

Abstract
Although alterations in μ-opioid receptor signaling mediate excitatory effects of opiates in opioid tolerance, the molecular mechanism for the excitatory effect of acute low dose morphine, as it relates to μ-opioid receptor coupling, is presently unknown. A pronounced coupling of μ-opioid receptor to the α subunit of G inhibitory protein emerged in periaqueductal gray from mice systemically administered with morphine at a dose producing acute thermal hyperalgesia. This coupling was abolished in presence of the selective μ-opioid receptor receptor antagonist CTOP administered at the periaqueductal gray site, showing that the low dose morphine effect is triggered by μ-opioid receptor activated G inhibitory protein at supraspinal level. When Gβγ downstream signalling was blocked by intra-periaqueductal gray co-administration of M119, a compound that inhibits Gβγ dimer-dependent signaling, a complete prevention of low dose morphine induced acute thermal hyperalgesia was obtained. Phospholipase C β3, an enzyme necessary to morphine hyperalgesia, was revealed to be associated with Gβγ in periaqueductal gray. Although opioid administration induces a shift in μ-opioid receptor-G protein coupling from Gi to Gs after chronic administration, our data support that this condition is not realized in acute treatment providing evidence that a separate molecular mechanism underlies morphine induced acute excitatory effect.
Keywords: hyperalgesia, G protein, M119, morphine, mu opioid receptor, phospholipase C
INTRODUCTION
Classically, morphine activates G inhibitory (Gi) protein coupled to μ-opioid receptors (μOR) to inhibit adenylyl cyclase activity and decrease neuronal cAMP levels (Uhl et al., 1994). However, recent studies suggest that opioids can exert stimulatory effects either at doses well below those producing neuronal inhibition or after chronic exposure. In cultured dorsal root ganglion neurons, nanomolar concentrations of opioid agonists increase action potential duration, whereas micromolar concentrations produce the opposite effect (Chen et al., 1988; Shen and Crain, 1989). This dual action of opioids has been explained on the basis of a bimodal opioid receptor model. In this model, ultra-low doses of an agonist activate a Gs-coupled mode of the opioid receptor to activate adenylyl cyclase and increase neuronal excitability. In contrast, higher doses of opioids activate a Gi/Go-coupled mode of the receptor to inhibit adenylyl cyclase activity and reduce neuronal excitability. This bimodal model of morphine action has been invoked to explain paradoxical hyperalgesia after chronic opioid administration. Accordingly, the predominance of the Gs-coupled mode of the μOR during chronic treatment opposes the analgesic response eliciting tolerance associated-hyperalgesia through adenylyl cyclase activation (Crain and Shen, 1990; Crain and Shen, 1992). Alternatively, different authors showed that excitatory signaling of opioid receptors in chronic morphine treatment can also occur by Gβγ activation of adenylyl cyclase (Wang and Gintzler, 1997; Gintzler and Chakrabarti, 2001). Yet, extremely low doses of morphine can induce acute excitatory effects. Peripheral application of a very low dose of morphine induces a flexor response (Ono et al, 2002). The stimulation of sensory nerve endings induced by morphine through peripheral μOR activation and its downstream mechanisms has been ascribed to activation of phospholipase C (PLC) through substance P release from polymodal C fibers (Ono et al, 2002). Extremely low doses of systemic morphine can elicit acute hyperalgesia in animal model of pain such as tail flick (Crain and Shen, 2001; Esmaeili-Mahani et al, 2008), Freund’s adjuvant-induced arthritic rats (Kayser et al, 1987), and hot plate tests (Galeotti et al, 2006). A specific signaling pathway for morphine-induced acute thermal hyperalgesia has been shown via μOR activation of PLC/protein kinase C inositol-lipid signalling pathway (Galeotti et al, 2006). Among the large PLC family, the β3 isoform of PLC appeared to be implicated in inducing this excitatory effect whereas adenylyl cyclase levels remained unmodified in CNS after low dose morphine exposure (Galeotti et al, 2006; Esmaeili-Mahani et al, 2008). Although alterations in μOR signaling mediate excitatory effects of opiates in opioid tolerance and dependence, these excitatory effects have not been directly examined in an in vivo acute treatment paradigm with respect to μOR-G protein coupling. Thus, this work investigates μOR-G protein coupling as well as coupling between Gβγ and PLC at supraspinal level in low dose morphine-induced acute hyperalgesia.
