

Abstract
A puzzling aspect of rapamycin-based therapeutic strategies is the wide disparity in the doses needed to suppress mTOR under different circumstances. A recent study revealing mechanistically how rapamycin suppresses mTOR provides two explanations for the differential sensitivities to rapamycin. First, mTOR exists as two functionally distinct complexes (mTORC1 and mTORC2), and while rapamycin suppresses both, it does so at very different concentrations. Whereas mTORC1 is suppressed by concentrations of rapamycin in the low nM range, mTORC2 generally requires low μM concentrations. Second, the efficacy of rapamycin is dependent on the level of phosphatidic acid (PA), which is required for the assembly of both mTORC1 and mTORC2 complexes. Rapamycin interacts with mTOR in a manner that is competitive with PA. Therefore, elevated levels of PA, which is common in cancer cells, increases the level of rapamycin needed to suppress both mTORC1 and mTORC2. A practical outcome of the recent study is that if PA levels are suppressed, mTORC2 becomes sensitive to concentrations of rapamycin that can be achieved clinically. Since mTORC2 is likely more critical for survival signals in cancer cells, the recent findings suggest new strategies for enhancing the efficacy of rapamycin-based therapeutic approaches in cancer cells.
Keywords: cancer, mTOR, phosphatidic acid, phospholipase D, rapamycin
Introduction
mTOR, the mammalian target of rapamycin, is a therapeutic target for a wide variety proliferative disorders. Rapamycin and rapamycin derivatives have long been employed for immunosuppression and more recently as an anti-cancer treatment. It has been known for many years that different effects of rapamycin often require concentrations that vary by more than 1,000-fold.1,2 Rapamycin has been observed to suppress the phosphorylation of the mTOR substrate S6 kinase at concentrations as low as 1 nM.1,2 In contrast, low μM concentrations rapamycin were required to suppress the proliferation of several breast cancer cells.1–3 While it has been speculated the high dose effects of rapamycin are due to non-specific effects, rapamycin-resistant mTOR mutants were able to reverse the effects seen with the high rapamycin concentrations.3 If high doses of rapamycin were affecting targets other than mTOR, the rapamycin-resistant mutants would not reverse the effect—indicating that the high doses of rapamycin were in fact due to an effect on mTOR. A recent report revealing the mechanism by which rapamycin suppresses mTOR has shed light on two mechanisms that explain the wide variations in the concentrations of rapamycin needed to suppress mTOR.4 The findings in this report have important implications for how rapamycin-based therapeutic strategies should be employed—especially in cancer where rapamycin has been widely employed in clinical trials with mostly disappointing results.
Elevated Phospholipase D Activity Confers Rapamycin Resistance
Chen and colleagues reported a phosphatidic acid (PA) requirement for mTOR activity and that PA interacted with the FKBP12-rapamycin-binding (FRB) domain of mTOR.5 This group has more recently demonstrated that PLD1 is activated by the GTPase Rheb, which has been implicated in signals that activate mTOR.6,7 It was subsequently shown that elevated phospholipase D (PLD) activity, which generates PA from phospahtidylcholine, increased the concentration of rapamycin needed to suppress mTOR.1 Similarly, in skeletal muscle, mechanical stimulation led to elevated PLD activity and also increased the IC50 for rapamycin.8 These studies were all consistent with the hypothesis that PA interacts with the FRB domain in a manner that is competitive with rapamycin. The NMR structure of the mTOR FRB domain bound to PA was recently reported and revealed an interaction between Arg2109 in the FRB domain and the phosphate group on PA.9 Arg2109 is at the end of an α-helical section of the FRB domain that is adjacent to another α-helix. The groove in between the two α-helices forms a hydrophobic pocket where rapamycin binds mTOR.9 The glycerol backbone and proximal aliphatic components of PA are proposed to insert into this pocket. This structural study is consistent with a model proposed by Chen and colleagues where PA and rapamycin compete for the same site on mTOR.5 Thus, current evidence suggests that rapamycin inhibits mTOR by preventing the interaction between mTOR and PA.
