

Abstract
To demonstrate protein modulation of metal cofactor reactivity through non-covalent interactions, pH-dependent sulfoxidation and ABTS oxidation reactivity of a designed myoglobin (Mb) containing non-native MnSalen complex (1) was investigated using H2O2 as the oxidant. Incorporation of 1 inside the Mb resulted in increase in turnover numbers through exclusion of water from the metal complex and prevention of MnSalen dimer formation. Interestingly, the presence of protein in itself is not enough to confer the increase activity as mutation of the distal His64 in Mb to Phe to remove hydrogen bonding interactions resulted in no increase in turnover numbers, while mutation His64 to Arg, another residue with ability to hydrogen bond interactions resulted in increase in reactivity. These results strongly suggest that the distal ligand His64, through its hydrogen bonding interaction, plays important roles in enhancing and fine-tuning reactivity of the MnSalen complex. Nonlinear least-squares fitting of rate vs. pH plots demonstrates that 1·Mb(H64X, X= H, R and F) and the control MnSalen 1 exhibit pKas varying from pH 6.4 to 8.3, and that the lower pKa of the distal ligand in 1·Mb(H64X), the higher the reactivity it achieves. Moreover, in addition to the pKa at high pH, 1·Mb displays another pKa at low pH, with pKa of 5.0±0.08. A comparison of the effect of different pH on sulfoxidation and ABTS oxidation indicates that, while the intermediate produced at low pH conditions could only perform sulfoxidation, the intermediate at high pH could oxidize both sulfoxides and ABTS. Such a fine-control of reactivity through hydrogen bonding interactions by the distal ligand to bind, orient and activate H2O2 is very important for designing artificial catalysts with dramatic different and tunable reactivity from catalysts without proteins.
Keywords: protein engineering, Mnsalen, oxidation, pH dependent reactivity
Introduction
Proteins are well known to be the best in fine-tuning reactivity and selectivity of the same metal cofactor inside them to achieve a wide range of functions. An excellent example is heme proteins where the same protoporphyrin IX can be modulated by different protein environments to display functions from electron transfer, to oxygen transport, catalase, peroxidase and oxidase.[1-4] Unlocking this secret of how proteins exert their fine controls of metal cofactors can enrich our fundamental knowledge of metalloprotein design and engineering and result in new (bio)catalysts with exceptional reactivity and selectivity.[5-13] Toward this goal, a number of biochemical and biophysical studies of metalloproteins and their variants have resulted in much insight into this question. A true test of our knowledge in this area is to apply the knowledge gained to design and fine-tune a metalloprotein that contains a metal cofactor that is similar to that in Nature. An even bigger challenge to go through this process using a metal cofactor that Nature has not yet to utilized, or we have not discovered from Nature yet. In this way, we can not only confirm what we have learned from studying native enzymes and their variants, but also reveal new principles that may not be apparent when investigating native systems.
MnSalen complexes (Salen =N,N’-bissalicylidene-1,2-ethanediamino anion, MnSalen, 1, Figure 1) is a good choice as a non-native metal cofactor to demonstrate fine-controls offered by proteins, because MnSalen complexes are well known inorganic catalysts with broad applications, including asymmetric catalysis.[14-17] Despite the tremendous successes, its reaction mechanism and how to control its reactivity is still not well understood. Most of efforts in this direction has so far focused on studying the effects of covalent modifications of the Salen ligand or replacing manganese with other metal ions.[18] Much less is known about the interaction of metal catalysts with H2O2, a cost-effective, stable, readily available, and produce only water as a by-product,[19, 20] because H2O2 is a poorer ligand than H2O, which is difficult to activate to the hydroperoxo or peroxo active intermediates. Recent work has shown that appending imidazole[21-23], urea[24], and carboxylate[25-28] functional groups to the Salen skeleton to introduce the distal or axial ligand in the microenvironment similar to that for heme in heme proteins can enhance their reactivity and selectivity toward oxidation (Left, figure 1). However, the roles of non-covalent interactions are much less understood, partly because it is much harder to pinpoint the exact location and their roles in complexes outside the protein. Therefore it is desirable to carry systematic studies on non-covalent modulations at more defined locations. Toward this goal, several model compounds have been made to position non-covalent interactions, such as hydrogen bonds at precise locations to demonstrate its roles in fine tuning reactivities.[25, 27, 29, 30] This strategy has not been applied to MnSalen family of compounds.
Figure 1.
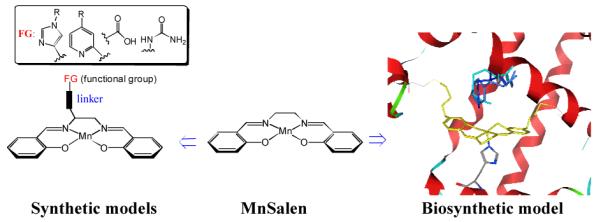
Synthetic MnSalen model vs. biosynthetic model (Computer model of Mb(L72C/T39C) with 1 covalently attached overlayed with heme) of 1. Mb(H64X, X=H, R and F) in this work.
Proteins prefer to use the same metal cofactor with minimal or no modification on the ligand because it causes more time and energy to synthesize or modify the metal cofactors in cells. Therefore proteins have developed a number of strategies using non-covalent interactions to fine-tune the same metal cofactors and activate mild oxidants such as H2O2 for a wide range of functions. However, the exact features and mechanisms by which proteins can control the properties are not clear. A major non-covalent modulation strategy used by proteins is site-specific control of pKa of specific residues around metal-binding site to activate mild oxidants such as H2O2 for reactions. [31] For example, pH dependent reactivity for heme peroxidases shows that the secondary coordination sphere, including the distal histidine facilitates the protonation/deprotonation equilibrium of H2O2 coordinated to the heme iron center and consequent pH-induced changeover in the mode of O-O bond cleavage in the Fe-OOH intermediates.[1-4, 32] Thus, learning Nature’s way to control the reactivity and selectivity of metal cofactor in activation of small molecules by exploiting their dependence on conditions such as pKa is important for further designing efficient metal catalysts.[29]
We previously reported the effectiveness of a dual covalent anchoring approach to inducing achiral MnSalen into heme-free apo-myoglobin (Mb) and the resulting artificial enzyme displayed good enantioselectivity and chemoselectivity toward sulfoxidation.[33, 34] With the metal cofactor firmly attached inside the protein, this artificial enzyme provides an ideal system to test our knowledge of fine-control catalytic reactivity and selectivity through non-covalent interactions and obtain new insights. MnSalen adopts a structural orientation similar to heme, allowing similar non-covalent interactions that are critical to heme enzymes to be utilized in MnSalen reactions. Furthermore, site-specific modulation of the primary and secondary coordination sphere can be easily realized by site-directed mutagenesis for the biosynthetic models.[9] Herein we investigate pH-dependent sulfoxidation and ABTS (2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) oxidation reactivity of MnSalen complex 1 inside Mb with different distal ligands presence. In the process, we demonstrate the ability of a protein scaffold to modulate the reactivity of a non-native metal catalyst through non-covalent interactions and have obtained new insights on how distal ligands, through hydrogen bonding interactions, activate H2O2 to generate different intermediates for different reactions.
