

Abstract
Neuroligins (NLs) are postsynaptic cell-adhesion molecules essential for normal synapse function. Mutations in neuroligin-4 (NL4) (gene symbol: NLGN4) have been reported in some patients with autism spectrum disorder (ASD) and other neurodevelopmental impairments. However, the low frequency of NL4 mutations and the limited information about the affected patients and the functional consequences of their mutations cast doubt on the causal role of NL4 mutations in these disorders. Here, we describe two brothers with classical ASD who carry a single amino-acid substitution in NL4 (R87W). This substitution was absent from the brothers' asymptomatic parents, suggesting that it arose in the maternal germ line. R87 is conserved in all NL isoforms, and the R87W substitution is not observed in control individuals. At the protein level, the R87W substitution impaired glycosylation processing of NL4 expressed in HEK293 and COS cells, destabilized NL4, caused NL4 retention in the endoplasmic reticulum in non-neuronal cells and neurons, and blocked NL4 transport to the cell surface. As a result, the R87W substitution inactivated the synapse-formation activity of NL4 and abolished the functional effect of NL4 on synapse strength. Viewed together, these observations suggest that a point mutation in NL4 can cause ASD by a loss-of-function mechanism.
Introduction
Autism spectrum disorders (ASDs) are neurodevelopmental diseases defined by impaired social interactions, deficient communication, restricted interests, and stereotyped activity patterns (American Psychiatric Association, 2000). ASDs probably represent a spectrum of disorders (Abrahams and Geschwind, 2008; Losh et al., 2008). Although most ASDs are sporadic, ASDs are primarily heritable (Beaudet, 2007). Multiple gene defects have been associated with ASDs, including mutations in the X-linked neuroligin-3 (NL3) and neuroligin-4 (NL4) genes (for review, see Südhof, 2008).
NLs are postsynaptic cell-adhesion molecules that participate in the formation, organization, and remodeling of synapses (Ichtchenko et al., 1995, 1996; Song et al., 1999; Scheiffele et al., 2000; Boucard et al., 2005; Nam and Chen, 2005; Chih et al., 2006; Varoqueaux et al., 2006; Berninghausen et al., 2007; Chubykin et al., 2007; Conroy et al., 2007; Dong et al., 2007; Futai et al., 2007). Multiple changes in NL genes were observed in patients with familial ASDs and related neuropsychiatric diseases (Jamain et al., 2003; Laumonnier et al., 2004; Yan et al., 2005; Chocholska et al., 2006; Talebizadeh et al., 2006; Macarov et al., 2007; Lawson-Yuen et al., 2008; Marshall et al., 2008). Furthermore, mutations in neurexin-1α [which binds to NLs extracellularly (Boucard et al., 2005)], and in SHANK3 [which binds to NLs intracellularly via PSD-95 (Naisbitt et al., 1999)] have also been associated with ASDs (Jeffries et al., 2005; Durand et al., 2007; Szatmari et al., 2007; Marshall et al., 2008; Morrow et al., 2008; Kim et al., 2009), suggesting a synaptic pathway for ASD pathogenesis involving trans-synaptic interactions of neurexins and neuroligins (the trans-synaptic interaction hypothesis of ASDs) (Südhof, 2008). In support of this hypothesis, mice carrying the R451C mutation of NL3 or a deletion of NL4 exhibit synaptic and behavioral phenotypes characteristic of ASDs (Tabuchi et al., 2007; Jamain et al., 2008).
However, several observations argue against a link of neuroligin mutations to ASDs and question the trans-synaptic interaction hypothesis. First, mutations in NLs, neurexin-1α, and SHANK3 are exceedingly rare in ASDs (Vincent et al., 2004; Gauthier et al., 2005; Blasi et al., 2006; Wermter et al., 2008). Second, at least some individuals with genetic changes in NL4 (Chocholska et al., 2006; Macarov et al., 2007), neurexin-1α (Marshall et al., 2008; Morrow et al., 2008), or SHANK3 (Durand et al., 2007) are asymptomatic. Third, although some NL4 mutations could be functionally significant because they ablate NL4 expression, other NL4 mutations may cause only minor structural changes (Yan et al., 2005) and could represent polymorphisms that are functionally insignificant.
Here, we have tested the possible role of NL4 mutations in ASDs by investigating a familial case of ASD in which two affected brothers carry a single amino-acid substitution (R87W) in the NL4 gene. We show that this mutation arose in the maternal germ line and causes a major impairment in the functional properties of NL4. Our data report a direct correlation of the functional effects of an NL4 point mutation with the clinical phenotype observed in human patients, strongly supporting the trans-synaptic interaction hypothesis of ASDs.
Materials and Methods
Clinical evaluation
SS and TS were born at term via normal spontaneous vaginal delivery with no reported prenatal or neonatal complications. Their mother does not report a history of potentially harmful exposures, including chemical, radiation, chemotherapy, unusual medications, herbal medications, or severe illnesses. She does not report any workplace exposures to chemicals. Neither child has a history of birth defects, major illnesses, surgeries, seizures, or hospitalizations. SS and TS have no reported vision problems, although they have not had formal ophthalmology evaluations. Neither has had a brain MRI. Both brothers have had normal audiology screenings. The probands were examined with a series of psychological tests (Tables 1, 2). SS had developmental evaluations through the Studies to Advance Autism Research and Treatment study at Boston University School of Medicine at 31, 43, and 52 months of age, and TS at 38 months of age.
Table 1.
Diagnostic scores on the ADOS and ADI-R
| SS |
TS | |||
|---|---|---|---|---|
| 31 months | 43 months | 52 months | 38 months | |
| ADOS | ||||
| Communication (cutoff 2 or higher) | 4 | 5 | 8 | 4 |
| Social interaction (cutoff 4 or higher) | 11 | 13 | 12 | 12 |
| Total (cutoff 7 or higher) | 15 | 18 | 20 | 16 |
| Play | 4 | 4 | 1 | 3 |
| Stereotyped behaviors | 5 | 4 | 6 | 3 |
| ADI-R | ||||
| Social interaction (cutoff 10 or higher) | 23 | 19 | 24 | 22 |
| Communication (cutoff 7/8 or higher) | 13 | 21 | 18 | 13 |
| Restricted/repetitive/stereotypedbehavior (cutoff 3 or higher) | 4 | 4 | 3 | 3 |
ADOS stands for Autism Diagnostic Observational Schedule (version G) (Lord et al., 2002) and ADI-R for Autism Diagnostic Interview-Revised (Lord et al., 1994). Both boys met criteria for a diagnosis of autism on both the tests at all time points. For a detailed description of the tests, see supplemental materials, available at www.jneurosci.org.
Table 2.
Developmental and adaptive behavior scores
| SS |
TS | |||||||
|---|---|---|---|---|---|---|---|---|
| 31 months |
43 months |
52 months |
38 months |
|||||
| T/SS | Age equivalent (in months) | T/SS | Age equivalent | T/SS | Age equivalent | T/SS | Age equivalent | |
| Mullen | ||||||||
| Visual reception | 33 | 24 | 33 | 34 | 22 | 36 | 20 | 21 |
| Fine motor | 20 | 18 | 20 | 22 | 20 | 28 | 20 | 21 |
| Receptive language | 20 | 13 | 20 | 23 | 20 | 30 | 20 | 13 |
| Expressive language | 20 | 13 | 39 | 36 | 28 | 36 | 20 | 13 |
| ELC | 53 | 60 | 52 | 49 | ||||
| VABS | ||||||||
| Communication | 68 | 16 | 68 | 21 | 65 | 23 | 56 | 11 |
| Daily living | 63 | 16 | 62 | 21 | 59 | 23 | 60 | 17 |
| Socialization | 63 | 11 | 55 | 11 | 58 | 15 | 56 | 10 |
| Motor skills | 70 | 20 | 55 | 23 | 58 | 29 | 62 | 20 |
| Composite | 61 | 16 | 55 | 19 | 55 | 23 | 54 | 15 |
Mullen denotes the Mullen Scales of Early Learning (Mullen, 1995) and VABS the Vineland Adaptive Behavior Scales-Expanded Form (Sparrow et al., 1984). All scores on the VABS are expressed as standard scores (mean ± SD, 100 ± 15). Again, both boys met criteria for a diagnosis of autism on both the tests at all time points. For a detailed description of the tests, see supplemental materials, available at www.jneurosci.org.
