries 1, 7, and 14 days after angioplasty.
3.7. Effect of bindarit on re-endothelialization in rat carotid arteries
Evans Blue staining identifies segments of injured carotid arteries that have not been re-endothelialized. As shown in Figure 4, the presence of intact endothelium in the carotid artery of naive rats was demonstrated by the absence of Evans Blue staining. Immunohistochemical staining with antibody to vWF verified the presence of endothelium. In contrast, the entire area of the artery harvested 1 day after injury was stained by Evans Blue. The absence of positive staining with antibody to vWF confirmed the observation. Analysis of samples at 2 weeks from angioplasty showed that bindarit treatment did not affect re-endothelialization of arteries when compared with control animals.
Figure 4.
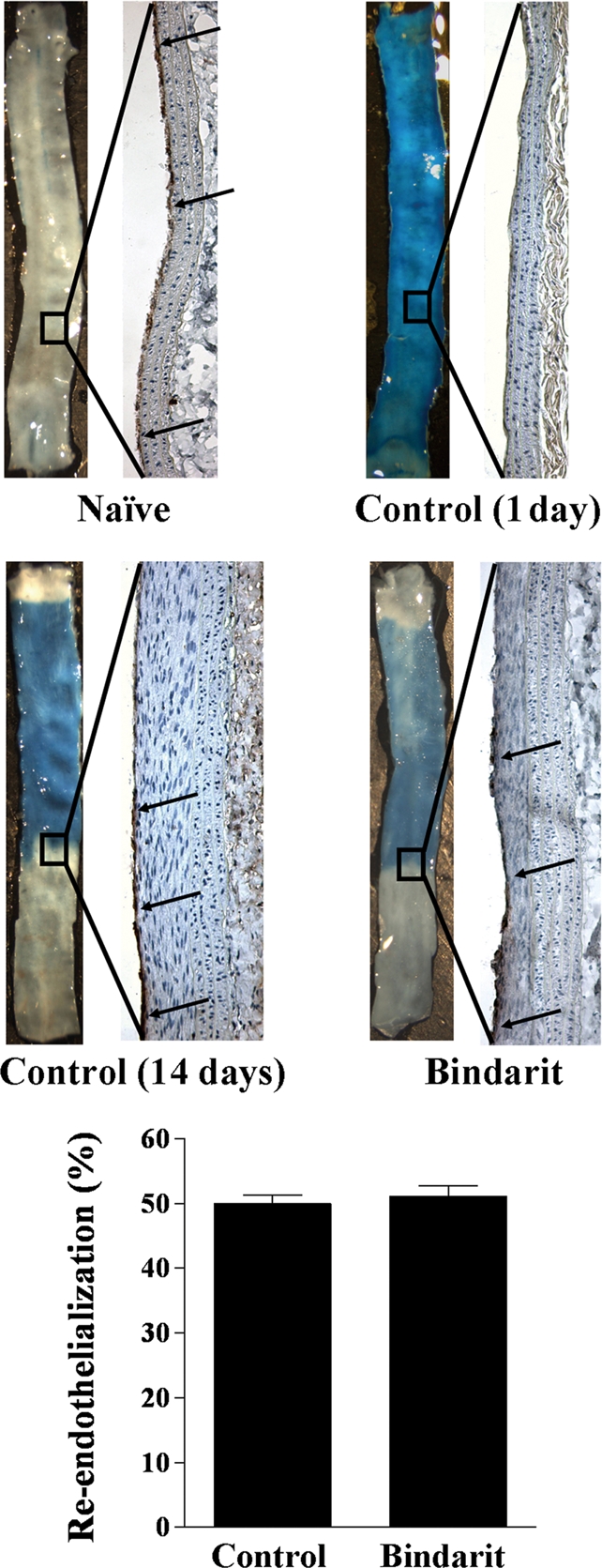
Extent of re-endothelialization in injured carotid arteries evaluated by Evans Blue staining. Representative arteries harvested from naive animals, as well as 1 and 14 days after carotid injury in vehicle- or bindarit-treated rats. Evans Blue staining identifies segments of each artery that have not been recovered by endothelium. Immunohistochemical staining with antibody to vWF verified the presence of endothelium. Re-endothelialization was expressed as the percentage of re-endothelialized area vs. the total denuded area (bottom panel) (n = 5).