Results
pH-Dependent Sulfoxidation with H2O2 Catalyzed by 1·Mb(H64X) (X= H, F or R)
Construction, expression, and purification of myoglobin (Mb) with T39C/L72C mutations (as dual anchor points for MnSalen complex 1) and its H64 variants (H64X, where X= F and R, to probe the role of the distal ligand) were carried out using procedures reported previously.[35] The heme in those Mbs were removed by extraction with butanone.[33, 36-38] To incorporate MnSalen complex (1) into those apo-Mbs, 10 equiv. of methanthiosulfonate-modified Mn(III)Salen complex (N,N’-bis(4-(2-methanesulfonylthioethoxy)salicylidene)-1,2-ethanediamino-manganese(III) bromide) in DMSO solution was added to 0.1 mM apo-Mb solution in 50 mM ammonia acetate buffer (pH= 5.1) at room temperature for 1-2 h, and the conjugation process was monitored by UV-vis spectroscopy until completion. UV/vis spectra of all MnSalen conjugates presented in this study display two absorption bands at 280 and 292 nm with the ratio of 1:1 (see supplemental information Figure S1), which is consistent with the attachment of a MnSalen moiety (with an absorption around 292 nm) into the protein (with an absorption around 280 nm); a shoulder was also observed near 340 nm, which is assignable to absorption by the MnSalen complex. These results suggest that the mutations introduced in this study did not perturb the active site structure grossly. The MnSalen incorporation into Mb was further confirmed by electrospray mass spectrometry; the molecular weights of the variants (called 1·Mb(H64X, X= H, F or R) from now on) are all consistent with MnSalen attached to Mb variants through dual Cys anchors (see supplemental information Figure S2).
Thioanisole and ABTS were used as mechanistic probes to detect the generation of possible intermediate(s) along a variety of pH profiles because sulfides can be oxidized by weak and strong oxidants at electron-rich sulfur atom[39, 40] whereas ABTS is only oxidized by strong oxidants such as metal-oxo or hydroxyl radical intermediates[32]. To evaluate the pH-dependence of sulfoxidation with H2O2, the reactivity in terms of turn over numbers (TON) and enantioselectivity in term of enantiomeric excess (e.e.) were determined at 21 different pH values between 4.0 and 9.0 at 25°C. To ensure buffer capacity over such a wide range of pH values, a mixed buffer (50 mM each of sodium acetate, MES, MOPS, TRIS) was used. MnSalen complex 1 without the protein scaffold was chosen as the control catalyst in this study. For each pH value studied, at least three independent experiments were conducted and the pH-dependences of the reaction rates (TONs) are illustrated Figure 2a. The MnSalen complex 1 exhibits very low activity in the low pH region between pH 4.0–7.0 (e.g., TON = 19.5±4.3 ×10−3 min−1 at pH 5.0, see Table 1), and its activity increased with increasing pH after pH 7 (e.g., TON = 758±74.9 ×10−3 min−1 at pH 8.0). No enantioselectivity was observed. A nonlinear least-squares fitting of the data suggests an apparent pKa of 7.5 for this pH dependent transition.
Figure 2.

(a) pH-dependent reactivity assay in catalytic sulfoxidation by hydrogen peroxide with biocatalysts 1·Mb(H64X, X=H (red line), R (pink line) and F (blue line)) and the control MnSalen 1 (black line).; (b) the observed reaction rates (kobss) for 1·Mb(H64X, X=H, F, and R) and the control MnSalen 1 in pH-dependent ABTS oxidations (5.0–9.0) with hydrogen peroxide; (c)-(f) pH effects of reactivity of 1·Mb(H64X, X=H, R, and F) and the control MnSalen 1 with H2O2 using ABTS (left axis) and thioanisole (right axis) as probes, respectively.
Table 1.
The reactivity and enantioselectivity of 1•Mb(H64X) and the control MnSalen 1 in catalytic sulfoxidiation at pH= 5.0 and 8.0.a
| Entry | Catalyst | pH=5.0 | pH=8.0 | ||
|---|---|---|---|---|---|
| TON×10−3 min−1b |
e.e.%b | TON×10−3 min−1b |
e.e.%b | ||
| 1 | 1 | 19.5±4.3 | — | 758±74.9 | — |
| 2 | 1•Mb | 405.5±7.8 | 67.0±2.2 | 1654.0±104.3 | 49.8±0.3 |
| 3 | 1•Mb(H64F) | 22.5±3.2 | —c | 597.2 ±33.6 | 38.7± 2.2 |
| 4 | 1•Mb(H64R) | 141.6±10.1 | 43.4±2.1 | 1724.0±184.3 | 47.7±1.1 |
Reactions were performed with 5 mM substrate, 10 mM H2O2, 0.5 mM catalyst in 200 μL mixed buffers (50 mM ammonia acetate, Mes, Heps and Tris, pH= 5.0 or 8.0) at 4°C for 10 mins.
Yield and enantioselectivity excess (56–60% for 1) were determined by GC with a chiral G-TA column with acetophenone as internal standard and std dev reported in parentheses.
For 1•Mb(H64F), in pH 5.0, the reactivity is too low to calculate e.e%.
When the same complex 1 is incorporated into the Mb protein, however, the biosynthetic complex 1·Mb shows higher activity even at low pH (e.g., TON: 405.5±7.8 ×10−3 min−1 see Table 1), and the reactivity increased even further with increasing pH (e.g., TON: 1654.0±104.3 min−1). Enantioselectivities were also observed at both high and low pH, with higher ee% (67.0±2.2%) at pH 5.0 than at pH 8.0 (49.8±0.3%). This result demonstrated that the protein scaffold can enhance metal complex reactivity and selectivity in water at all physiologically relevant pH values. More interestingly, there are two pH dependent transitions, one at low pH with an apparent pKa of 5.0, and another at high pH with an apparent pKs of 7.3. This observation suggests that the protein can not only enhance but also can fine-tune metal complex activity in water.