The parents of SS and TS were of Scottish and Irish descent. Family history is notable for a paternal first cousin with cerebral palsy. A paternal uncle has learning disabilities and mental illness since his 20s. There was no other reported history of mental retardation, ASDs, neuropsychiatric disorders, multiple miscarriages, genetic disorders, or known chromosome abnormalities on either side of the family.
On physical exam, SS has some mild dysmorphic features, including epicanthal folds, broad nasal bridge, smooth philtrum, and mild clinodactyly. Head circumference is 50th centile, weight is 50–75th centile, and height is 50–75th centile. TS has abnormal hair patterning and a widow's peak. He has a broad forehead, broad nasal bridge, and slight midface hypoplasia. He has epicanthal folds. He has finger pads, mild syndactyly, and mild fifth finger clinodactyly. TS's head circumference is 75th centile, weight is >95th centile, and height is 90–95th centile. Neurologic exam of both brothers was nonfocal.
Clinical psychological tests
Autism Diagnostic Observational Schedule (Lord et al., 2002).
The Autism Diagnostic Observational Schedule (ADOS) is a semi-structured, interactive observation designed to assess social and communicative functioning in individuals suspected of having an autism spectrum disorder. The assessment involves a variety of social “presses” designed to elicit behaviors relevant to a diagnosis of autism. Behaviors are scored from 0 (no atypical presentation) to 2 (definitely atypical). A standardized diagnostic algorithm can be calculated, consistent with autism criteria in Diagnostic and Statistical Manual of Mental Disorders-IV/International Classification of Diseases-10. Established cutoff scores in the social and communication domains are used to differentiate autism, autism spectrum, and non-autism spectrum disorders. In addition, a child receives scores for atypical play and stereotyped behaviors that are not included in the total algorithm score.
Autism Diagnostic Interview-Revised (Lord et al., 1994).
Autism Diagnostic Interview-Revised (ADI-R) This is a semi-structured caregiver interview designed to gather information necessary to assign a diagnosis of autism. Like the ADOS, each item is scored from 0 to 2, with higher scores denoting greater impairment or atypical behavior. Items have been shown to be reliable and the accompanying algorithm adequately discriminates individuals with autism non-autistic peers. Both boys met criteria for a diagnosis of autism on both the ADOS and ADI at all time points. Their scores were above the threshold cutoff scores for a diagnosis of autism in each subdomain and for the algorithm totals on the ADOS and ADI-R. Table 1 presents the scores on the ADOS and ADI-R for both children.
Mullen Scales of Early Learning (Mullen, 1995).
This is an assessment of developmental functioning for children from birth to 5 years 8 months. The Mullen provides an overall score (“early learning composite” expressed as a standard score, with a mean ± SD of 100 ± 15) and subtest scores (expressed as t scores, with a mean ± SD of 50 ± 10) for gross and fine motor skills, visual reception (which reflects nonverbal problem solving), and receptive and expressive language.
Vineland Adaptive Behavior Scales-Expanded Form (Sparrow et al., 1984).
This interview was administered to the boys' mother to assess personal and social sufficiency in four domains: communication (receptive, expressive, written), daily living skills (personal, domestic, community), socialization (interpersonal relationships, play and leisure time, coping skills), and motor skills (gross, fine). The Vineland Adaptive Behavior Scales (VABS) also yields a summary score, the “adaptive behavior composite.” All scores on the VABS are expressed as standard scores (mean ± SD of 100 ± 15).
Molecular analysis of patient's genome
Normal or negative genetic testing performed on both brothers included high-resolution chromosomes, 500K single nucleotide polymorphism (SNP) microarray, Fragile X syndrome, and NLGN3 sequence analysis. Fluorescence-labeled PCR primers in exon flanking regions were used to amplify and sequence the five coding exons in the NLGN4 gene in both directions. This analysis was performed on samples from SS and TS. For maternal, paternal, and 300 healthy control samples, fluorescence-labeled PCR primers in exon flanking regions were used to amplify and sequence exon 1 of the NLGN4 gene in both directions. The paternal sample was analyzed to rule out the presence of an alteration on the Y-chromosome homolog of NLGN4. Both maternal blood and buccal samples were analyzed. Maternity and paternity testing was performed by analyzing DNA samples from SS, TS, mother, and father at 15 unlinked DNA marker loci.
X-inactivation assay was performed on a maternal sample by the restriction fragment length polymorphism analysis using a methylation-sensitive enzyme, HpaII, followed by PCR amplification of a (CAG)n triplet repeat region in the androgen receptor gene on the X chromosome. Methylation-sensitive enzymes cut the androgen receptor repeat region on the active X chromosome but leave this site intact on the inactive X chromosome. DNA fragments were resolved by POP4 on the ABI genetic analyzer 3100 (Applied Biosystems). The X-inactivation ratio is obtained by comparing quantitative PCR results with and without digestion by the methylation-sensitive enzyme.
Construction of expression vectors
Vectors expressing unmodified human Neuroligin 4 (hNL4, in pcDNA1 backbone) was described previously (Bolliger et al., 2001). Human NL4R87W plasmid was generated by introducing a point mutation at residue 87 (arginine to tryptophan) using the QuickChange kit (Stratagene) at pcDNA1 human NL4 plasmid with the following primers (JK08202, CCCCACTGGAGAGAGGTGGTTTCAGCCCCC AGAAC; JK08203, GTTCTGGGGGCTGAAACCACCTCTCTCCAGTGGGG) and verified by nucleotide sequencing. Monomeric Venus (mVenus)-tagged hNL4 and hNL4R87W plasmids (in pCMV5 backbone) were generated by inserting the cDNA encoding mVenus into the EcoRV site of cytoplasmic tail of human NL4 (insertion sequence, PSPQRNTTND–mVenus–IAHIQNEEIM).
Antibodies
Polyclonal antibodies to synapsin (E028) and pan-neuroligins, monoclonal antibody to microtubule-activated protein 2 (MAP2) were described previously (Chubykin et al., 2005; Bolliger et al., 2008). The following antibodies were commercially obtained: GM130 (mouse monoclonal; BD Biosciences Transduction Laboratories), GFP (mouse monoclonal; Invitrogen), calnexin (mouse monoclonal; BD Biosciences Transduction Laboratories), and synaptotagmin-1 (clone 41.1; Synaptic Systems).
Neuronal cultures and Ca2+-phosphate transfections
Primary hippocampal neurons were cultured on poly-d-lysine-coated glass coverslips from newborn mice pups (Maximov et al., 2007). Cultured neurons were transfected with various cDNA constructs at days in vitro 10 (DIV10) using Ca2+-phosphate transfection method. Briefly, for each coverslip in a 24-well plate, 4 μg of total plasmid was mixed with 3.1 μl of 2 m CaCl2 solution, dH2O was added to a final volume of 25 μl, and the DNA/CaCl2 solution was added slowly to 25 μl of 2× HBS. Then DNA/CaCl2/HBS solution was incubated at room temperature for 30 min and then added to neuronal culture and incubated for 30 min in incubator. Afterward, the culture was washed three times with MEM and kept in the incubator for 4 d before recording. All transfections used the same total amount of plasmids [3 μg of test or control plasmids plus 1 μg of enhanced GFP (EGFP) plasmid for identifying transfected neurons]; for control transfections, empty pCMV5 vector was used to substitute for the expression vectors. As visualized by the cotransfected EGFP, 1–2% of neurons were typically transfected.