3.8. Effect of bindarit on MCP-1 serum levels
A time-dependent increase in MCP-1 serum concentration was observed in rats subjected to angioplasty (Table 2). Bindarit caused a significant inhibition of MCP-1 serum levels at day 1 by 20% (P < 0.05, n = 10) and at days 7 and 14 by 30% (P < 0.001, n = 10) compared with their respective control groups. In naive animals, the MCP-1 serum level was 29.4 ± 3.0 ng/mL (n = 5).
Table 2.
Effect of bindarit on MCP-1 serum levels in rats
| Group | MCP-1 (ng/mL) |
||
|---|---|---|---|
| 1-day | 7-day | 14-day | |
| Sham-operated | 37.1 ± 2.4 | 35.4 ± 1.2 | 32.8 ± 2.7 |
| Control | 69.2 ± 3.5°° | 51.4 ± 2.2°° | 53.7 ± 3.9°° |
| Bindarit (200 mg/kg/day) | 55.6 ± 3.0* | 36.2 ± 2.4*** | 37.2 ± 2.1*** |
Results are expressed as mean ± SEM, where n = 5–10 rats per each time point considered.
°°P < 0.01 vs. sham-operated animals.
*P < 0.05.
***P < 0.001 vs. control group.
3.9. Effect of bindarit on neointima formation in apoE−/− mice
To evaluate the effect of bindarit after arterial injury in hyperlipidaemic animals, carotid endothelial denudation was performed in apoE−/− mice fed an atherogenic diet. Seven days after injury, the number of PCNA-positive cells was significantly reduced (42%; P < 0.05, n = 10) by treatment with bindarit (200 mg/kg/day) compared with control mice (Figure 5A). Neointimal area was reduced by 47% in apoE−/− mice treated with bindarit compared with control mice 28 days after injury (Figure 5B). Moreover, the apoE−/− mice receiving bindarit showed a 66% reduction in the relative content of F4/80-positive macrophages and a 30% reduction in the number of VSMCs in neointimal lesion (Figure 5C and D).
Figure 5.

(A) Photomicrographs showing the effect of bindarit (200 mg/kg/day) on PCNA-positive cells in apoE−/− mice 7 days after injury (magnification ×400). Results are expressed as mean ± SEM of the percentage of total PCNA-positive cells as described in Section 2, where n = 10 rats for each group. *P < 0.05 vs. control group. (B) Photomicrographs showing the effect of bindarit (200 mg/kg/day) on neointima formation in apoE−/− mice 28 days after carotid injury (magnification ×100). Results are expressed as mean ± SEM, where n = 10 for each group; **P < 0.01 vs. control group. (C) Photomicrographs showing the effect of bindarit (200 mg/kg/day) on macrophage content in carotid arteries 28 days after injury (magnification ×400). Results are expressed as mean ± SEM of the percentage of F4/80 immunostained area per total neointima area, where n = 10 for each group; ***P < 0.001 vs. control group. (D) Photomicrographs showing the effect of bindarit (200 mg/kg/day) on neointimal VSMCs content in carotid arteries 28 days after injury (magnification ×100). Results are expressed as mean ± SEM of the calponin-immunostained cell number, where n = 10 for each group; *P < 0.05 vs. control group. (E) Immunofluorescence visualization of α-SMA (green) and MCP-1 (red) in mouse carotid arteries 28 days after injury. DAPI (blue) was used to locate nuclei. Co-localization of MCP-1 and α-actin (yellow) was evident in some cells in both media and neointima (magnification ×200).
Injured carotid arteries stained strongly for MCP-1 detectable in media and neointima 28 days after injury. Co-localization of MCP-1 and α-actin was evident in both media and neointima. MCP-1 localization was not modified by bindarit, but again the drug reduced both medial and neointimal MCP-1 expression (Figure 5E).
Total cholesterol levels did not differ between groups (1036 ± 38 mg/dL in bindarit-treated group, n = 10 vs. 1167 ± 50 mg/dL in control group, n = 10), whereas triglyceride levels were significantly lower (P < 0.05) in bindarit-treated animals (191.6 ± 21.3 mg/dL, n = 10) than in the control group (260.7 ± 20.6 mg/dL, n = 10). Moreover, bindarit caused a significant inhibition (P < 0.01) of MCP-1 serum levels by 42% (131.0 ± 15.6 pg/mL in bindarit-treated group, n = 10 vs. 224.8 ± 28.3 pg/mL in control group, n = 10; P < 0.01).