Mechanism of Rapamycin Suppression of mTOR
While the studies above suggest that rapamycin works by interfering with an interaction between mTOR and PA, it does indicate how this interferes with mTOR activity. mTOR exists in two complexes, mTORC1 and mTORC2, which have different components—most significantly Raptor for mTORC1, and Rictor for mTORC2.10 Toschi et al.4 have now demonstrated that PA impacts on mTOR by facilitating the assembly of mTOR complexes. Suppression of PA production by PLD blocks the association between mTOR and Raptor and the association between mTOR and Rictor. Consistent with these findings, rapamycin, which competes with PA for binding mTOR,5 similarly blocked the association between mTOR and Raptor,11 and between mTOR and Rictor.12 These studies reveal a mechanism for the action of rapamycin on mTOR activity by preventing the interaction between PA and mTOR and thusly, preventing the PA-dependent assembly of mTOR complexes (Fig. 1). This model is consistent with the previous observation that PA did not appear to affect the kinase activity of mTOR directly.5 Significantly, there was a substantial difference in the stability of the two mTOR complexes with mTORC2 being far more stable than mTORC1. This differential stability explains the relative resistance of mTORC2 to rapamycin in that it takes higher concentrations of rapamycin to interfere with the more stable mTORC2 complex that rarely dissociates (Fig. 1). However, reducing the level of PA generated by PLD increased the sensitivity of mTORC2 to rapamycin, which as discussed below could have important clinical implications for targeting mTORC2 in cancer cells where mTORC2 is likely the more relevant target.
Figure 1.
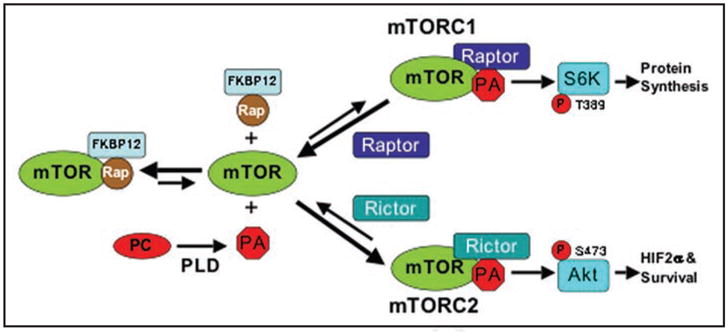
Model for the differential effects of rapamycin on mTORC1 and mTORC2. The stability of mTORC2 is much stronger than mTORC1, and therefore, there are fewer dissociations of mTORC2 to PA, Rictor and free mTOR. And it is the free mTOR that binds rapamycin-FKBP12.12 The ability of rapamycin-FKBP12 to suppress mTORC1 and mTORC2 would be dependent on the frequency that free mTOR becomes available to bind rapamycin-FKBP12. There would be far more dissociated mTOR generated from mTORC1 than from mTORC2 and therefore less rapamycin would be required to compete with PA for binding to the mTOR derived from mTORC1. In contrast, the rare dissociations of mTORC2 would require much more rapamycin-FKBP12 to compete with PA to capture the rare mTOR proteins derived from mTORC2. Reducing PA levels by suppressing the hydrolysis of phosphatidylcholine (PC) to PA shifts the equilibrium in favor of dissociation of the mTORC2 complex and therefore reduces the concentration of rapamycin-FKBP12 needed to bind to and suppress mTORC2. The larger arrows reflect relative differences in the direction for the equilibrium between free mTOR and the mTOR complexes. This model is clearly an over-simplification in that there are other components of the mTOR complexes that are not considered such as mSin1 and Protor that are part of mTORC2,8 and which could contribute to the greater stability of mTORC2.