To search for structural feature(s) responsible for this interesting pH dependent behavior of 1·Mb, we changed the distal His64, which has been shown to play a critical role in modulating the oxygen-binding ability of heme proteins through hydrogen-bonding interaction, into either phenylalanine to remove the hydrogen bond, or arginine to introduce a different hydrogen bond donor. Mutation of His64 to Phe (1·Mb(H64F)) resulted in much lower activity at both low pH (e.g., TON: 22.5±3.2 ×10−3 min−1 at pH 5.0, see Table 1) and high pH (e.g., TON: 597.2 ±33.6 × 10−3 min−1 at pH 8.0) than those observed for 1·Mb, and in a single pH dependent transition (Figure 2a), similar to that of complex 1 without the protein. This result indicates that His64 plays an important role in enhancing and fine-tuning MnSalen 1 activities. Since this distal His64 has been proposed to influence dioxygen binding and activity in myoglobin through hydrogen bonding network,[32, 41, 42] we investigated a mutant of His64 to Arg, which has a different hydrogen bonding interaction.[24, 43] The variant (1·Mb(H64R) also resulted a single pH dependent transition, and yet the reactivity is higher than those of either 1·Mb(H64F) or 1 by itself (e.g., for 1·Mb(H64R) TON: 141.6±10.1 ×10−3 min−1 at pH 5.0, and 1724.0±184.3 × 10−3 min−1 at pH 8.0, see Table 1). For 1·Mb(H64R), the enantioselectivity remained similar at both high and low pH’s (from 43.4±2.1at pH 5.0 to 47.7±1.1% at pH 8.0). These results strongly suggest that the distal His64, through its hydrogen bonding interaction, plays important roles in enhancing and fine-tuning reactivity of the MnSalen complex.
The above investigation used H2O2 as an oxidant, in which protonation and deprotonation of H2O2 through hydrogen bonding interactions can be important. To confirm that is the case, we choose to use another terminal oxidant, idosobenzene PhIO, where oxidant protonation and deprotonation is not as important (supporting information, figure S3). Repeating the same reactions using PhIO at different pH indicates little pH dependent reactivity for 1·Mb (TONs: from 859.2±12.6 at pH 5.0 to 683.5±56.5×10−3 min−1 at pH 9.0). This result is in contrast to the results obtained when H2O2 is the oxidant (TONs: from 405.5±7.8 at pH 5.0 to 1654.0±104.3×10−3 min−1 at pH 8.0). In addition, enantioselectivity is much lower when using PhIO as the oxidant (e.e.: 15–20%) than that when using H2O2 is the oxidant. These results demonstrate that this strong pH effect only occurs when H2O2 was used as an oxidant.
All the variants investigated in this study, including the control MnSalen 1 without the protein, display a pH dependent reactivity transition at high pH, which suggests a protonation/deprotonation of H2O2 may be responsible for this phenomenon both in the presence and in the absence of the protein scaffold. Different environments, however, do influence the pKa of the high pH transitions; 1·Mb and 1·Mb(H64R) with polar distal ligands (H or R) show pKas (7.3 and 6.6, respectively) that are much lower than that of a less polar ligand (F) in 1·Mb(H64F) (8.1), indicating that the polar distal ligand facilitates the binding and deprotonation of H2O2. Indeed, comparison of the reactivities and pKas of 1·Mb(H64X, X=H, R and F) shows that the lower pKa of the distal ligand in 1·Mb(H64X), the higher the reactivity it achieves.
Interestingly, 1·Mb with distal His64 present has an extra pKa transition at 5.0, close to the reported pKa (4.0–6.0) of His64 in native myoglobin,[44] indicating that this pKa reflects the effect of the ionization state of His 64 on the interaction with H2O2. The other mutants 1·Mb(H64F) and 1·Mb(H64R) only show one pKa because F and R have no change in ionization within the pH range from 4.0–9.0. These results clearly suggests that pH modulates the H2O2-dependent reactivity of MnSalen inside the Mb scaffold, not only directly through controlling protonation/deprotonation equilibrium of H2O2, but also through changing ionization states of the distal ligand.
pH-Dependent ABTS Oxidation Catalyzed by 1·Mb(H64X, X= H, F or R)
To explore whether pH affects generation of different active intermediates at different pHs, we chose 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) (ABTS) as a probe because it is commonly applied for the quantification of the enzymatic activity of peroxidases, and because it serves to trap any hypervalent metal porphyrin[45-47] or Salen species[48, 49]. The experiments for pH-dependent ABTS oxidation were carried out in the presence of H2O2 at 25°C with the ratio of the [catalyst]: [H2O2]: [ABTS] being 2.5 μM: 250 μM: 250 μM (1: 100: 100), and the kobs was obtained by monitoring the change of absorption at 734 nm. A mixed buffer of 50 mM each of sodium acetate, Mops, Mes and Tris buffer was used to maintain pH 6.0–9.0, while a 200 mM Mes buffer was used in pH 5.0 to 6.0, because the carboxylate buffer resulted in higher background reactivity below pH 6.0. We found that ABTS oxidation was catalyzed by 1·Mb(H64X, X=H, F and R) and the control MnSalen 1 also exhibits strong pH-dependence (Figure 2b), as reaction rate (kobs) increases with increasing pH.
The most striking difference between the sulfoxidation reaction presented in Figure 2a and ABTS oxidation presented in Figure 2b is the lack of ABTS oxidation reactivity of all variants in the low pH range (5.0–6.0 (kobs <0.05×10−3 s−1), which is close to the value observed in control experiment where no catalyst was present. Increasing pH from 6.0 to 8.0 resulted in higher reactivity toward ABTS oxidation, with the most dramatic increase in the reaction rates of 1·Mb(H64R) (from 0.06±0.006 to 4.7±0.05 × 10−3 s−1), followed by 1·Mb (from 0.11±0.01 to 3.1±0.05×10−3 s−1) and then by 1·Mb(H64F) (from 0.005±0.001 to 0.83±0.04×10−3 s−1). From these results, we conclude that generation of a reactive intermediate such as Mn-oxo intermediate or the other hydroxyl radicals is highly pH-dependent and that breaking the O-O bond is more favored at high pH than at low pH.
Through a nonlinear least-square fit of rate (kobss) vs. pHs (pH=5.0–9.0) in Figure 2b, we obtained pKas of the reactivity transition for 1·Mb(H64X) (X= H, F or R). With the polar distal ligands such as His64 and Arg 64, the pKa’s (7.8 and 7.6) are lower than the pKa (8.4) obtained with hydrophobic distal ligand Phe64; this trend is similar to that observed for the sulfoxidation reaction shown in Figure 2a.