4. Discussion
The results provided in this study show that bindarit given systemically significantly reduced neointimal formation in animal models of arterial injury by inhibiting VSMC proliferation/migration, and macrophage infiltration; these effects correlated with a reduction in MCP-1 synthesis.
Bindarit is an original compound selected by screening a series of indazolic derivatives endowed with peculiar anti-inflammatory activity associated with a selective inhibition of a subfamily of CC inflammatory chemokines, including MCP-1/CCL2, MCP-3/CCL7, and MCP-2/CCL8, showing no effect on other CC and CXC chemokines such as MIP-1α/CCL3, MIP-1β/CCL4, MIP-3/CCL23, RANTES/CCL5, and IL8/CXCL8.1–5
After vascular damage, the rat carotid artery develops neointimal formation, mainly due to proliferation and migration of VSMCs, that causes a clear narrowing of the vessel lumen.
Neointima formation contributes to the development of restenosis after coronary artery angioplasty, with or without stenting, in which a pivotal mechanism is represented by the loss of differentiation of VSMCs that were able to proliferate and migrate.22 It is well known that among pro-inflammatory CC chemokines, MCP-1 is implicated in all these processes and the source of this chemokine is likely to include the major cells in injured arteries, such as endothelial cells, VSMCs, and macrophages.23
In injured carotid arteries from animals treated with bindarit, a significant inhibition of neointima formation associated with a reduced MCP-1 production was observed. Bindarit did not modify MCP-1 localization but reduced MCP-1 expression in both media and neointima. Moreover, bindarit caused significant inhibition of MCP-1 serum levels. Increased levels of circulating MCP-1 in animals subjected to vascular injury are in keeping with an active role for this cytokine in tissue pathogenesis and correlate with epidemiological evidence showing higher MCP-1 plasma levels associated with human restenosis.13,24
Bindarit showed both in vivo and in vitro antiproliferative effects. For example, bindarit diminished the number of PCNA-positive proliferating cells in the media and intima 7 days after angioplasty, concomitantly with the beginning of neointimal formation, without affecting re-endothelialization evaluated 14 days after injury. In vitro studies further demonstrated inhibitory effects of bindarit on PDGF-BB-stimulated rat VSMC proliferation and migration. These effects were associated with a significant and concentration-related inhibition of MCP-1 amounts measured in the supernatants of stimulated cells treated with bindarit. It is well known that MCP-1 activity is in part due to recruitment of monocytes/macrophages that are responsible for local production of cytokines, but MCP-1 may also directly induce VSMC proliferation and migration through cell cycle proteins and intracellular proliferative signals.14–16
Our results also showed that bindarit reduced monocytes/macrophages recruitment in injured rat carotid arteries. The rat balloon angioplasty model could be considered ideal to study the proliferation of VSMCs in vivo; however, it is not an ideal model for the study of monocytes/macrophages recruitment, considering the injury is performed in a non-atherosclerotic arterial bed.25,26
It is well known that hypercholesterolaemia, a potent trigger of vessel wall inflammation in atherosclerosis, induces MCP-1 expression in VSMCs and upregulates CCR2 in human monocytes, enhancing monocyte recruitment after arterial injury and thus mediates the exacerbation of neointimal growth.27–29 In our experiments, no clear atherosclerotic lesions were detectable in the aortic root of mice 28 days after injury. However, we and others have demonstrated that lymphocytes already reside into the adventitia of arterial wall of apoE−/− mice even before the onset atherosclerosis.30,31 In this light, the hyperlipidaemic mice tool represents a step forward to the rat balloon angioplasty model, allowing us to study the effect of bindarit in the contest of increased vascular inflammation. Our results clearly demonstrated that bindarit reduced neointima formation by reducing proliferating rate and macrophage infiltration, effects associated with a reduced expression of MCP-1 in injured vessels. In hypercholesterolaemic rabbits, gene transfer of a plasmid coding for a mutant form of MCP-1 appeared to reduce neointima formation after balloon injury, mainly inhibiting macrophage infiltration to the injured vessels.32 Similarly, in hypercholesterolaemic apoE−/− mice also deficient in CCR2, neointimal lesions after wire injury of the carotid artery were diminished and showed a marked reduction in macrophage content compared with apoE−/−/CCR2+/+ mice.33
In conclusion, this study demonstrates that bindarit is effective in reducing neointima formation both in a non-hyperlipidaemic animal model of vascular injury, mainly by a direct effect on VSMC proliferation/migration, and in hyperlipidaemic animals by reducing macrophage infiltration.