Differential Doses of Rapamycin are Required to Suppress mTORC1 and mTORC2
There was a puzzling aspect to a previous study where it was demonstrated that elevated PLD activity increased the concentration of rapamycin needed to suppress mTOR.1 In this study, higher concentrations of rapamycin were needed to suppress cell proliferation and viability than were needed to suppress S6 kinase phosphorylation. Rapamycin suppressed the proliferation of MCF7 breast cancer cells with an IC50 of 20 nM, whereas rapamycin suppressed S6 kinase phosphorylation with an IC50 of 0.5 nM (1). In MDA-MB-231 breast cancer cells where for suppression of there are high levels of PLD activity, the IC50 proliferation was 10 μM, whereas suppression of S6 kinase phosphorylation in these cells was 20 nM.1 It is well established that S6 kinase is phosphorylated by mTORC1.10 The higher concentrations of rapamycin needed to suppress proliferation were consistent with an effect of rapamycin on mTORC2. Toschi et al.4 have now demonstrated similar differences for the effect of rapamycin on the phosphorylation of the mTORC2 substrate S6 kinase and the phosphorylation of Akt at the mTORC2 site at Ser473 in the renal cancer cell line 786-O. These data are consistent with a model whereby low concentrations of rapamycin (0.5–100 nM) target mTORC1 and higher concentrations (0.2–20 μM) target mTORC2. We have seen dose dependent effects up to 20 μM. Beyond this concentration of rapamycin, there does not seem to be an increased effect (our unpublished results). This could be due to an FKBP12 requirement in order for rapamycin to work effectiviely. Thus, the effect of rapamycin beyond a certain concentration would be due to rapamycin alone—not rapamycin-FKBP12. It was recently reported that the rapamycin analog CCI-779 could suppress protein synthesis independent of FKBP12 in the μM range,13 indicating that rapamycin can suppress mTORC1 at high concentrations in the absence of FKBP38. Based on the finding that rapamycin can suppress mTORC1 at μM concentrations, it is possible that rapamycin alone could also block mTORC2 at even greater concentration, but this would not likely have any practical consequences because these doses would likely be far beyond that tolerated in humans. However, while the lower concentrations of rapamycin apparently work well to suppress mTORC1 and block S6 kinase phosphorylation, it is the higher concentrations of rapamycin-FKBP12 that block proliferation and lead to apoptosis in some cancer cells. Thus, the ability to target mTORC2, which requires the higher concentrations of rapamycin, may prove to be more important than targeting mTORC1.
Implications and Directions
mTOR has been implicated in survival signals in cancer cells,10 and therefore, rapamycin has been widely and rationally employed in clinical trials for many cancers.14,15 In a recent trial for refractory pediatric solid tumors, plasma concentrations of the rapmycin analog everolimus (RAD-001) of 200 nM were achieved and well tolerated.16 At this concentration, this group observed a reduction of Akt phosphorylation at the mTORC2 site at Ser473 in peripheral-blood mononuclear cells. Prolonged treatment with 200 nM rapamycin was previously shown to block mTORC2 by preventing the formation of new mTORC2 complexes.12 While this concentration of rapamycin should easily suppress mTORC1, a problem not considered was that many, if not most, tumors have elevated PLD activity and therefore higher levels of PA.17 High levels of PA in the cancer cells would make these cells, specifically, more resistant to rapamycin than the peripheral-blood mononuclear cells that were used to determine whether rapamycin was effective.16 Thus, while treatment with rapamycin may look like it is working in normal tissues, the elevated PLD activity in the cancer cells will make these cells resistant to rapamycin.
The finding that that rapamycin suppresses mTORC2 as well as mTORC1 has additional therapeutic implications. There have been some successes with rapamycin-based therapeutic strategies in renal cancer. Of significance, renal cancer is commonly dependent on the constitutive expression of hypoxia-inducible factor 2α (HIF2α).18 And, it was recently demonstrated that HIF2α expression in the 786-O renal cancer cell line is dependent on mTORC2 and not on mTORC1.19 The 786-O cells are especially resistant to rapamycin,12 and have high levels of PLD activity.20 However, suppressing the level of PLD-generated PA in these cells made mTORC2 sensitive to 200 nM rapamycin. 4 Therefore a strategy for suppressing the level of PA in combination with rapamycin-based therapeutic strategies may offer new opportunities to target survival signals mediated by mTORC2, which may be especially important in renal cancers where HIF2α is critical. Alternatively, it may be possible to suppress mTORC2 by blocking the PLD activity that generates the PA necessary for the formation of mTORC2 complexes. Suppressing PLD-generated PA directly with primary alcohols,4 including ethanol, killed 786-O renal cancer cells when they were deprived of serum.20 The concentrations of primary alcohols needed to kill the 786-O cells was beyond that tolerated by humans; however, as little as 0.2% 1-butanol made the mTORC2 in 786-O cells sensitive to 200 nM rapamycin.4 While this concentration of ethanol would prohibit the operation of a motor vehicle, it could be tolerated in a clinical setting to enhance the efficacy of rapamycin. In the absence of an effective way to target PLD activity directly, targeting the signals that activate PLD might also be an attractive alternative. It was recently reported that the natural product honokiol suppresses PLD activity in several human cancer cell lines including 786-O cells.21 Honokiol induced apoptosis in several human cancer cell lines with elevated PLD activity and has been well tolerated in mouse xenograft studies.22 Thus, there may be options for therapeutic strategies that suppress PA levels such that mTORC2 can be targeted with rapamycin or rapamycin derivatives.