Interestingly, the control MnSalen 1, without protein scaffold, shows different pH effects on the reaction rates: reaction rates increase with pH increasing from pH 7.0 to 8.0, and then the rates began to decrease from 0.69±0.02 to 0.06±0.005×10−3s−1 from pH 8.0 to 9.0 (Figure 2b). Similar decrease in sulfoxidation is also observed (see Figure 2a), although not as pronounced. Such a bell-shape curve vs pH is probably due to the less steric bulky structure of MnSalen permitting the formation of the low reactive μ-oxo MnSalen dimer species in high pH. To confirm this hypothesis, we collected electrospray mass spectra of Mnsalen complex 1 at pH 5, 7, 8 and 9, and found that the extent of μ-oxo dimer formation increases with increasing pH (see supporting information Figure S4). This observation suggests that the protein scaffold is beneficial for the MnSalen to prevent formation of less active μ-oxo MnSalen dimer. More interestingly, 1·Mb only shows one pKa (7.8) in this ABTS oxidation, in contrast to two pKa’s observed in sulfoxidation, which suggests that the ionization state of His64 does not affect the reactivity of the MnSalen cofactor in ABTS oxidation.
Comparison of pH effects in sulfoxidation and ABTS oxidation
To reveal whether pH modulates generation of different intermediates in the 1·Mb and its variants, a side-by-side comparison of pH effects on sulfoxidation and ABTS oxidation is shown in Figure 2c-f. All biocatalysts 1·Mb(H64X) (X= H, F or R) show higher pKa’s and narrower pH-dependent regions in ABTS oxidation than those in sulfoxidation. This observation clearly suggests that pH modulates at least two different active intermediates: one has low reactivity only for sulfoxidation of thioanisole at low pH; another has high reactivity for oxidation of both thioanisole and ABTS at high pH. This is consistent with the proposed role of base in catalyzing cleavage of O-O bond to generate active Mn-oxo intermediates for MnSalen or porphyrin catalysts with peroxides as oxidants in high pHs.[16, 50, 51]
Producing different active intermediates is highly dependent on the type of distal ligands. To measure different pH effects on sulfoxidation or ABTS oxidation, we use the maximum ΔpKa between the pH dependent sulfoxidation and ABTS oxidation curves (Figure 2c-f). The MnSalen complex 1 without the protein exhibited small ΔpKa (0.3, Figure 2c) for both sulfoxidation and ABTS oxidation, indicating that the active intermediates for this complex in both reactions are very similar, if not identical. The 1·Mb, on the other hand, has two ΔpKa’s (Figure 2d); a similarly small ΔpKa of 0.5 in the high pH region as the MnSalen complex 1 suggests they share a similar active intermediate, while a new large ΔpKa of 2.8 in the low pH region where no ABTS oxidation could be observed, indicating a different active intermediate from MnSalen complex 1. Replacing the distal His64 with Phe to remove the hydrogen bond interaction 1·Mb(H64F) abolished the large ΔpKa in the low pH region, while the ΔpKa in the high pH region (0.3) is identical to that of MnSalen complex 1 (Figure 2e). The 1·Mb(H64R) displays larger ΔpKas of 1.0 in the high pH region (Figure 2f). Therefore, it is reasonable to conclude that the dominant active intermediate in sulfoxidation at low pH is different from that at high pH, and the distal ligand plays a critical role in fine-tuning which type of active intermediates is generated.
To support the above conclusion further, we carried out the competitive sulfoxidation catalyzed by 1·Mb in the presence of different amount of ABTS (0, 0.1, 0.2, 0.3, 0.4 and 0.5 equivalents with respective to that of thioanisole) at pH 5.0 and 8.0. 1·Mb is an excellent system to address this issue because it shows dramatically different reactivity for sulfoxidation and ABTS oxidation at low pH. If increasing the amount of ABTS inhibits the reactivity in sulfoxidation at a given pH, it supports that the same intermediate is indeed involved in both sulfoxidation and ABTS oxidation. As shown in Figure 3, addition of increasing amount of ABTS at pH 8.0 inhibits the reactivity of sulfoxidation; when the ratio of ABTS to thioanisole is up to 0.5 equivalents, the reaction rate decrease 50% compared to that without ABTS. These results suggest a common active intermediate for both reactions at high pH. On the other hand, addition of ABTS has limited effects on the reactivity of sulfoxidation (±5%) at pH 5.0, which strongly indicates that the dominant active intermediate in sulfoxidation at low pH is different from that in high pH.
Figure 3.
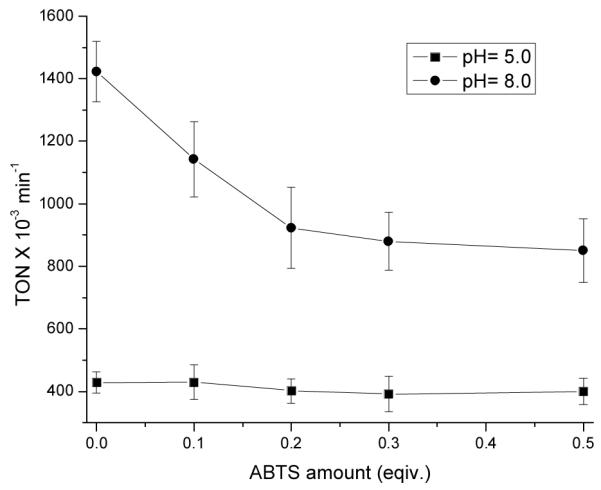
Inhibition of 1·Mb reactivity of thioanisole sulfoxidation in the presence of different amounts of ABTS (0–0.5 equiv. with respective to thioanisole) at pH 5.0 and 8.0.
Discussions
Non-covalent modulations of metal complex reactivity is very important in designing better catalysts, and it has been difficult to pinpoint the exact structural features and roles the structural features play in fine-tuning the reactivity. By incorporating a non-native metal cofactor MnSalen Complex 1 into myoglobin and its variants, and comparing their functional properties with and without proteins, we have gained the following new insights in this aspect.
In the absence of proteins, the MnSalen Complex 1 has exhibited a low reactivity in aqueous solution and no enantioselectivity (Table 1, entry 1). This is not surprising because water is not an innocent solvent like many organic solvents used in most MnSalen reactivity studies, and can interfere with many organic transformations including sulfoxidation investigated here. So the first benefit of proteins is to surround the complex with an environment to exclude water or to direct water to the metal-binding site in a controlled manner so that organic transformations can be carried efficiently in water. This is evident by the higher reactivity at both low and high pH when 1 is inside the myoglobin (Table 1, entry 2). This result is consistent with findings from many other groups who incorporated metal complexes inside proteins.[52-60] A new finding from this study is that simple incorporation of metal complexes inside protein does not result in automatic enhancement of reactivities because the His64 to Phe variant (1·Mb(H64F)) resulted in no increase of TON over 1 without the protein (Table 1, entry 3). Therefore other structural features must be important in controlling the reactivity of the complex.