Electrophysiological recordings
Electrophysiological recordings were performed as described previously (Maximov and Südhof, 2005; Maximov et al., 2007). Synaptic responses were evoked by 0.5 ms current injections (90–100 μA) with an Isolated Pulse Stimulator (model 2000; A-M Systems) using a concentric bipolar electrode (CBAEC75; FHC). Data were digitized at 10 kHz with a 2 kHz low-pass filter. The intracellular pipette solution contained the following (in mm): 145 CsCl, 5 NaCl, 10 HEPES, 5 EGTA, 5 QX-314 [2(triethylamino)-N-(2,6-dimethylphenyl) acetamine], 4 MgATP, and 0.3 Na2GTP, pH 7.2 (305 mOsm). The bath solution contained the following (in mm): 150 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.4 (315 mOsm). Evoked IPSCs were pharmacologically isolated by adding 10 μm CNQX to the bath solution. Evoked EPSCs were pharmacologically isolated by adding 100 μm picrotoxin to the bath solution. Tetrodotoxin (1 μm) was added to the bath to block evoked synaptic responses for miniature EPSC (mEPSC)/mIPSC recordings. The readily-releasable pool was measured by background perfusion of hypertonic sucrose (0.5 m) into the bath solution at a speed of 2 ml/min. Series resistance was compensated to 60–70%, and recordings with series resistances of >20 MΩ were rejected. Data were analyzed using Clampfit 9.02 (Molecular Devices), Excel (Microsoft), and Igor 4.0 (Wavemetics).
Immunocytochemistry and imaging analysis
Primary hippocampal cultures were prepared from embryonic day 18 rat hippocampi. Cultured neurons were transfected with CalPhos transfection kit (Clontech) at DIV10, fixed with 4% paraformaldehyde/4% sucrose, permeabilized with 0.2% Triton X-100 in PBS, and incubated with the indicated primary and Alexa Fluor 488- or 546-labeled goat anti-mouse or anti-rabbit antibodies (Invitrogen) at DIV14. For immunostainings, GFP (1:500) and synapsin (1:1000) antibodies were used. COS-7 cells were maintained as described previously (Chubykin et al., 2005). FuGENE-6 (Roche) was used to transfect COS-7 cells, and immunostaining was performed after 24 h with calnexin [1:150; endoplasmic reticulum (ER) marker] or GM130 (1:200; cis-Golgi marker) antibodies. Images were captured using Leica TCS2 confocal microscope. Images were collected with the identical confocal setting for all of the samples of an experiment using Leica TCS2 confocal microscope. Z-stacked images were projected with maximal projection mode using Leica Confocal Software. The contours of the transfected COS-7 cells were randomly chosen, and their fluorescence intensity was quantified for both red and green channels in the region of interest with MetaMorph Software. For the analysis of neuron transfection experiments, Z-stacked images were obtained, converted to maximally projected images, and analyzed using MetaMorph Software to measure the numbers of dendritic spines and presynaptic terminals per 50 μm dendrites or puncta cluster area.
Artificial synapse formation assays
Coculture assays were performed as described previously (Chubykin et al., 2005). COS-7 cells transfected with NL4 plasmids (hNL4 or hNL4R87W) or EGFP were cultured for 24 h, trypsinized, plated onto the hippocampal neurons, cocultured for 2 d (from DIV6 to DIV8), and immunostained for pan-neuroligin (19C; 1 μg/ml) and synaptotagmin-1 antibodies (1:200; Synaptic Systems). Images were captured by Z-stacking with Leica TCS2 microscope.
Purification of membrane protein from HEK293 cells and affinity chromatography experiments
HEK293 cells were transfected with indicated plasmid using FuGENE-6 (Roche), and 2 μg of plasmid with 6 μl of FuGENE was used for one well in a six-well plate. After 48 h, HEK293 cells were washed with PBS, kept at −80°C overnight, and thawed at 37°C for 1 min. Lysed cell membranes and nuclei were pelleted by centrifugation (2000 × g for 15 min at 4°C), and membrane proteins were extracted from the pellet for 1 h at 4°C in 20 mm HEPES–NaOH, pH 7.4, containing 1% Triton X-100, 0.1 mm EDTA, 2 mm CaCl2, 1 mm MgCl2, and 50 mm NaCl with protease inhibitors (1 mm PMSF, 1 mg/L pepstatin, 1 mg/L leupeptin, and 2 mg/L aprotinin). Insoluble material was removed by centrifugation (10,000 × g for 30 min), and the supernatant was used for various pull-downs. All pull-down experiments were done overnight at 4°C, and bound proteins were eluted with sample buffer and analyzed by immunoblotting with specific antibodies.
Glycosylation assay
Solubilized proteins (10 μg) from HEK293 cells transfected with hNL4 or hNL4R87W were first denatured by adding 10× denaturing buffer (New England Biolabs) and heated to 100°C for 10 min. For endoglycosidase H (Endo H) treatment, denatured protein was treated with 1 μl of enzyme and incubated at 37°C for 1 h. For PNGase F treatment, denatured protein was treated with 1 μl of enzyme plus 2 μl of NP-40 (10%) and incubated at 37°C for 1 h. After enzyme treatment, proteins, together with equal amount of untreated proteins, were analyzed by immunoblotting with indicated antibodies and followed by ECL detection.
Membrane biotinylation assay
Transfected HEK293 cells were washed with ice-cold PBS (with 1 mm MgCl2 and 0.1 mm CaCl2) three times. Then sulfo-NHS-LC-biotin (1 mg/ml) was added to the cells, and cells were kept at 4°C for 30 min. After incubation, cells were washed six times with PBS plus glycine (100 mm) to quench and remove excess biotin. Purified membrane protein (100 μg) was then incubated with Neutravidin (Thermo Fisher Scientific) for overnight at 4°C. After six washes with PBS, protein was eluted with sample buffer and analyzed by immunoblotting.
Miscellaneous
SDS-PAGE and immunoblotting experiments, including quantitation with 125I-labeled secondary antibodies followed by PhosphorImager detection (Molecular Dynamics), were performed as described previously (Tabuchi et al., 2007).
Statistical analyses
All data shown are means ± SEMs. Statistical significance was determined by the Student's t test or ANOVA Tukey's test.
Results
Case report
The probands are brothers (SS, 5 years; TS, 3 years) who were referred to Boston University School of Medicine with a diagnosis of developmental delays (especially language), atypical behaviors, and ASD. SS and TS were born to healthy, non-consanguineous parents without a family history of neuropsychiatric disorders (see Materials and Methods). Developmentally, TS began walking at 14½ months and received a diagnosis of pervasive developmental disorder–not otherwise specified at 21 months. An Arena developmental evaluation at a chronological age of 17 months revealed fine motor skills at a 19 months level, cognitive functioning at an 18 months level, expressive language at a 7 months level, receptive language at an 11 months level, social-emotional skills at a 26 months level, self-care and feeding at a 33 months level, toileting at a 13 months level, dressing and hygiene at a 17 months level, and gross motor skills at a 22 months level. TS exhibited atypical behaviors, including poor social interaction, decreased eye contact, biting, and spinning. Developmentally, SS reportedly sat at 8 months, crawled at 10 months, and began walking at 18 months. He said his first word at 31 months and began speaking in two to three word phrases by 34 months. SS also displays atypical behaviors, including perseveration, temper tantrums, unusual play and social skills, minimal eye contact, and echolalia. SS received a diagnosis of ASD at 23 months of age.