A key implication from the early studies showing that higher concentrations of rapamycin were required to suppress proliferation and induce apoptosis than were required to suppress S6 kinase1,3 clearly indicates that mTORC2 is the more critical target for strategies that desire to retard or kill cancer cells. These earlier studies were reinforced by studies with renal cancer cells where rapamycin suppressed HIF2α expression only when PA levels were suppressed19—again implicating mTORC2. And since HIF2α is required for tumorigenesis in the renal cancer cells,18 any significant effect of rapamycin-based strategies would have to take into account the need to suppress mTORC2. This would require either very high levels of rapamycin, or a combined strategy that suppresses PA levels to make mTORC2 sensitive to levels of rapamycin that are achievable in a clinical setting.
Another approach for circumventing the problems with rapamycin would be the development of drugs that inhibit mTOR by another mechanism. There have been several notable successes with small molecules that block the kinase activity—usually ATP analogues. However, ATP analogues have the disadvantage that they are competing with ATP and that means that there are a large number of side effects possible. In fact, most kinase inhibitors suppress more than one kinase—most famously Gleevec, which in addition to inhibiting the Bcr-Abl kinase found in chronic myelogenous leukemia, also inhibits Kit and the platelet-derived growth factor receptor.23 Thus, the development inhibitors that target kinase activity have their drawback as well. In this regard, two inhibitors of phosphatidylinositol-3-kinase (PI3K) that compete with ATP, LY294002 and wortmannin, also inhibit mTOR,24 which is a related protein kinase—a poorly known fact that has generated a large body of evidence that has mislead investigators investigating signals mediated by PI3K. Thus, while it would be useful to discover or develop other drugs that target mTOR, the high specificity of rapamycin for mTOR still offers strong promise for suppressing survival signals mediated by mTOR in a large number of human cancers. However, the problems associated with the doses of rapamycin needed to suppress mTORC1 and mTORC2 and the competition with PA needs to be acknowledged and dealt with.
Rapamycin at low doses has been shown to induce feedback mechanisms that activate an IGF1 receptor-dependent activation of mTORC2,25,26 and the MAP kinase signaling pathway.27 With regard to the activation of mTORC2, high doses of rapamycin where mTORC2 is suppressed would overcome this problem. As for the MAP kinase pathway, it has been proposed that a combination therapy that targets both mTOR and MAP kinase signals might be more effective.28 Thus, there may be additional ways to make rapamycin based strategies that involve both the suppression of PLD activity and other signaling pathways that impact on survival and cell cycle progression.
Summary
It is becoming apparent that mTOR signals promote cell cycle progression and suppress apoptosis in what is perhaps a majority of human cancers.10,14,17 Therefore, targeting mTOR has received much attention as an anti-cancer therapeutic strategy. The recent report by Toschi et al.4 reveals two major problems for rapamycin-based therapies. The first being that mTORC2 requires much higher concentrations of rapamycin than mTORC1, and that mTORC2 may be the more critical target for suppressing proliferation and promoting apoptosis.1,3,19 The second and perhaps more difficult problem is that most cancers have elevated PLD activity and therefore high levels of PA—resulting in rapamycin resistance. It is therefore critical that as clinical trials progress, the level of PLD activity in the target cancer be considered and perhaps a combination therapy be considered whereby both PA levels and mTOR are targeted.
Acknowledgments
We thank Darin Saloum and Amy Goldschmidt for comments on the manuscript. The work described here was supported by grants from the National Cancer Institute CA46677 and a SCORE grant from the National Institutes of Health GM60654. Research Centers in Minority Institutions award RR-03037 from the National Center for Research Resources of the National Institutes of Health, which supports infrastructure and instrumentation in the Biological Sciences Department at Hunter College, is also acknowledged.