As shown in Figure 2b, MnSalen 1 exhibits a bell-shaped pH dependence of oxidation activity, i.e., the activity increases with increasing pH until ~pH 8, after which the activity decreases with increasing pH. The activity decrease at high pH can be attributed to formation of less active μ-oxo MnSalen dimer. In contrast, no such bell-shaped dependence curve was observed when the MnSalen complex 1 is incorporated into Mb and its variants. This result suggests a second benefit of protein; through site isolation, the protein can prevent dimer formation. Such a benefit has been shown in heme proteins in preventing protoporphyrin IX dimer formation.[1, 2] We have now demonstrated the benefit in a non-native MnSalen complex.
In addition to water exclusion and dimer prevention, perhaps the biggest benefit of proteins is that they allow fine control of functional properties through site-specific non-covalent interactions that are difficult to achieve in non-protein environment except in a few well-defined model systems. [21, 24, 28, 29] One such control demonstrated here is the control of strong pH-dependence reactivity towards sulfoxidation and ABTS oxidation, and different reaction intermediates. This strong pH-dependence was observed only when H2O2 was used as oxidant instead of PhIO, indicating that the observed pH effect is related to the protonation/deprotonation equilibrium of the oxidant.[46, 47, 61, 62] All biocatalysts including the control MnSalen complex 1 without the protein exhibits a pKa in the high pH region from 6.5 to 8.5 for both sulfoxidation and ABTS oxidation. Similar pKa’s of 7.0–8.0 were previously observed when soluble iron and manganese porphyrins were used as catalysts with H2O2 as oxidant. [46, 47, 61, 62] Therefore it is reasonable to assign these pKa’s in high pH region to correspond to the equilibrium of protonation/deprotonation of H2O2 to form hydroperoxo MnSalen intermediate.
More interestingly, in addition to the pH-dependent transition at high pH for both sulfoxidation and ABTS oxidation reactions discussed above, 1·Mb alone shows an additional pKa at 5.0 in the sulfoxidation reaction, but not ABTS oxidation. This pKa is very close that of distal histidine in native myoglobion (His64), suggesting deprotonation of hydrogen peroxide is related to ionization of distal histidine in this pH range (4.0–6.0). Under these conditions, the basicity of His64 increases along with increasing pH, which is favored to form a hydrogen bond with H atom of H2O2, facilitating binding and activating H2O2 through deprotonation. This structural feature is responsible for this additional pH-dependent transition at low pH, and for the high reactivity of 1·Mb at low pH in comparison with 1 without protein and 1·Mb(H64F) when His64 was changed to Phe and thus eliminated the hydrogen bonding interaction. Reactivity of 1·Mb(H64R) further supports this conclusion, as Arg64 provides a different hydrogen bonding interaction and thus is responsible for its higher reactivity at both low and high pH (Table 1, entry 4). On the other hand, because Arg has a much higher intrinsic pKa that is beyond the pH range investigated here, it explains why no second pKa transition was observed at low pH, unlike in the case of 1·Mb when His 64 is present. These results suggest that that non-covalent interactions, specifically the presence of hydrogen bonding by the distal ligand, is critical in fine-tuning the reactivity of the metal catalysts.
Through comparison of pH effects on sulfoxidation and ABTS oxidation by 1·Mb(H64X, X=H, F and R), we also found that different intermediates may be involved in reactions at low or high pHs. It has been shown that sulfide can be oxidized by both weak and strong oxidants because of the electron-rich sulfur atom[39, 40] whereas ABTS can be oxidized only by strong oxidants such as Mn-oxo or hydroxyl radical intermediates.[32] ABTS oxidation was observed only at high pH for all biocatalysts investigated here while sulfoxidation was observed at both low and high pHs for 1·Mb. Competition experiments for oxidation of ABTS and sulfide in pH 5.0 and 8.0 catalyzed by 1·Mb show the ABTS inhibition of sulfoxidation occurred at pH 8.0, but not pH 5.0, which confirms that changing pH generates different active intermediates: at low pH, it only oxidizes sulfide whereas at high pH, it could oxidize both sulfide and ABTS. Reactivity modulated by pH has been reported for heme peroxidases[32] and iron or manganese porphyrins,[45, 47, 61, 63-65] and has been ascribed to induce changeover in the mode of O-O bond cleavage of the same M-OOH intermediates from heterolysis at low pH to homolysis at high pH. In contrast, in the MnSalen catalytic system investigated here, we observed pH could modulate different active intermediates, as the active intermediate could not oxidize ABTS at low pH, indicating that the active intermediate at this pH is not the highly active Mn-oxo species. We propose that the intermediate(s) with relatively low activity could be either manganese(III) hydroperoxide deprotonated from Mn-H2O2 without the cleavage of O-O bond;[66] or less reactive manganese high valent intermediates such as Mn-oxo with a different spin-state or interconversion to less reactive intermediate such as MnIV-OH through acid-base equilibrium.[67]
Taken the results together, we proposed that the reason behind the distal ligand modulation of pH dependent reactivity of MnSalen complex 1 is through control of generation of different intermediates at different pH through hydrogen bonding interactions. In the absence of hydrogen bonding interaction, such as in the case of His64 to Phe mutant, 1·Mb(H64F) exhibits similar pH-dependent behavior and pKas in both sulfoxidation and ABTS oxidation, indicating the same active intermediate is generated. On the other hand, His64 and Arg64 are capable of hydrogen bond interaction and thus could play important roles in binding, orientation, activation of H2O2 in the distal pocket of myoglobins. Although both protonated and deprotonated states of histidine could bind H2O2, the binding modes are different. When His64 is protonated, the hydrogen bond formation is between oxygen atoms of H2O2 with the ε-proton of histidine (Figure 4b), while when His 64 is deprotonated His64 plays a role of base and bind α-H of H2O2, which facilitates removing hydrogen peroxide’s α-proton to form the Mn-hydroperoxide intermediate (Figure 4c). In the case of 1·Mb(H64R), Arg remains an anion under all pH investigated here, and is a well-known H-donor, forming hydrogen-bonds with the β-O or both of O atoms of H2O2 as shown in Figure 4d and 6e. For the indirect contact with α-H of H2O2, Arg is less effective than the deprotonated His in deprotonation of H2O2 to form Mn-OOH. Thus, although Arg could also generate a strong hydrogen bond to H2O2, 1·Mb(H64R)’s reactivity in sulfoxidation is lower than 1·Mb’s at low pH. When pH increases, 1·Mb(H64R) exhibits higher reactivity than 1·Mb, because its hydrogen bond between β-O atom and distal ligand polarizes the O-O bond more effectively, promoting the cleavage of O-O bond to form the Mn-oxo active intermediate.