Evaluations of SS and TS with the ADOS and ADI-R showed that both SS and TS met the criteria for a diagnosis of autism on both the ADOS and the ADI-R scale (Table 1). On the ADOS, both boys had atypical scores on many items, including frequency of vocalizations directed to others, gestures, eye contact, facial expressions toward others, shared enjoyment in interaction, showing, joint attention, and quality of social overtures. Both brothers also had atypical behaviors in functional play with objects, imagination, unusual sensory interests, and repetitive interests or stereotyped behaviors. SS and TS also went through developmental/cognitive and adaptive behavior assessments using the Mullen Scales of Early Learning and the VABS. The composite scores for each brother on both tests indicate that they are >2 SDs below the mean in the range of mild-to-moderate mental retardation (Table 2). Both SS and TS scored below age expectations in motor, cognitive, language/communication, socializations, and daily living skills.
Genomic analyses revealed that both SS and TS were hemizygous for a point mutation, the R87W substitution, in exon 1 of the NL4 gene (Fig. 1). Maternal blood, maternal buccal swab, and paternal blood samples were negative for the R87W substitution. Samples from 300 healthy control individuals were negative for the R87W substitution, and no changes in R87 were detected in SNP databases. Maternal X-inactivation studies revealed an abnormal, mildly skewed X-inactivation pattern (75:25%) of unknown significance. Maternity and paternity testing confirmed that SS and TS share alleles with their mother and father at all 15 tested loci, thus confirming true maternity and paternity.
Figure 1.

Identification of a nonconservative arginine-to-tryptophan substitution (R87W) in the NL4 gene in two brothers with ASD. A, Representative sequencing gel chromatograms from PCR-amplified genomic DNA of the NL4 gene from the individuals indicated on the left. B, Alignment of the sequences of human NLs in the region corresponding to R87 in NL4 (boxed).
The R87W mutation likely impairs folding of NL4
Inspection of the crystal structure of NL4 (Fabrichny et al., 2007) reveals that R87 is at a region that is on the opposite side of the neurexin-binding and the dimerization surface of NL4. This region is mostly composed of rigid loop-like sequences that may be important for nucleating the overall fold of NL4. In the NL4 structure, R87 forms four hydrogen bonds (one each with K321 and L318 and two with E144). Not only R87 but also its hydrogen bonds appear to be conserved in all neuroligins (Araç et al., 2007; Chen et al., 2008). Thus, the R87 mutation to tryptophan has a high potential to disrupt the structure of NL4.
To test this hypothesis, we transfected wild-type and R87W-mutant human NL4 into HEK293 cells. Immunoblotting demonstrated that mutant NL4 was expressed at much lower levels than wild-type NL4 and migrated at a different position (Fig. 2A). Quantitations showed that the levels for the immature precursor protein of NL4 were significantly increased, whereas those for mature NL4 were significantly decreased, by the R87W mutation (Fig. 2B). Note that, in these experiments, wild-type and R87W-mutant neuroligin were expressed with identical vectors that only differed in the R87W substitution. To determine the relationship of mature and immature NL4 in wild-type and R87W-mutant-expressing HEK293 cells, we deglycosylated the protein with endoglycosidase H (which only cleaves immature sugars attached to proteins in the ER) or PNGase F (which cleaves N-linked sugars). Endoglycosidase H did not alter the electrophoretic mobility of wild-type NL4 expressed in HEK293 cells, but completely shifted R87W-mutant NL4 to a lower molecular weight, suggesting that R87W-mutant but not wild-type NL4 carries Endo H-sensitive sugars (Fig. 2C). PNGase F, in contrast, cleaved both wild-type and mutant NL4. After PNGase F cleavage, wild-type NL4 still migrated at a higher molecular weight than R87W-mutant NL4, presumably because mature NL4 contains O-linked sugars that are not present in immature R87W-mutant NL4 and that are not cleaved by PNGase F (which only cleaves N-linked sugars).
Figure 2.

Impaired glycosylation and destabilization of R87W-mutant NL4. A, Representative immunoblot of wild-type and R87W-mutant human NL4 (hNL4WT and hNL4R87W, respectively) expressed in transfected HEK293 cells. The migrations of mature and immature NL4 are indicated on the right. A blot for valosin-containing protein (VCP) as a loading control is shown on the bottom. B, Quantitations of wild-type and R87W-mutant NL4 expression levels in HEK293 cells. Equal amounts of plasmids that were identical, except for the point mutation in NL4, were transfected. Expression levels were measured with quantitative immunoblotting using 125I-labeled secondary antibodies and PhosphoImager detection; levels were normalized for VCP as an internal control (see A). Total NL4 and mature and immature glycosylation forms of NL4 were quantified based on their migration on SDS gels. C, Analysis of wild-type and R87W-mutant NL4 glycosylation. Image shows a representative immunoblot of NL4 expressed in HEK293 cells, before and after treatment with Endo H and PNGase F. Untreated control samples were examined without incubation (Untreated-a) or after incubation in control buffer (Untreated-b). D, Destabilization of NL4 by the R87W mutation. The rate of NL4 degradation in HEK293 cells was monitored by quantitative immunoblotting after blocking additional protein synthesis with 0.35 mm cycloheximide (CHX). NL4 levels are normalized for the starting value. Data in B and D are means ± SEMs (B, n = 4; D, n = 3 independent experiments). Statistical significance was evaluated by Student's tailed t test (**p < 0.01; ***p < 0.001).
The glycosylation data and expression levels of R87W-mutant NL4 suggest that the R87W mutation leads to the retention of NL4 in the ER, with subsequent rapid degradation. To test this hypothesis, we measured the rate of degradation of wild-type and R87W-mutant NL4 by adding cycloheximide (0.1 g/L) to transfected HEK293 cells. Cycloheximide stops protein translation, allowing us to monitor the degradation rate of the NL4 protein synthesized up to that point (Fig. 2D) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We observed a rapid loss of R87W-mutant NL4, whereas wild-type NL4 was relatively stable. Thus, the R87W substitution destabilizes NL4.
The R87W mutation inhibits surface expression of NL4
If the R87W substitution impairs maturation of NL4 by trapping it in the ER, it should block surface expression of NL4. To test this hypothesis, we examined the localization of wild-type and R87W-mutant NL4 in transfected COS cells whose large size facilitates localization of proteins. Whereas wild-type NL4 was predominantly localized to the plasma membrane in COS cells, the majority of R87W-mutant NL4 localized to the ER in which it was present in the same compartment as calnexin, distinct from the Golgi marker GM130 (Fig. 3A,B).
Figure 3.

R87W mutation blocks ER export and surface transport of NL4. A, B, Fluorescence images of COS-7 cells transfected with wild-type and R87W-mutant human NL4 (hNL4WT and hNL4R87W, respectively), double labeled for the ER marker calnexin (top images) or the cis-Golgi marker GM130 (bottom images) and for NL4 using the pan-NL antibody 19C. Scale bars on the right (20 μm) apply to all images in a set. C, Analysis of NL4 surface exposure in transfected HEK293 cells using surface biotinylation, analyzed by immunoblotting of affinity-purified surface-biotinylated proteins. The membrane protein Na,K–ATPase was used as a positive control for surface biotinylation, and the intracellular protein VCP was used as a negative control.
To confirm that the R87W mutation impaired the surface exposure of NL4, we performed surface biotinylation experiments, using Na,K–ATPase as an internal control. Surface proteins were biotinylated in transfected HEK293 cells and purified on avidin columns, and the relative amount of biotinylated surface protein was measured by quantitative immunoblotting (Fig. 3C). We found that the R87W mutation severely impaired the surface expression of NL4 protein (percentage of surface-biotinylated NL4 protein of the total NL4 protein: 4.54 ± 0.70% for wild-type NL4 vs 0.23 ± 0.03% for R87W-mutant NL4; p < 0.01; n = 3). As an internal control, we measured the surface Na,K–ATPase levels, which remained constant in both groups (1.34 ± 0.58 vs 1.27 ± 0.59% for cells expressing wild-type vs R87W-mutant NL4, respectively; p > 0.05; n = 3 independent experiments). This assay confirmed that the R87W mutation severely impaired transport of NL4 to the cell surface.