METHODS and MATERIALS
Animals
Pathogen free sexually mature male albino Swiss Webster mice (Charles River) weighing 23–30g were used. Five-six mice were housed per cage. The animals were fed a standard laboratory diet and water ad libitum and kept at 23±1°C with a 12-h light/dark cycle. All experiments were carried out in accordance with the European Community Council Directive of November 24 1986 for experimental animal care. All efforts were made to minimize the number of animals used and their suffering. All the animals were previously habituated to the laboratory according to Abbot (Abbott, 1986).
Drug treatment
The following drugs were used: D-Phe–Cys–Tyr–D-Trp–Orn–Thr–Pen–Thr–NH2 (CTOP), morphine HCl (Sigma Chemicals St Louis, MO, USA); 2-(3,4,5-trihydroxy-6-oxoxanthen-9-5 yl)cyclohexane-1-carboxylic acid (M119). Drugs were administered in a volume of 0.2 µL per mouse by intracranial infusion, and 10mL/kg by intraperitoneal (i.p.) injection. Morphine and CTOP were dissolved in isotonic (NaCl 0.9%) saline solution immediately before use. M119 was initially solubilized in DMSO and subsequently diluted in distilled water. Morphine was administered i.p.; CTOP and M119 were administered intra-PAG immediately before morphine administration; saline was administered i.p. and DMSO vehicle intra-PAG.
Different groups of mice received: i.p. control saline or intra-PAG vehicle; 1µg/Kg morphine in presence or absence of CTOP (80ng/mouse) administered intra-PAG immediately before morphine;1µg/Kg morphine or saline in the presence or absence of intra-PAG M119 (5–40ng/mouse). CTOP and M119 were also administered alone. Other different groups received i.p. injection of saline, morphine (7mg/Kg) or twice daily morphine (10mg/Kg) for 7 days.
Surgery and Microinjection
Cannula implantation was performed as previously described (Carvalho-Netto et al, 2007). Briefly, mice were implanted with stainless steel guide cannula (32-gauge) under anesthesia. Stereotaxic coordinates (Paxinos and Franklin, 2001) for the PAG were, respectively, 4.2 mm posterior to bregma, 1.3 mm lateral to the midline, and 2.2 mm ventral to the skull surface, with the guide cannula angled 26° to the vertical. A dummy cannula was inserted into each guide-cannula immediately after each surgery to reduce the incidence of occlusion. Five to seven days after surgical recovery, solutions were injected into the PAG by microinjection unit which extended 1.0 mm beyond the tips of the guide cannula. Each microinjection unit was attached to a 5-µl Hamilton microsyringe via polyethylene tubing and administration was controlled by an infusion pump programmed to deliver a volume at rate of 0.1 µl over a period of 30 s. At the conclusion of the experiments 1% Evans blue dye was administered to mice according to the microinjection procedure described above for intra-PAG administration. A post-mortem histological control of the injection site was performed on cryostat sections of unfixed brains previously frozen. The data of any mice were excluded from statistical analysis if the cannula tip was outside the PAG or if the region had sustained extensive damage. The brains from mice which were further submitted to Western blot were extracted leaving the cannula implanted in the brain. The location of cannula inside PAG was observed under stereomicroscope (Leica MZ12.5, Leica, Solms, Germany). Only specimens from mice with cannula path inside the PAG were used for Western blot experiments.
Hot plate test
Mice were placed inside a stainless steel container, which was set thermostatically at 52.5±0.1 °C in a precision water-bath. Here we have used lower temperature in hot plate test (52°C instead of 54°C) to reveal possible subtle alterations that may occur in basal nociception. The licking latency was measured immediately prior i.p. morphine injection. Hot plate test started 15 min after morphine administration and licking latencies were measured at 15 min intervals for 60min after starting time. A 30s cut-off to prevent tissue damage was used. The endpoint for the licking response was the first paw lick of the front paw. Increased nociception was seen as shorter latencies to the responses evaluated. The test was performed in a blind fashion. Mice pretreated with drugs administered as previously described, were evaluated for licking latency basal value.