The exploitation of the chemokine system as a drug target in vascular pathology has relied mainly on the development of receptor antagonists and blocking antibodies.23 However, the attempt to block chemokines and their receptors in humans is more complex.6 Here, we report the use of bindarit, an inhibitor of MCP-1 synthesis, as a potentially viable approach to control neointimal formation even if the clinical effects cannot be immediately predicted and further experiments, as well as clinical trials, will be necessary.
Funding
This work was supported by the Angelini (ACRAF, Italy; 004VPO7085; 004VPO7186) awarded to A.I. P.M. is supported by an IMB Capacity Building Award. Funding to pay the Open Access publication charges for this article was provided by ACRAF.
Acknowledgements
The authors thank Dr Gary Dever (Centre for Biophotonics, SIPBS, University of Strathclyde, Glasgow, UK) for the insightful English revision and observations.
Conflict of interest: A.I. received the project grants (004VPO7085; 004VPO7186) for this study. A.G., G.M., and G.B. are Angelini employees.
References
- 1.Zoja C, Corna G, Morigi M, Donarelli R, Guglielmotti A, Pinza M, et al. Bindarit retards renal disease and prolongs survival in murine lupus autoimmune disease. Kidney Int. 1998;53:726–734. doi: 10.1046/j.1523-1755.1998.00804.x. [DOI] [PubMed] [Google Scholar]
- 2.Guglielmotti A, D'Onofrio E, Coletta I, Aquilini L, Milanese C, Pinza M. Amelioration of rat adjuvant arthritis by therapeutic treatment with bindarit, an inhibitor of MCP-1 and TNF-α production. Inflamm Res. 2002;51:252–258. doi: 10.1007/pl00000301. [DOI] [PubMed] [Google Scholar]
- 3.Bhatia M, Devi Ramnath RD, Chevali L, Guglielmotti A. Treatment with bindarit, a blocker of MCP-1 synthesis, protects mice against acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1259–G1265. doi: 10.1152/ajpgi.00435.2004. [DOI] [PubMed] [Google Scholar]
- 4.Bhatia M, Landolfi C, Basta F, Bovi G, Devi Ramnath RD, Capezzone de Joannon A, et al. Treatment with bindarit, an inhibitor of MCP-1 synthesis, protects mice against trinitrobenzene sulfonic acid-induced colitis. Inflamm Res. 2008;57:1–8. doi: 10.1007/s00011-008-7210-y. [DOI] [PubMed] [Google Scholar]
- 5.Mirolo M, Fabbri M, Sironi M, Vecchi A, Guglielmotti A, Mangano G, et al. Impact of the anti-inflammatory agent on the chemokinome: selective inhibition of the monocyte chemotactic proteins. Eur Cytokine Netw. 2008;19:119–122. doi: 10.1684/ecn.2008.0133. [DOI] [PubMed] [Google Scholar]
- 6.Perico N, Benigni A, Remuzzi G. Present and future drug treatments for chronic kidney diseases: evolving targets in renoprotection. Nat Rev Drug Discov. 2008;7:936–853. doi: 10.1038/nrd2685. [DOI] [PubMed] [Google Scholar]
- 7.The Pharmaprojects Database, Informa UK Ltd 2008. www.pharmaprojects.com. (accession no. 17736)
- 8.Schober A. Chemokines in vascular dysfunction and remodeling. Arterioscler Thromb Vasc Biol. 2008;28:1950–1959. doi: 10.1161/ATVBAHA.107.161224. [DOI] [PubMed] [Google Scholar]
- 9.Furukawa Y, Matsumori A, Ohashi N, Shioi T, Ono K, Harada A, et al. Anti-monocyte chemoattractant protein-1/monocyte chemotactic and activating factor antibody inhibits neointimal hyperplasia in injured rat carotid arteries. Circ Res. 1999;84:306–314. doi: 10.1161/01.res.84.3.306. [DOI] [PubMed] [Google Scholar]