To ensure that the R87W mutation has a similar effect on the transport of NL4 out of the ER in neurons, we transfected cultured hippocampal neurons and analyzed them by immunocytochemistry (Fig. 4). Whereas wild-type NL4 was efficiently transported into dendrites, R87W-mutant NL4 was completely retained in the cell body of the transfected neurons in a compartment that appears to be pre-Golgi, because it was not stained by GM130, and thus most likely represents the ER (Fig. 4). This finding suggests that the R87W mutation has a similar effect on the processing and transport of NL4 in non-neuronal and neuronal cells.
Figure 4.
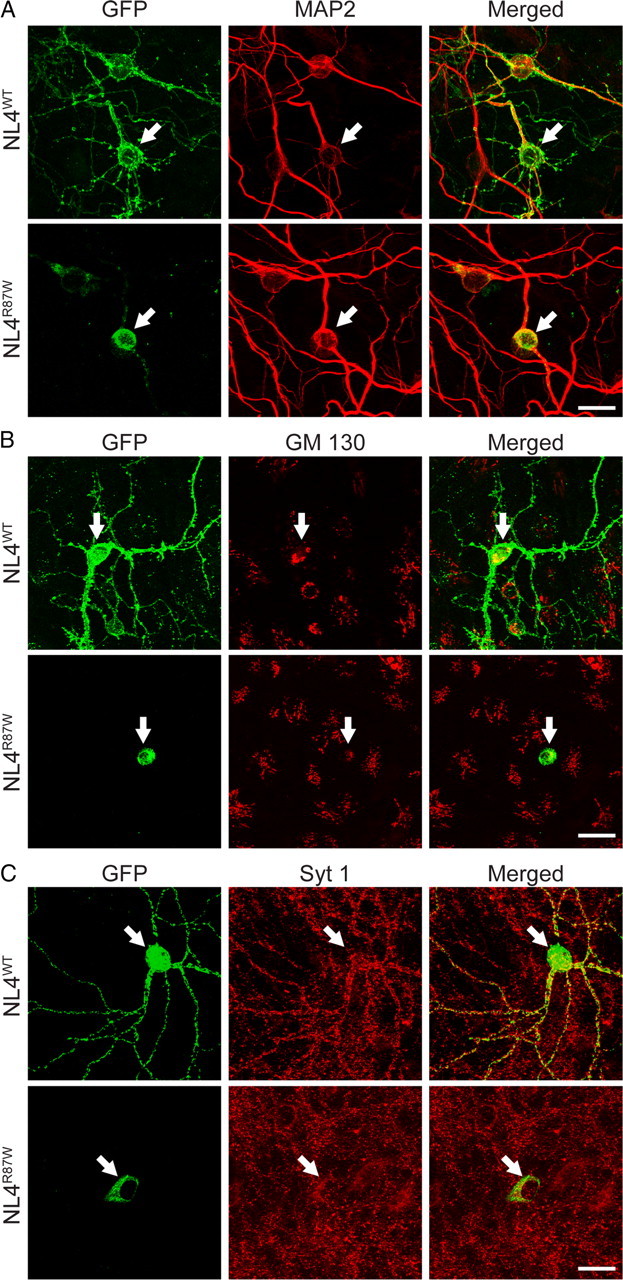
Wild-type but not R87W-mutant NL4 is transported to dendrites in transfected neurons. A–C, Fluorescence images of hippocampal neurons transfected with wild-type and R87W-mutant NL4 that were tagged with mVenus (hNL4WT and hNL4R87W, respectively). Neurons were double labeled for the dendritic marker MAP2 (A), the cis-Golgi marker GM130 (B), or the synaptic vesicle protein synaptotagmin-1 (Syt 1; C); transfected NL4 was visualized with a GFP-directed antibody reacting to its Venus moiety. Arrows point to the soma of transfected neurons. Scale bars (20 μm) apply to all images in a set.
The R87W mutation blocks the ability of NL4 to induce synapse formation
The effect of the R87W mutation on the maturation and ER export of NL4 resembles that of the R451C mutation in NL3 (Comoletti et al., 2004; De Jaco et al., 2006). However, although the R451C mutation impairs folding and surface export of NL3, it does so incompletely. As a result, 10–20% of R451C-mutant NL3 escapes to the cell surface, still binds to neurexins, and induces synapse formation when transfected COS cells expressing R451C-mutant NL3 are cocultured with neurons (Chubykin et al., 2005). This is important because, in knock-in mice, the R451C mutation in NL3 acts as a dominant mutation and not as loss-of-function mutation (Tabuchi et al., 2007). Thus, it is crucial to test whether the R87W mutation in NL4 also allows a small percentage of NL4 to escape to the surface and to induce synapse formation.
To test this question, we first measured the ability of R87W-mutant NL4 to bind to recombinant neurexin-2β and observed that neurexin binding was not impaired by the R87W mutation (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). We then measured its ability to induce synapse formation by coculturing transfected COS cells expressing wild-type or R87W-mutant NL4 with neurons (Fig. 5). We found that, whereas wild-type NL4 was fully active in this assay, R87W-mutant NL4—different from R451C-mutant NL3—was unable to induce synapse formation. Synapse formation was quantified as the average fluorescence intensity of synaptotagmin-1-positive nerve terminals over the transfected COS cells (wild-type NL4 transfected cells, 46 ± 5; R87W-mutant NL4 transfected cells, 4 ± 1; p < 0.001; n = 19–21 cells/3 independent experiments; values are in arbitrary fluorescence units) normalized for the expression of wild-type and R87W-mutant NL4 (wild-type NL4, 149 ± 11; R87W-mutant NL4, 89 ± 12; p < 0.01). This normalization yields the ratio of synapses for expressed NL4 (wild-type NL4, 0.31 ± 0.08; R87W-mutant NL4, 0.05 ± 0.007; p < 0.001). Thus, as expected from the lack of surface transport, R87W-mutant NL4 is functionally inactive. This was additionally confirmed in experiments testing the effect of overexpressed NL4 directly in transfected neurons. We found that overexpression of wild-type NL4 in transfected neurons increased the density of synapses on the neurons (Fig. 6), similar to other neuroligin isoforms (Chih et al., 2004; Boucard et al., 2005). R87W-mutant NL4, however, was unable to do so.
Figure 5.

Wild-type but not R87W-mutant NL4 induces synapse formation. Representative fluorescence images of COS-7 cells expressing wild-type or R87W-mutant human NL4 (hNL4WT and hNL4R87W, respectively) or Venus alone (mVenus) as a negative control, as indicated. Transfected cells were cocultured with primary rat hippocampal neurons (Chubykin et al., 2005), fixed, and stained by immunofluorescence for synaptotagmin-1 to visualize presynaptic terminals (red) and for NL4 (using pan-NL antibodies) to visualize the transfected COS cells (green). Two examples for each condition are shown. Scale bar on the bottom right corner (15 μm) applies to all images.
Figure 6.