Rota-rod and hole-board test
For both tests, animals were pretreated with saline (i.p.), intra-PAG vehicle or M119(40ng/mouse), 1µg/Kg morphine (i.p.) in presence or absence of M119 and submitted to rota-rod and hole-board (Ghelardini et al, 2008). Twelve mice per group were tested. The rota-rod test apparatus consists of a base platform and a rotating rod (3×30cm) placed at a height of 15 cm from the base and divided into five equal sections by six disks. Up to five mice were tested simultaneously on the apparatus, with a rod-rotation speed of 16 r.p.m. The integrity of motor coordination was assessed on the basis of the number of falls from the rod in 30 s, according to Vaught et al. (1985). Performance time was measured before and 15, 30 and 45 min after the administration of the investigated compounds. The hole board test consisted of a 40 cm square plane with 16 flush-mounted cylindrical holes (3 cm diameter) distributed four by four in an equidistant, grid-like manner. Mice were placed on the center of the board one by one and allowed to move about freely for a period of 10 min each. Two electric eyes, crossing the plane from midpoint to midpoint of opposite sides, thus dividing the plane into four equal quadrants, automatically signaled the movement of the animal (counts in 5 min) on the surface of the plane (spontaneous motility). Miniature photoelectric cells, in each of the 16 holes, recorded (counts in 5 min) the exploration of the holes (inspection activity) by the mice. Mice pretreated with 1µg/Kg morphine (i.p.) in presence or absence of intra-PAG CTOP (80ng/mouse) were previously submitted to both tests (Ghelardini et al, 2008).
μOR-G protein coupling assay
Mice used for these experiments were sacrified 15 min after 1µg/Kg or 7mg/Kg morphine administration at which time maximum thermal hyperalgesic/analgesic effect is obtained (Galeotti et al, 2006) or seven days after repeated morphine administration at the doses previously described. μOR-G protein coupling assay data after acute and chronic high-dose morphine administration are largely known (Sánchez-Blázquez et al, 2001; Askari et al, 2008; Wang et al, 2005) but were used to support the method utilized in this study. The animals were anesthetized with CO2, cervically dislocated, decapitated and the brain dissected, put immediately in liquid nitrogen and then stored at −80°C. PAG brain area from control and treated mice was dissected on a cold plate. Enriched synaptic membranes were prepared from PAG of mice from each treatment group as described by Gray and Whittaker (Gray and Whittaker, 1962). Protein concentration was determined according to Lowry (Lowry et al, 1951). The association of G protein coupled receptors with G proteins was investigated using coimmunoprecipitation procedure as previously described (Wang et al, 2005). Briefly, PAG membranes were incubated with Krebs–Ringer and then solubilized in immunoprecipitation buffer (25mM HEPES, pH 7.5, 200mM NaCl, 2mM MgCl2, 1mM EDTA, 0.2% 2-mercaptoethanol, 50µg/ml leupeptin, 25µg/ml pepstatin A, 0.01 U/ml soybean trypsin inhibitor, 0.04mM phenylmethylsulfonyl fluoride) containing 0.5% digitonin, 0.2% sodium cholate and 0.5% NP-40 emulsifying agent with end-over-end rotation for 60 min at 4°C and further centrifugated at 50,000xg. The supernatant was divided for separate passage through immunoaffinity columns containing immobilized antibodies to Gαs, Gαi, Gαo, Gα11, Gαq or Gβ proteins. Anti-G protein antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were covalently cross-linked to protein-G-conjugated resin in Seize-X protein G immunoprecipitation kit (Pierce-ENDOGEN, Rockford, IL, USA) according to the manufacturer instructions. G protein complexes in solubilized brain lysates were isolated by immunoprecipitation in which 200µg solubilized brain membrane extracts were incubated with immobilized anti-G-protein G-resin at 4°C overnight. After centrifugation and three washes with phosphate-buffered saline (pH 7.2) at 4°C, the G protein complexes were eluted with 190 µl of antigen elution buffer (pH 2.8) and immediately neutralized by adding 20µl of 1.5M Tris buffer (pH 8.8). The neutralized G complexes were combined with 180µl of 2× PAGE sample preparation buffer (62.5mM Tris–HCl, pH 6.8; 20% glycerol, 4% SDS; 10% 2-mercaptoethanol, 0.1% Bromophenol Blue), boiled for 5 min and then submitted to Western blotting using a specific antibody directed against the amino-terminal region of the μOR for Gα and PLCβ1–4 for Gβ (Santa Cruz Biotechnology, Santa Cruz, CA, USA). 18% and 4–15% or 4–20%Tris-HCl gels were used respectively for G-protein-μOR complex and specificity assay of antibodies or Gβ-PLCβ1–4 coimmunoprecipitation experiments. The specificity of the anti-Gα, anti-Gβ and PLCβ1–4 antibodies was determined by Western blotting using 100µl of mouse whole brain homogenate with or without antigen peptide (25µg) pre-absorption for 30 min. The specificity of anti-μOR antibody was assayed on brain tissue from knockout brain mouse (GR21−/−; a generous gift of Dr Brigitte Kieffer, Institut de Génétique et de Biologie Moléculaire et Cellulaire, Département Neurobiologie et génétique, Illkirch, France). A highly acidic (pH 2.8) or neutral elution buffer was used to elute antigens from the Gα immuno-complexes in order to establish if the experimental procedure yielded μOR with an approximate molecular weight of respectively 53 and 67 kD (Chen et al, 1995; Wang et al, 2005).