- 10.Egashira K, Koyanagi M, Kitamoto S, Ni W, Kataoka C, Moroshita R, et al. Anti-monocyte chemoattractant protein-1 therapy inhibits vascular remodeling in rats: blockade of MCP-1 activity after intramuscular transfer of a mutant gene inhibits vascular remodeling induced by chronic blockade of NO synythesis. FASEB J. 2000;14:1974–1978. doi: 10.1096/fj.00-0141com. [DOI] [PubMed] [Google Scholar]
- 11.Egashira K, Nakano K, Ohtani K, Funakoshi K, Zhao G, Ihara Y, et al. Local delivery of anti-monocyte chemoattractant protein-1 by gene-eluting stents attenuates in-stent stenosis in rabbits and monkeys. Arterioscler Thromb Vasc Biol. 2007;27:2563–2568. doi: 10.1161/ATVBAHA.107.154609. [DOI] [PubMed] [Google Scholar]
- 12.Nakano K, Egashira K, Ohtani K, Zhao G, Funakoshi K, Ihara Y, et al. Catheter-based adenovirus-mediated anti-monocyte chemoattractant gene therapy attenuates in-stent neointima formation in cynomolgus monkeys. Atherosclerosis. 2007;194:309–316. doi: 10.1016/j.atherosclerosis.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 13.Cipollone F, Marini M, Fazia M, Pini B, Iezzi A, Reale M, et al. Elevated circulating levels of monocyte chemoattractant protein-1 in patients with restenosis after coronary angioplasty. Arterioscler Thromb Vasc Biol. 2001;21:327–334. doi: 10.1161/01.atv.21.3.327. [DOI] [PubMed] [Google Scholar]
- 14.Selzman CH, Miller SA, Zimmerman MA, Gamboni-Robertson F, Harken AH, Banerjee A. Monocyte chemotactic protein-1 directly induces human vascular smooth muscle proliferation. Am J Physiol Heart Circ Physiol. 2002;283:H1455–H1461. doi: 10.1152/ajpheart.00188.2002. [DOI] [PubMed] [Google Scholar]
- 15.Massberg S, Vogt F, Dickfeld T, Brand K, Page S, Gawaz M. Activated platelets trigger an inflammatory response and enhance migration of aortic smooth muscle cells. Thromb Res. 2003;110:187–194. doi: 10.1016/s0049-3848(03)00342-6. [DOI] [PubMed] [Google Scholar]
- 16.Parenti A, Bellik L, Brogelli L, Filippi S, Ledda F. Endogenous VEGF-A is responsible for mitogenic effects of MCP-1 on vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2004;286:H1978–H1984. doi: 10.1152/ajpheart.00414.2003. [DOI] [PubMed] [Google Scholar]
- 17.Schepers A, Eefting D, Bonta PI, Grimbergen JM, de Vries MR, van Weel V, et al. Anti-MCP-1 gene therapy inhibits vascular smooth muscle cells proliferation and attenuates vein graft thickening both in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2006;26:2063–2069. doi: 10.1161/01.ATV.0000235694.69719.e2. [DOI] [PubMed] [Google Scholar]
- 18.Maffia P, Grassia G, Di Meglio P, Carnuccio R, Berrino L, Garside P, et al. Neutralization of interleukin-18 inhibits neointimal formation in a rat model of vascular injury. Circulation. 2006;114:430–437. doi: 10.1161/CIRCULATIONAHA.105.602714. [DOI] [PubMed] [Google Scholar]
- 19.Lindner V, Fingerle J, Reidy MA. Mouse model of arterial injury. Circ Res. 1993;73:792–796. doi: 10.1161/01.res.73.5.792. [DOI] [PubMed] [Google Scholar]
- 20.Asahara T, Chen D, Tsurumi Y, Kearney M, Rossow S, Passeri J, et al. Accelerated restitution of endothelial integrity and endothelium-dependent function after phVEGF165 gene transfer. Circulation. 1996;94:3291–3302. doi: 10.1161/01.cir.94.12.3291. [DOI] [PubMed] [Google Scholar]