Wild-type but not R87W-mutant NL4 increases synapse numbers in transfected neurons. A, Representative fluorescence images of neurons cotransfected with plasmids expressing mVenus and wild-type or R87W-mutant human NL4 (hNL4WT and hNL4R87W, respectively) or Venus alone (mVenus). Neurons were transfected at 10 d in vitro, fixed at 14 d in vitro, and visualized by the Venus fluorescence (left column) or by synapsin immunofluorescence (middle column; merged images are shown in the right column). Scale bar in the bottom right corner (5 μm) applies to all images. B, Quantitation of the number of synapses per 50 μm of dendrite (synapse density), analyzed as the density of postsynaptic (Postsyn.) spines (left; visualized via Venus fluorescence) or as of presynaptic (Presyn.) nerve terminals (right; visualized via synapsin staining). C, Same as B, except that not the synapse density but the fluorescence signal strength per punctum (interpreted as synapse area) was analyzed. Data shown in B and C are means ± SEMs (n = 3 independent experiments; statistical analysis was performed with Student's t test; *p < 0.05; **p < 0.01).
The R87W mutation abolishes the NL4-induced suppression of excitatory synaptic transmission
The synapse number quantifications strongly suggest that R87W-mutant NL4 is inactive, but it is possible that a functional effect by the mutant NL4, as opposed to a morphological change, could have been missed. To test this hypothesis, we transfected neurons with wild-type NL4, R87W-mutant NL4, or a control plasmid and recorded from the transfected neurons that were identified via cotransfected EGFP.
We first recorded spontaneous mEPSCs and mIPSCs. Surprisingly, we found that, although overexpression of wild-type NL4 increased synapse numbers in transfected neurons, in physiological measurements it selectively suppressed the frequency of mEPSCs but not mIPSCs, without altering the amplitude of either mEPSCs or mIPSCs (Fig. 7). Moreover, when we monitored EPSCs and IPSCs evoked by action potentials, we found that wild-type NL4 overexpression selectively decreased the amplitude of EPSCs but not of IPSCs (Fig. 8A,B). The same effect was observed for EPSCs induced by application of hypertonic sucrose (which induces exocytosis of all readily-releasable vesicles at a synapse in a Ca2+- and action potential-independent manner) (Fig. 8C). In all cases, R87W-mutant NL4 was inactive. Together, these findings indicate that the R87W mutation inactivates NL4.
Figure 7.

Wild-type but not R87W-mutant NL4 suppresses excitatory but not inhibitory spontaneous synaptic events. A, Representative traces (left) and summary graphs (right) of mEPSCs recorded in transfected neurons expressing pCMV5 empty vector alone (control) or untagged wild-type or R87W-mutant human NL4 (hNL4WT and hNL4R87W, respectively; all plasmids were cotransfected with EGFP to identify transfected cells). mEPSCs were monitored in the presence of 1 μm tetrodotoxin and 0.1 mm picrotoxin, and the frequency and amplitude of mEPSCs were quantified. B, Same as A, except that mIPSCs were recorded in the presence of 1 μm tetrodotoxin and 10 μm CNQX. All data are means ± SEMs (A: control, n = 21 cells/3 cultures; hNL4WT, n = 15/3; hNL4R87W, n = 12/3; B: control, n = 17/3; hNL4WT, n = 16/3; hNL4R87W, n = 18/3; statistical analysis was performed with Student's t test; **p < 0.01). Calibration below a set of representative traces applies to all traces in a set. Amp., Amplitude; Freq., frequency.
Figure 8.

Wild-type but not R87W-mutant NL4 suppresses evoked excitatory synaptic transmission. A, Representative traces (left) and summary graphs (right) of EPSCs recorded in transfected neurons transfected with an empty vector (control) or untagged wild-type or R87W-mutant human NL4 (hNL4WT and hNL4R87W, respectively; all plasmids were cotransfected with EGFP to identify transfected cells). EPSCs were evoked by isolated action potentials in the presence of 0.1 mm picrotoxin, and the amplitudes of evoked responses were quantified (Maximov et al., 2007). B, Same as A, except that, instead of EPSCs, IPSCs were monitored in the presence of 10 μm CNQX, and the amplitudes of evoked responses were quantified. C, Same as A, except that EPSCs were evoked by a 30 s application of 0.5 m sucrose. All data are means ± SEMs (A: control, n = 22 cells/3 cultures; hNL4WT, n = 21/3; hNL4R87W, n = 15/3; B: control, n = 16/3; hNL4WT, n = 16/3; hNL4R87W, n = 14/3; C: control, n = 14/3; hNL4WT, n = 13/3; hNL4R87W, n = 12/3; statistical analysis was performed with Student's t test; *p < 0.05). Calibration below a set of representative traces applies to all traces in a set. Amp., Amplitude.
Discussion
Although accumulating evidence suggested that mutations in NL4 may cause ASDs and other neuropsychiatric diseases (for review, see Südhof, 2008), previously described mutations in NL4 could also be interpreted as polymorphisms. Here, we describe two brothers who presented with classical ASD and who were found to carry a mutation in NL4 that likely caused their ASD based on the following evidence. (1) R87 is conserved in all neuroligins, and no mutations in R87 were detected in a large number of control individuals. (2) The genetically validated mother and father did not carry the R87W substitution, suggesting that the mutation arose in the maternal germ line, which is presumably mosaic for the mutation. (3) The R87W substitution causes retention and degradation of NL4 in the ER, with an accompanying loss of normal NL4 glycosylation, suggesting that the substitution impairs folding of NL4. (4) The R87W mutations abolishes the effects of NL4 on synapse formation and on synaptic transmission.
Thus, the R87W substitution represents a loss-of-function mutation that interferes with the normal folding of NL4, similar to what was previously described for the R451C substitution of NL3 (Comoletti et al., 2004; De Jaco et al., 2006). However, the R87W substitution in NL4 critically differs from the R451C substitution in NL3 in that the R451C substitution still allows some NL3 to escape the ER and be displayed on the cell surface (Comoletti et al., 2004), whereas the R87W substitution completely traps NL4 in the ER (Figs. 2, 3). As a result, the NL4 R87W substitution abolishes the ability of NL4 to induce synapse formation in transfected neurons (Fig. 6) and blocks the effect of transfected NL4 on synaptic transmission (Figs. 7, 8). In contrast, the R451C substitution in NL3 does not block its synapse-inducing activity (Chubykin et al., 2005) but instead acts as a dominant-positive mutation (or a dominant-negative, depending on the perspective) because it alters synaptic properties of neurons, which is not observed for the simple deletion of NL3 (Tabuchi et al., 2007). Thus, the R87W substitution has all the characteristics of a classical loss-of-function mutation, similar to other mutations in NL4 that were associated with ASDs (Jamain et al., 2003; Laumonnier et al., 2004; Yan et al., 2005; Chocholska et al., 2006; Talebizadeh et al., 2006; Macarov et al., 2007; Lawson-Yuen et al., 2008; Marshall et al., 2008). In summary, our data suggest a clear association of an NL4 mutation with a defined ASD phenotype and an alteration in protein folding, strongly supporting the notion that inactivation of NL4 function can be a cause of ASDs.
From a human genetics standpoint, this is, to our knowledge, the first report of germ-line mosaicism for an NL mutation, although germ-line mosaicism has been reported for alterations in other genes associated with ASDs. Szatmari et al. (2007) described two sisters with typical autism (including characteristic developmental delays) that had apparently identical 300 kb deletions on chromosome 2p16 not detected in either parent (Szatmari et al., 2007). The chromosomal deletions eliminated coding exons from the neurexin-1 gene. Microsatellite analysis showed the identical maternal chromosomal segment but no paternal DNA in the siblings, providing a likely explanation of paternal gonadal mosaicism. Neither parent of these sisters with the neurexin-1 deletion had clinically significant findings. Durand et al. (2007) described two brothers with ASD, severe speech impairment, and severe mental retardation who had a de novo insertion of a G nucleotide in exon 21 of the SHANK3 gene, which leads to a frame shift and presumed loss of function (Durand et al., 2007). Using 14 informative SNPs, it was determined that the mutation was located on the same maternal haplotype in the two affected brothers. This alteration was not observed in an unaffected brother, the unaffected parents (both maternal blood and buccal samples were analyzed), or control individuals. Given these findings, the mutation was thought to be a result of maternal germ-line mosaicism.