Figure 4.
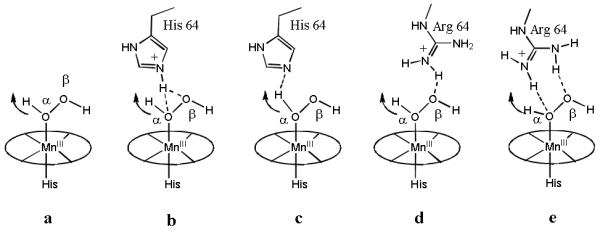
Distal ligand’s effect on the deprotonation of [MnSalen-H2O2] (a) along with the variant pHs. “b” and “c” show the possible H-bond formation between H2O2 (α-H) and His64 in acidic and basic sides of pKa (5.0) in 1·Mb, respectively. “d” shows the possible H-bond formation between H2O2 (β-O) and Arg64 in 1·Mb(H64R).
Conclusion
In conclusion, we have demonstrated the crucial roles of distal ligands in modulating pH dependent sulfoxidation and ABTS oxidation reactivity of MnSalen complex 1 in aqueous media when 1 is incorporated into apo-myoglobin. The modulation is achieved through hydrogen bonding interactions by the distal ligand to bind, orient and activate H2O2 through perturbation of H2O2 protonation/deprotonation equilibrium, which then resulted in different reaction intermediates at different pH’s and thus dramatic changes in the reactivity.
Experimental Section
Protein Expression and Biocatalysts Preparation
Introduction of mutations at the desired locations in the Mb scaffold was accomplished via quick change mutagenesis and the variant proteins were expressed and purified as reported previously.[68, 69] The heme was extracted and MnSalen was incorporated via covalent attachment as reported previously.[33, 36, 70] The successful incorporation of the desired site direct mutation was verified by DNA sequence analysis and ESI-MS.
Generation of the heme free apo variants was accomplished via a combination of Teale[33] and Fisher[37] methods. Addition of a solution of N,N’-bis(4-(2-methanesulfonylthioethoxy)salicylidene)-1,2-ethanediamino-manganese(III) bromide in dimethylsulfoxide to the apo Mb variant resulted in 100% conversion to the dually anchored MnSalen artificial biocatalysts for all mutants as assessed by ESI mass spectrometry (see supporting information).
Sulfoxidation Reaction
To examine the pH effect on the reaction, a mixed buffer system tunable to various pH values was used. The mixed buffer comprised to 50 mM each of sodium acetate, 2-(N-morpholino)ethanesulfonic acid (MES), 3-(N-morpholino) propanesulfonic acid (MOPS), tris(hydroxymethyl)aminomethane (TRIS), adjusted to the indicated pH.[38] The protein was exchanged into this buffer and the pH adjusted on aliquots by addition of small amounts of acid or base. A pH micro electrode was used to monitor the pH adjustment. To 180 μL of 0.144 mM catalyst in buffer was added 10 μL 100 mM of the thioether substrate in MeOH. The resulting mixture was equilibrated for 30 min while stirring at 4°C. The reaction was initiated by addition of 10 μL of 100 mM oxidant (H2O2 in this work) in buffer. Final reaction volume was 200 μL with final concentrations of 0.130 mM catalyst, 5 mM (39 equivalents) substrate and 5 mM (39 equivalents) oxidant. The reaction was allowed to proceed with stirring at 4°C for 10 minutes at which point 10 μL of 100 mM internal standard in MeOH were added. The reaction was quenched with addition of 10 βL of 1M NaS2O3 (a 10 fold excess of the oxidant added) and 250 μL of CH2Cl2. After vigorous mixing the entire solution (both aqueous and organic phases) was passed over a column of 1:1 Na2SO3: Na2SO4 to remove the water. The later method of quenching and sample work up was found to be more rapid than methods previously reported[33, 34]. The resulting organic extracts were then evaporated to just dryness under gentle argon flow. The residue was dissolved in 50 μL of ethyl acetate that had been dried over molecular sieves. The reaction rate is calculated by the yield of sulfoxide vs reaction time and enantioselectivity represented as e.e is obtained from gas chromatography analysis, performed on a Hewlett-Packard model 6890 equipped with a Hewlett-Packard 7683 auto injector using a 40 meter Astec Chiraldex G-TA capillary column.[71]
ABTS (2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) Oxidation
1·Mb(H64X) (X= H, F or R) or Mnsalen 1 (2.5 βM) was m[72]ixed with 250 βM 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) (ABTS) in 50 mM each of sodium acetate, MES, MOPS, TRIS at the indicated pH (5.0–9.0). The reaction was initiated by adding 250 βM (1 equiv) H2O2. The spectral changes were monitored at 734 nm for ABTS+· with the kinetic software (Chemstation, version A10.01) provided with the UV-vis spectrometer. The rates were calculated by fitting changes in absorbance versus time to a single-exponential equation of formation [eq 1].
| (1) |
Inhibition of Sulfoxidation by ABTS
To 180 μL of 0.144 mM catalyst in buffer was added 10 μL 100 mM of thioanisole in MeOH and a 0–0.5 equiv. of ABTS. The resulting mixture was equilibrated for 30 min while stirring at 4°C. The reaction was initiated by addition of 10 μL of 100 mM oxidant (H2O2 in this work) in buffer. The work-up procedure is the same as sulfoxidation described above.
Supplementary Material
Acknowledgements
This material is based upon work supported by the National Science Foundation (CHE-05-52008) and the National Institute of Health (GM062211).