- 21.Yue TL, Vickery-Clark L, Louden CS, Gu JL, Ma XL, Narayanan PK, et al. Selective estrogen receptor modulator idoxifene inhibits smooth muscle cell proliferation, enhances reendothelialization, and inhibits neointimal formation in vivo after vascular injury. Circulation. 2000;102:III281–III288. doi: 10.1161/01.cir.102.suppl_3.iii-281. [DOI] [PubMed] [Google Scholar]
- 22.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 23.Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95:858–866. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- 24.Martinovic I, Abegunewardene N, Seul M, Vosseler M, Horstick G, Buerke M, et al. Elevated monocyte chemoattractant protein-1 serum levels in patients at risk for coronary artery disease. Circ J. 2005;69:1484–1489. doi: 10.1253/circj.69.1484. [DOI] [PubMed] [Google Scholar]
- 25.Reidy MA, Fingerle J, Lindner V. Factors controlling the development of arterial lesions after injury. Circulation. 1992;86:III-43–III-46. [PubMed] [Google Scholar]
- 26.Roque M, Fallon JT, Badimon JJ, Zhang WX, Taubman MB, Reis ED. Mouse model of femoral artery denudation injury associated with the rapid accumulation of adhesion molecules on the luminal surface and recruitment of neutrophils. Arterioscler Thromb Vasc Biol. 2000;20:335–342. doi: 10.1161/01.atv.20.2.335. [DOI] [PubMed] [Google Scholar]
- 27.Yu X, Dluz S, Graves DT, Zhang L, Antoniades HN, Hollander W, et al. Elevated expression of monocyte chemoattractant protein 1 by vascular smooth muscle cells in hypercholesterolemic primates. Proc Natl Acad Sci USA. 1992;89:6953–6957. doi: 10.1073/pnas.89.15.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han KH, Tangirala RK, Green SR, Quehenberger O. Chemokine receptor CCR2 expression and monocyte chemoattractant protein-1-mediated chemotaxis in human monocytes. Arterioscl Thromb Vasc Biol. 1998;18:1983–1991. doi: 10.1161/01.atv.18.12.1983. [DOI] [PubMed] [Google Scholar]
- 29.Oguchi S, Dimayuga P, Zhu J, Chyu KY, Yano J, Shah PK, et al. Monoclonal antibody against vascular cell adhesion molecule-1 inhibits neointimal formation after periadventitial carotid artery injury in genetically hypercholesterolemic mice. Arterioscl Thromb Vasc Biol. 2000;20:1729–1736. doi: 10.1161/01.atv.20.7.1729. [DOI] [PubMed] [Google Scholar]
- 30.Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K. Lynphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J Exp Med. 2006;203:1273–1282. doi: 10.1084/jem.20052205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maffia P, Zinselmeyer BH, Ialenti A, Kennedy S, Baker A, McInnes IB, et al. Multiphoton microscopy for 3-dimensional imaging of lymphocyte recruitment into apolipoprotein-E-deficient mouse carotid artery. Circulation. 2007;115:e326–e328. doi: 10.1161/CIRCULATIONAHA.106.658492. [DOI] [PubMed] [Google Scholar]
- 32.Mori E, Komori K, Yamaoka T, Tanii M, Kataoka C, Takeshita A, et al. Essential role of monocyte chemoattractant protein-1 in development of restenotic changes (neointimal hyperplasia and constrictive remodeling) after balloon angioplasty in hypercholesterolemic rabbits. Circulation. 2002;105:2905–2910. doi: 10.1161/01.cir.0000018603.67989.71. [DOI] [PubMed] [Google Scholar]
- 33.Schober A, Zernecke A, Liehn EA, von Hundelshausen P, Knarren S, Kuziel WA, et al. Crucial role of the CCL2/CCR2 axis in neointimal hyperplasia after arterial injury in hyperlipidemic mice involves early monocyte recruitment and CCL2 presentation on platelets. Circ Res. 2004;95:1125–1133. doi: 10.1161/01.RES.0000149518.86865.3e. [DOI] [PubMed] [Google Scholar]