Our finding of germ-line mosaicism for a likely deleterious alteration in the NL4 gene has implications for genetic counseling. Because mutations, and thus germ-line mosaicism, in NLs and other ASD-linked genes are rare, no precise estimates of recurrence risk for a family with such a mutation can be made. However, recurrence risk data from better-described conditions in which germ-line mosaicism occurs, such as Duchenne muscular dystrophy, are available. In Duchenne muscular dystrophy, the recurrence risk for a proven new mutation was estimated to be 14–18% when the haplotype at risk is known (Bakker et al., 1987). A more recent study from this group (Helderman-van den Enden et al., 2009) has indicated that the recurrence risk is 4.3% when the haplotype at risk is not known. Based on this information, it can be postulated that the recurrence risk in this family may range from 4.3 to 15%.
Collectively, the finding of mutations in genes encoding neurexins, neuroligins, and SHANK3 in patients with neuropsychiatric disorders suggests an impairment in a synaptic pathway as a cause of ASDs, referred to as the trans-synaptic interaction hypothesis of ASDs. Clearly, mutations in genes encoding this synaptic pathway are exceedingly rare, with the relatively high frequency of germ-line mosaicism for such mutations (i.e., relative to their overall rare occurrence), consistent with the notion that their consequences are rather severe.
Our results raise a number of new questions. Our finding that wild-type NL4 increases synapse numbers in transfected neurons and simultaneously decreases EPSC size and mEPSC frequency is puzzling. The effect of wild-type NL4 is selective, because IPSC size and mIPSC frequency were not changed. The paradoxical effect of NL4 on synapse numbers and synaptic strength does not fit with previous observations, raising the question whether NL4 is intrinsically different from other neuroligins and what the mechanistic basis for this difference is. At present, we cannot answer these questions. Independent of what the answers to these questions are, however, the existence of this difference suggests a possible rationale for the fact that mutations in the NL4 gene are repeatedly observed in patients with ASDs, whereas mutations in other neuroligin genes are not. Future studies will also have to analyze this possibility systematically.
Footnotes
This study was supported by National Institutes of Health Grants U54 MH 66398 STAART Center Grant (H.T.-F.), R37 MH52804-08 (T.C.S.), and R01 MH081164A (C.M.P.), the Simons Foundation (T.C.S.), and gifts from BRAINS for Autism/Debra Caudy and Clay Heighten Founders, The Crystal Charity Ball, and Hartwell Foundation (C.M.P.). J.K. was partly supported by the Korea Research Foundation funded by the Korean Government (Ministry of Education and Human Resources Development, Basic Research Promotion Fund, Grant KRF-2007-357-C00093) and by a long-term fellowship from the Human Frontier Science Program. We thank the parents of SS and TS for the support in conducting this study.
References
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. Washington, DC: American Psychiatric Association; 2000. DSM-IV-TR. Diagnostic and statistical manual of mental disorders, Ed 4, text revision. [Google Scholar]
- Araç D, Boucard AA, Ozkan E, Strop P, Newell E, Südhof TC, Brunger AT. Structures of neuroligin-1 and the neuroligin-1/neurexin-1 beta complex reveal specific protein-protein and protein-Ca2+ interactions. Neuron. 2007;56:992–1003. doi: 10.1016/j.neuron.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Bakker E, Van Broeckhoven C, Bonten EJ, van de Vooren MJ, Veenema H, Van Hul W, Van Ommen GJ, Vandenberghe A, Pearson PL. Germline mosaicism and Duchenne muscular dystrophy mutations. Nature. 1987;329:554–556. doi: 10.1038/329554a0. [DOI] [PubMed] [Google Scholar]
- Beaudet AL. Autism: highly heritable but not inherited. Nat Med. 2007;13:534–536. doi: 10.1038/nm0507-534. [DOI] [PubMed] [Google Scholar]
- Berninghausen O, Rahman MA, Silva JP, Davletov B, Hopkins C, Ushkaryov YA. Neurexin Ibeta and neuroligin are localized on opposite membranes in mature central synapses. J Neurochem. 2007;103:1855–1863. doi: 10.1111/j.1471-4159.2007.04918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi F, Bacchelli E, Pesaresi G, Carone S, Bailey AJ, Maestrini E. Absence of coding mutations in the X-linked genes neuroligin 3 and neuroligin 4 in individuals with autism from the IMGSAC collection. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:220–221. doi: 10.1002/ajmg.b.30287. [DOI] [PubMed] [Google Scholar]
- Bolliger MF, Frei K, Winterhalter KH, Gloor SM. Identification of a novel neuroligin in humans which binds to PSD-95 and has a widespread expression. Biochem J. 2001;356:581–588. doi: 10.1042/0264-6021:3560581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolliger MF, Pei J, Maxeiner S, Boucard AA, Grishin NV, Südhof TC. Unusually rapid evolution of Neuroligin-4 in mice. Proc Natl Acad Sci U S A. 2008;105:6421–6426. doi: 10.1073/pnas.0801383105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucard AA, Chubykin AA, Comoletti D, Taylor P, Südhof TC. A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron. 2005;48:229–236. doi: 10.1016/j.neuron.2005.08.026. [DOI] [PubMed] [Google Scholar]
- Chen X, Liu H, Shim AH, Focia PJ, He X. Structural basis for synaptic adhesion mediated by neuroligin-neurexin interactions. Nat Struct Mol Biol. 2008;15:50–56. doi: 10.1038/nsmb1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Afridi SK, Clark L, Scheiffele P. Disorder-associated mutations lead to functional inactivation of neuroligins. Hum Mol Genet. 2004;13:1471–1477. doi: 10.1093/hmg/ddh158. [DOI] [PubMed] [Google Scholar]
- Chih B, Gollan L, Scheiffele P. Alternative splicing controls selective trans-synaptic interactions of the neuroligin-neurexin complex. Neuron. 2006;51:171–178. doi: 10.1016/j.neuron.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Chocholska S, Rossier E, Barbi G, Kehrer-Sawatzki H. Molecular cytogenetic analysis of a familial interstitial deletion Xp22.2–22.3 with a highly variable phenotype in female carriers. Am J Med Genet A. 2006;140:604–610. doi: 10.1002/ajmg.a.31145. [DOI] [PubMed] [Google Scholar]
- Chubykin AA, Liu X, Comoletti D, Tsigelny I, Taylor P, Südhof TC. Dissection of synapse induction by neuroligins: effect of a neuroligin mutation associated with autism. J Biol Chem. 2005;280:22365–22374. doi: 10.1074/jbc.M410723200. [DOI] [PubMed] [Google Scholar]
- Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, Südhof TC. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron. 2007;54:919–931. doi: 10.1016/j.neuron.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comoletti D, De Jaco A, Jennings LL, Flynn RE, Gaietta G, Tsigelny I, Ellisman MH, Taylor P. The Arg451Cys-neuroligin-3 mutation associated with autism reveals a defect in protein processing. J Neurosci. 2004;24:4889–4893. doi: 10.1523/JNEUROSCI.0468-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy WG, Nai Q, Ross B, Naughton G, Berg DK. Postsynaptic neuroligin enhances presynaptic inputs at neuronal nicotinic synapses. Dev Biol. 2007;307:79–91. doi: 10.1016/j.ydbio.2007.04.017. [DOI] [PubMed] [Google Scholar]
- De Jaco A, Comoletti D, Kovarik Z, Gaietta G, Radic Z, Lockridge O, Ellisman MH, Taylor P. A mutation linked with autism reveals a common mechanism of endoplasmic reticulum retention for the alpha, beta-hydrolase fold protein family. J Biol Chem. 2006;281:9667–9676. doi: 10.1074/jbc.M510262200. [DOI] [PubMed] [Google Scholar]