References
- [1].Dawson J. Science. 1988;240:433–439. doi: 10.1126/science.3358128. [DOI] [PubMed] [Google Scholar]
- [2].Ortiz de Montellano PR. Cytochrome P-450: Structure, Mechanism, and Biochemistry. Plenum PressOrtiz de Montellano, Paul R; New York, N. Y.: 2005. p. 556. [Google Scholar]
- [3].Turano P, Lu Y. In: Iron in Heme and Related Proteins. Bertini I, Sigel H, Sigel A, editors. Marcel Dekker, Inc; New York, NY: 2001. pp. 269–356. [Google Scholar]
- [4].Reedy CJ, Gibney BR. Chem. Rev. 2004;104:617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- [5].Kennedy ML, Gibney BR. Curr. Opin. Struct. Biol. 2001;11:485–490. doi: 10.1016/s0959-440x(00)00237-2. [DOI] [PubMed] [Google Scholar]
- [6].Lu Y, Berry SM, Pfister TD. Chem. Rev. 2001;101:3047–3080. doi: 10.1021/cr0000574. [DOI] [PubMed] [Google Scholar]
- [7].Qi D, Tann C-M, Haring D, Distefano MD. Chem. Rev. 2001;101:3081–3111. doi: 10.1021/cr000059o. [DOI] [PubMed] [Google Scholar]
- [8].Lu Y. Curr. Opin. Chem. Biol. 2005;9:118–126. doi: 10.1016/j.cbpa.2005.02.017. [DOI] [PubMed] [Google Scholar]
- [9].Lu Y. Angew. Chem., Int. Ed. 2006;45:5588–5601. doi: 10.1002/anie.200600168. [DOI] [PubMed] [Google Scholar]
- [10].Letondor C, Ward TR. ChemBioChem. 2006;7:1845–1852. doi: 10.1002/cbic.200600264. [DOI] [PubMed] [Google Scholar]
- [11].Ueno T, Abe S, Yokoi N, Watanabe Y. Coord. Chem. Rev. 2007;251:2717–2731. [Google Scholar]
- [12].Goodman CM, Choi S, Shandler S, DeGrado WF. Nat. Chem. Biol. 2007;3:252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Steinreiber J, Ward TR. Coord. Chem. Rev. 2008;252:751–766. [Google Scholar]
- [14].Jacobsen EN, Zhang W, Muci AR, Ecker JR, Deng L. J. Am. Chem. Soc. 1991;113:7063–7064. [Google Scholar]
- [15].Irie R, Noda K, Ito Y, Matsumoto N, Katsuki T. Tetrahedron: Asymmetry. 1991;2:481–494. [Google Scholar]
- [16].Katsuki T. Coord. Chem. Rev. 1995;140:189–214. [Google Scholar]
- [17].Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis I-III. Vol. 1. 1999. p. 487. [Google Scholar]
- [18].Matsumoto K, Saito B, Katsuki T. Chem. Commun. (Cambridge, U. K.) 2007:3619–3627. doi: 10.1039/b701431g. [DOI] [PubMed] [Google Scholar]
- [19].Lane BS, Burgess K. Chem. Rev. 2003;103:2457–2473. doi: 10.1021/cr020471z. [DOI] [PubMed] [Google Scholar]
- [20].Bäckvall J-E. Modern Oxidation Methods. Wiley-VCH; 2004. p. 350. [Google Scholar]
- [21].Shitama H, Katsuki T. Chem.--Eur. J. 2007;13:4849–4858. doi: 10.1002/chem.200601420. [DOI] [PubMed] [Google Scholar]
- [22].Berkessel A, Frauenkron M, Schwenkreis T, Steinmetz A, Baum G, Fenske D. J. Mol. Catal. A: Chemical. 1996;113:321–342. [Google Scholar]
- [23].Shitama H, Katsuki T. Tetrahedron Lett. 2006;47:3203–3207. [Google Scholar]
- [24].Watanabe Y, Namba A, Umezawa N, Kawahata M, Yamaguchi K, Higuchi T. Chem. Commun. (Cambridge, U. K.) 2006:4958–4960. doi: 10.1039/b608846e. [DOI] [PubMed] [Google Scholar]
- [25].Liu SY, Soper JD, Yang JY, Rybak-Akimova EV, Nocera DG. Inorg. Chem. 2006;45:7572–7574. doi: 10.1021/ic0602087. [DOI] [PubMed] [Google Scholar]
- [26].Rosenthal J, Nocera DG. Acc. Chem. Res. 2007;40:543–553. doi: 10.1021/ar7000638. [DOI] [PubMed] [Google Scholar]
- [27].Yang JY, Bachmann J, Nocera DG. J. Org. Chem. 2006;71:8706–8714. doi: 10.1021/jo0613075. [DOI] [PubMed] [Google Scholar]
- [28].Yang JY, Nocera DG. J. Am. Chem. Soc. 2007;129:8192–8198. doi: 10.1021/ja070358w. [DOI] [PubMed] [Google Scholar]
- [29].Borovik AS. Acc. Chem. Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- [30].MacBeth GA, Young VG, Yang C, Kuczera K, Hendrich MP, Borovik AS. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]
- [31].Gray HB, Stiefel EI, Valentine JS, Bertini I. Biological Inorganic Chemistry: Structure and Reactivity. University Science Book; 2006. p. 739. [Google Scholar]
- [32].Dunford HB. Heme Peroxidases. Wiley-VCH; New York: 1999. [Google Scholar]
- [33].Carey JR, Ma SK, Pfister TD, Garner DK, Kim HK, Abramite JA, Wang Z, Guo Z, Lu Y. J. .Am. Chem. Soc. 2004;126:10812–10813. doi: 10.1021/ja046908x. [DOI] [PubMed] [Google Scholar]
- [34].Zhang J-L, Garner DK, Liang L, Chen Q, Lu Y. Chem. Commun. (Cambridge, U. K.) 2008:1665–1667. doi: 10.1039/b718915j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Anderson JL, Ding J, McCulla RD, Jenks WS, Armstrong DW. J.Chromatogr., A. 2002;946:197–208. doi: 10.1016/s0021-9673(01)01526-6. [DOI] [PubMed] [Google Scholar]
- [36].Springer BA, Sligar SG. Proc.Natl.Acad.Sci.U.S.A. 1987;84:8961–8965. doi: 10.1073/pnas.84.24.8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Teale FWJ. Biochim.Biophys.Acta. 1959;35:543. doi: 10.1016/0006-3002(59)90407-x. [DOI] [PubMed] [Google Scholar]
- [38].Fisher WR, Taniuchi H, Anfinsen CB. J. Biol. Chem. 1973;248:3188–3195. [PubMed] [Google Scholar]