References
- 1.Chen Y, Zheng Y, Foster DA. Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene. 2003;22:3937–42. doi: 10.1038/sj.onc.1206565. [DOI] [PubMed] [Google Scholar]
- 2.Yu K, Toral-Barza L, Discafani C, Zhang WG, Skotnicki J, Frost P, et al. mTOR, a novel target in breast cancer: the effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocr Relat Cancer. 2001;8:249–58. doi: 10.1677/erc.0.0080249. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Rodrik V, Foster DA. Alternative phospholipase D/mTOR survival signal in human breast cancer cells. Oncogene. 2005;24:672–9. doi: 10.1038/sj.onc.1208099. [DOI] [PubMed] [Google Scholar]
- 4.Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid—a competition with rapamycin. Mol Cell Biol. 2009;29:1411–1420. doi: 10.1128/MCB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–5. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 6.Sun Y, Fang Y, Yoon MS, Zhang C, Roccio M, Zwartkruis FJ, et al. Phospholipase D1 is an effector of Rheb in the mTOR pathway. Proc Natl Acad Sci USA. 2008;105:8286–91. doi: 10.1073/pnas.0712268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun Y, Chen J. mTOR signaling: PLD takes center stage. Cell Cycle. 2008;7:3118–23. doi: 10.4161/cc.7.20.6881. [DOI] [PubMed] [Google Scholar]
- 8.Hornberger TA, Chu WK, Mak YW, Hsiung JW, Huang SA, Chien S. The role of phospholipase D and phosphatidic acid in the mechanical activation of mTOR signaling in skeletal muscle. Proc Natl Acad Sci USA. 2006;103:4741–6. doi: 10.1073/pnas.0600678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veverka V, Crabbe T, Bird I, Lennie G, Muskett FW, Taylor RJ, et al. Structural characterization of the interaction of mTOR with phosphatidic acid and a novel class of inhibitor: compelling evidence for a central role of the FRB domain in small molecule-mediated regulation of mTOR. Oncogene. 2008;27:585–95. doi: 10.1038/sj.onc.1210693. [DOI] [PubMed] [Google Scholar]
- 10.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 11.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–75. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 12.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 13.Shor B, Zhang WG, Toral-Barza L, Lucas J, Abraham RT, Gibbons JJ, et al. A new pharmacologic action of CCI-779 involves FKBP12-independent inhibition of mTOR kinase activity and profound repression of global protein synthesis. Cancer Res. 2008;68:2934–43. doi: 10.1158/0008-5472.CAN-07-6487. [DOI] [PubMed] [Google Scholar]
- 14.Sawyers CL. Will mTOR inhibitors make it as cancer drugs? Cancer Cell. 2003;4:343–8. doi: 10.1016/s1535-6108(03)00275-7. [DOI] [PubMed] [Google Scholar]
- 15.Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene. 2006;25:6436–46. doi: 10.1038/sj.onc.1209886. [DOI] [PubMed] [Google Scholar]
- 16.Fouladi M, Laningham F, Wu J, O’Shaughnessy MA, Molina K, Broniscer A, et al. Phase I study of everolimus in pediatric patients with refractory solid tumors. J Clin Oncol. 2007;25:4806–12. doi: 10.1200/JCO.2007.11.4017. [DOI] [PubMed] [Google Scholar]
- 17.Foster DA. Phospholipase D survival signals as a therapeutic target in cancer. Curr Signal Transduct Ther. 2006;1:295–303. [Google Scholar]
- 18.Kondo K, Kim WY, Lechpammer M, Kaelin WG., Jr Inhibition of HIF2α is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003;1:83. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toschi A, Lee E, Gadir N, Ohh M, Foster DA. Differential dependence of HIF1α and HIF2α on mTORC1 and mTORC2. J Biol Chem. 2008;283:34495–9. doi: 10.1074/jbc.C800170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toschi A, Edelstein J, Rockwell P, Ohh M, Foster DA. HIFα expression in VHL deficient renal cell carcinoma cells is dependent on phospholipase D. Oncogene. 2008;27:2746–53. doi: 10.1038/sj.onc.1210927. [DOI] [PubMed] [Google Scholar]
- 21.Garcia A, Zheng Y, Zhao C, Toschi A, Fan J, Schreibman N, et al. Honokiol suppresses survival signals mediated by Ras-dependent phospholipase D activity in human cancer cells. Clinical Cancer Res. 2008;14:4267–74. doi: 10.1158/1078-0432.CCR-08-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bai X, Cerimele F, Ushio-Fukai M, Waqas M, Campbell PM, Govindarajan B, et al. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J Biol Chem. 2003;278:35501–7. doi: 10.1074/jbc.M302967200. [DOI] [PubMed] [Google Scholar]
- 23.Druker BJ. Imatinib as a paradigm of targeted therapies. Adv Cancer Res. 2004;91:1–30. doi: 10.1016/S0065-230X(04)91001-9. [DOI] [PubMed] [Google Scholar]
- 24.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 25.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–40. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- 27.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–74. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carracedo A, Baselga J, Pandolfi PP. Deconstructing feedback-signaling networks to improve anticancer therapy with mTORC1 inhibitors. Cell Cycle. 2008;7:3805–9. doi: 10.4161/cc.7.24.7244. [DOI] [PMC free article] [PubMed] [Google Scholar]