- Dong N, Qi J, Chen G. Molecular reconstitution of functional GABAergic synapses with expression of neuroligin-2 and GABAA receptors. Mol Cell Neurosci. 2007;35:14–23. doi: 10.1016/j.mcn.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Rogé B, Héron D, Burglen L, Gillberg C, Leboyer M, Bourgeron T. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrichny IP, Leone P, Sulzenbacher G, Comoletti D, Miller MT, Taylor P, Bourne Y, Marchot P. Structural analysis of the synaptic protein neuroligin and its beta-neurexin complex: determinants for folding and cell adhesion. Neuron. 2007;56:979–991. doi: 10.1016/j.neuron.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futai K, Kim MJ, Hashikawa T, Scheiffele P, Sheng M, Hayashi Y. Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95-neuroligin. Nat Neurosci. 2007;10:186–195. doi: 10.1038/nn1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J, Bonnel A, St-Onge J, Karemera L, Laurent S, Mottron L, Fombonne E, Joober R, Rouleau GA. NLGN3/NLGN4 gene mutations are not responsible for autism in the Quebec population. Am J Med Genet B Neuropsychiatr Genet. 2005;132B:74–75. doi: 10.1002/ajmg.b.30066. [DOI] [PubMed] [Google Scholar]
- Helderman-van den Enden AT, de Jong R, den Dunnen JT, Houwing-Duistermaat JJ, Kneppers AL, Ginjaar HB, Breuning MH, Bakker E. Recurrence risk due to germ line mosaicism. Duchenne and Becker muscular dystrophy. Clin Genet. 2009;75:465–472. doi: 10.1111/j.1399-0004.2009.01173.x. [DOI] [PubMed] [Google Scholar]
- Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Südhof TC. Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell. 1995;81:435–443. doi: 10.1016/0092-8674(95)90396-8. [DOI] [PubMed] [Google Scholar]
- Ichtchenko K, Nguyen T, Südhof TC. Structures, alternative splicing, and neurexin binding of multiple neuroligins. J Biol Chem. 1996;271:2676–2682. doi: 10.1074/jbc.271.5.2676. [DOI] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Radyushkin K, Hammerschmidt K, Granon S, Boretius S, Varoqueaux F, Ramanantsoa N, Gallego J, Ronnenberg A, Winter D, Frahm J, Fischer J, Bourgeron T, Ehrenreich H, Brose N. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc Natl Acad Sci U S A. 2008;105:1710–1715. doi: 10.1073/pnas.0711555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffries AR, Curran S, Elmslie F, Sharma A, Wenger S, Hummel M, Powell J. Molecular and phenotypic characterization of ring chromosome 22. Am J Med Genet A. 2005;137:139–147. doi: 10.1002/ajmg.a.30780. [DOI] [PubMed] [Google Scholar]
- Kim HW, Cho SC, Kim JW, Cho IH, Kim SA, Park M, Cho EJ, Yoo HJ. Family-based association study between NOS-I and -IIA polymorphisms and autism spectrum disorders in Korean trios. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:300–306. doi: 10.1002/ajmg.b.30798. [DOI] [PubMed] [Google Scholar]
- Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, Raynaud M, Ronce N, Lemonnier E, Calvas P, Laudier B, Chelly J, Fryns JP, Ropers HH, Hamel BC, Andres C, Barthélémy C, Moraine C, Briault S. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004;74:552–557. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson-Yuen A, Saldivar JS, Sommer S, Picker J. Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur J Hum Genet. 2008;16:614–618. doi: 10.1038/sj.ejhg.5202006. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, LeCouteur A. Autism Diagnostic Interview- Revised: revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, DiLavore PC, Risi S. Los Angeles: Western Psychological Services; 2002. Autism Diagnostic Observation Schedule-Generic (ADOS-G) manual. [Google Scholar]
- Losh M, Sullivan PF, Trembath D, Piven J. Current developments in the genetics of autism: from phenome to genome. J Neuropathol Exp Neurol. 2008;67:829–837. doi: 10.1097/NEN.0b013e318184482d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macarov M, Zeigler M, Newman JP, Strich D, Sury V, Tennenbaum A, Meiner V. Deletions of VCX-A and NLGN4: a variable phenotype including normal intellect. J Intellect Disabil Res. 2007;51:329–333. doi: 10.1111/j.1365-2788.2006.00880.x. [DOI] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, Thiruvahindrapduram B, Fiebig A, Schreiber S, Friedman J, Ketelaars CE, Vos YJ, Ficicioglu C, Kirkpatrick S, Nicolson R, Sloman L, Summers A, Gibbons CA, Teebi A, Chitayat D, Weksberg R, Thompson A, Vardy C, Crosbie V, Luscombe S, Baatjes R, Zwaigenbaum L, Roberts W, Fernandez B, Szatmari P, Scherer SW. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Südhof TC. Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron. 2005;48:547–554. doi: 10.1016/j.neuron.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Maximov A, Pang ZP, Tervo DG, Südhof TC. Monitoring synaptic transmission in primary neuronal cultures using local extracellular stimulation. J Neurosci Methods. 2007;161:75–87. doi: 10.1016/j.jneumeth.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, Al-Saad S, Ware J, Joseph RM, Greenblatt R, Gleason D, Ertelt JA, Apse KA, Bodell A, Partlow JN, Barry B, Yao H, Markianos K, Ferland RJ, Greenberg ME, Walsh CA. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen EM. Circle Pines, MN: American Guidance Service; 1995. Mullen scales of early learning. [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- Nam CI, Chen L. Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc Natl Acad Sci U S A. 2005;102:6137–6142. doi: 10.1073/pnas.0502038102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Song JY, Ichtchenko K, Südhof TC, Brose N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc Natl Acad Sci U S A. 1999;96:1100–1105. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow S, Balla D, Cicchetti D. Circle Pines, MN: American Guidance Service; 1984. Vineland adaptive behavior scales. [Google Scholar]
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, Vincent JB, Skaug JL, Thompson AP, Senman L, Feuk L, Qian C, Bryson SE, Jones MB, Marshall CR, Scherer SW, Vieland VJ, Bartlett C, Mangin LV, Goedken R, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, Südhof TC. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talebizadeh Z, Lam DY, Theodoro MF, Bittel DC, Lushington GH, Butler MG. Novel splice isoforms for NLGN3 and NLGN4 with possible implications in autism. J Med Genet. 2006;43:e21. doi: 10.1136/jmg.2005.036897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Südhof TC, Brose N. Neuroligins determine synapse maturation and function. Neuron. 2006;51:741–754. doi: 10.1016/j.neuron.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Vincent JB, Kolozsvari D, Roberts WS, Bolton PF, Gurling HM, Scherer SW. Mutation screening of X-chromosomal neuroligin genes: no mutations in 196 autism probands. Am J Med Genet B Neuropsychiatr Genet. 2004;129B:82–84. doi: 10.1002/ajmg.b.30069. [DOI] [PubMed] [Google Scholar]
- Wermter AK, Kamp-Becker I, Strauch K, Schulte-Körne G, Remschmidt H. No evidence for involvement of genetic variants in the X-linked neuroligin genes NLGN3 and NLGN4X in probands with autism spectrum disorder on high functioning level. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:535–537. doi: 10.1002/ajmg.b.30618. [DOI] [PubMed] [Google Scholar]
- Yan J, Oliveira G, Coutinho A, Yang C, Feng J, Katz C, Sram J, Bockholt A, Jones IR, Craddock N, Cook EH, Jr, Vicente A, Sommer SS. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol Psychiatry. 2005;10:329–332. doi: 10.1038/sj.mp.4001629. [DOI] [PubMed] [Google Scholar]