- [39].Sharma PK, de Visser SP, Shaik S. J. Am. Chem. Soc. 2003;125:8698–8699. doi: 10.1021/ja035135u. [DOI] [PubMed] [Google Scholar]
- [40].Volz TJ, Rock DA, Jones JP. J. Am. Chem. Soc. 2002;124:9724–9725. doi: 10.1021/ja026699l. [DOI] [PubMed] [Google Scholar]
- [41].Ozaki SI, Roach MP, Matsui T, Watanabe Y. Acc. Chem. Res. 2001;34:818–825. doi: 10.1021/ar9502590. [DOI] [PubMed] [Google Scholar]
- [42].Nagano S, Tanaka M, Watanabe Y, Morishima I. Biochem. Biophys .Res. Commun. 1995;207:417–423. doi: 10.1006/bbrc.1995.1204. [DOI] [PubMed] [Google Scholar]
- [43].Rodriguez-Lopez JN, Lowe DJ, Hernandez-Ruiz J, Hiner ANP, Garcia-Canovas F, Thorneley RNF. J. Am. Chem. Soc. 2001;123:11838–11847. doi: 10.1021/ja011853+. [DOI] [PubMed] [Google Scholar]
- [44].Yang F, Phillips GN., Jr J.Mol.Biol. 1996;256:762–774. doi: 10.1006/jmbi.1996.0123. [DOI] [PubMed] [Google Scholar]
- [45].Bruice TC, Balasubramanian PN, Lee RW, Smith JRL. J.Am.Chem.Soc. 1988;110:7890–7892. [Google Scholar]
- [46].Arasasingham RD, Jeon S, Bruice TC. J.Am.Chem.Soc. 1992;114:2536–2544. [Google Scholar]
- [47].Bruice TC, Zipplies MF, Lee WA. Proc.Natl.Acad.Sci.U.S.A. 1986;83:4646–4649. doi: 10.1073/pnas.83.13.4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Maneiro M, Bermejo MR, Fernandez MI, Gomez-Forneas E, Gonzalez-Noya AM, Tyryshkin AM. New J.Chem. 2003;27:727–733. [Google Scholar]
- [49].Bermejo MR, Fernandez MI, Gonzalez-Noya AM, Maneiro M, Pedrido R, Rodriguez MJ, Garcia-Monteagudo JC, Donnadieu B. J. Inorg. Biochem. 2006;100:1470–1478. doi: 10.1016/j.jinorgbio.2006.04.012. [DOI] [PubMed] [Google Scholar]
- [50].Nakagaki PC, Calderwood TS, Bruice TC. Proc.Natl.Acad.Sci.U.S.A. 1988;85:5424–5428. doi: 10.1073/pnas.85.15.5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Nam W, Kim I, Lim MH, Choi HJ, Lee JS, Jang HG. Chem.--Eur. J. 2002;8:2067–2071. doi: 10.1002/1521-3765(20020503)8:9<2067::AID-CHEM2067>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- [52].Kuang H, Brown ML, Davies RR, Young EC, Distefano MD. J.Am.Chem.Soc. 1996;118:10702–10706. [Google Scholar]
- [53].Ohashi M, Koshiyama T, Ueno T, Yanase M, Fujii H, Watanabe Y. Angew. Chem., Int. Ed. 2003;42:1005. doi: 10.1002/anie.200390256. [DOI] [PubMed] [Google Scholar]
- [54].Ueno T, Koshiyama T, Ohashi M, Kondo K, Kono M, Suzuki A, Yamane T, Watanabe Y. J.Am.Chem.Soc. 2005;127:6556–6562. doi: 10.1021/ja045995q. [DOI] [PubMed] [Google Scholar]
- [55].Collot J, Gradinaru J, Humbert N, Skander M, Zocchi A, Ward TR. J.Am.Chem.Soc. 2003;125:9030–9031. doi: 10.1021/ja035545i. [DOI] [PubMed] [Google Scholar]
- [56].Collot J, Humbert N, Skander M, Klein G, Ward TR. J. Organomet. Chem. 2004;689:4868–4871. [Google Scholar]
- [57].Creus M, Ward TR. Org. Biomol. Chem. 2007;5:1835–1844. doi: 10.1039/b702068f. [DOI] [PubMed] [Google Scholar]
- [58].Reetz MT, Jiao N. Angew. Chem., Int. Ed. 2006;45:2416–2419. doi: 10.1002/anie.200504561. [DOI] [PubMed] [Google Scholar]
- [59].Reetz MT, Peyralans JJP, Maichele A, Fu Y, Maywald M. Chem. Commun. (Cambridge, U. K.) 2006:4318–4320. doi: 10.1039/b610461d. [DOI] [PubMed] [Google Scholar]
- [60].Mahammed A, Gross Z. J.Am.Chem.Soc. 2005;127:2883–2887. doi: 10.1021/ja045372c. [DOI] [PubMed] [Google Scholar]
- [61].Panicucci R, Bruice TC. J.Am.Chem.Soc. 1990;112:6063–6071. [Google Scholar]
- [62].Wolak M, van Eldik R. J.Am.Chem.Soc. 2005;127:13312–13315. doi: 10.1021/ja052855n. [DOI] [PubMed] [Google Scholar]
- [63].Wolak M, van Eldik R. Chem.--Eur. J. 2007;13:4873–4883. doi: 10.1002/chem.200601148. [DOI] [PubMed] [Google Scholar]
- [64].Lee YJ, Goh YM, Han SY, Kim C, Nam W. Chem. Lett. 1998:837–838. [Google Scholar]
- [65].Yang SJ, Lee HJ, Nam W. Bull. Korean Chem. Soc. 1998;19:276–278. [Google Scholar]
- [66].Venkataramanan NS, Kuppuraj G, Rajagopal S. Coord. Chem. Rev. 2005;249:1249–1268. [Google Scholar]
- [67].Kurahashi T, Kikuchi A, Tosha T, Shiro Y, Kitagawa T, Fujii H. Inorg. Chem. 2008;47:1674–1686. doi: 10.1021/ic702061y. [DOI] [PubMed] [Google Scholar]
- [68].Sigman JA, Kwok BC, Lu Y. J.Am.Chem.Soc. 2000;122:8192–8196. [Google Scholar]
- [69].Zhao X, Yeung N, Russell BS, Garner DK, Lu Y. J.Am.Chem.Soc. 2006;128:6766–6767. doi: 10.1021/ja058822p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Sigman JA, Kwok BC, Gengenbach A, Lu Y. J.Am.Chem.Soc. 1999;121:8949–8950. [Google Scholar]
- [71].Hwang HJ, Lu Y. Proc.Natl.Acad.Sci.U.S.A. 2004;101:12842–12847. doi: 10.1073/pnas.0403473101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Mac Leoda TCO, Barros VP, Faria AL, Schiavon MA, Yoshida IVP, Queiroz MEC, Assis MD. J. Mol. Catal. A: Chemical. 2007;273:259–264. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.