Western blot analysis
Western blot was performed as previously described in detail (Pan et al, 1995). In summary, immunoprecipitates (from 1µg/µl protein lysate) of Gαi, Gαo, Gαq, Gα11, Gαs and Gβ protein from PAG of morphine, CTOP, M119, saline and vehicle pretreated mice were solubilized in SDS buffer and separated on polyacrylamide gels (1.5mm). Proteins were transferred to nitrocellulose (1.5 h at 190 mA) and the membranes were blocked in PBS containing 3% BSA for 1h before addition of anti-μOR or anti-Gβ antibody at 1:500 dilution. The blots were stripped and reprobed with antibodies against various G proteins using the antisera against Gαi, Gαo, Gαq, Gα11, Gαs and Gβ protein as probes at 1:1,000 dilution. The blotting was visualized using a chemiluminescence detection system (Super Signal West Fento, Pierce Biotechnology Inc., Rockford, IL, USA) and quantified with the Versa Doc 1000 Imaging System (Bio-Rad Laboratories, Hercules, CA, USA). Three independent experiments were done at the same protein concentration for each experimental condition. Specific bands were quantitated by densitometric scanning.
Immune complex PLC activity measurement
PLC enzyme activity in anti-Gβ immunoprecipitates was measured as described in Allan (Allan et al, 1997). Immune complexes for Gβ (30µL) were assayed for 15 min at 37°C in the presence of 35 mM sodium phosphate, pH 6.8, 70 mM KCl, 0.8 mM EGTA, 0.8 mM CaCl2, 0.20mM [3H]phosphatidylinositol 4,5-bisphosphate (8Ci/nmol, Perkin-Helmer Massachusetts, USA), and 2.86 mM (0.18%, v/v) Triton X-100 in a final volume of 50 µL. Enzyme activity was quantitated as the release of [3H] Ins(1,4,5)P3 measured by liquid scintillation spectroscopy. Isozyme-specific activity was calculated by subtracting background [3H] Ins(1,4,5)P3 (release present in no antibody control samples) from the activity measured in immune complexes. Data were calculated as nanomoles Ins(1,4,5)P3 product formed per minute per milligram protein present in the fraction from which the enzyme was immunoprecipitated. All conditions were run in triplicate.
Statistical analysis
All experimental results are given as mean ±S.E.M. Analysis of variance followed by Fisher protected least significant difference (PLSD) procedure for post-hoc comparisons was used to verify significance between two means for data obtained from co-immunoprecipitation of μOR-G protein or Gβγ-PLCβ complexes after hyperalgesic/analgesic dose and chronic morphine administration. Unpaired repeated measures ANOVA followed by Scheffè test paired post hoc was applied to hot plate and rota-rod test results. Data were analysed with the Statview Software for the Macintosh (1992).
RESULTS
Co-immunoprecipitation of μOR-G protein complexes after hyperalgesic or analgesic ose and chronic morphine administration
Under non-denaturing conditions that keep μOR-G protein complexes intact, specific G proteins (Gi, Go, Gq, G11 and Gs) together with their coupled receptors were immunoprecipitated with selective anti-Gα antibodies from solubilized synaptic membranes obtained from PAG of the different treatment groups under both basal and morphine-stimulated conditions. μOR coupling to each of the G protein subtypes in the different treatment groups is shown in Western blots of the Gα immunoprecipitates probed with the anti-μOR-specific antibody (fig.1a). In PAG, μOR coupled exclusively to Go in saline and CTOP treated mice, and to both Go and Gi in low dose morphine-treated mice. In PAG of mice treated with morphine in presence of CTOP, coupling to Gi was markedly decreased from that in the morphine-treated animals (fig.1a). Morphine i.p. administration at analgesic dose dramatically increased μOR-Gi coupling (fig.1a). Go and Gs coupling by μOR weakly increased with respect to control; however these were not statistically significant (fig.1a). Chronic morphine decreased Gi coupling by μOR. A pronounced coupling to Gs appeared in PAG from mice submitted to chronic morphine treatment (fig.1a). Densitometric scanning of immunoprecipitated Gα proteins was used as loading control. The relative amount of each of these proteins was not significantly different with respect to saline in all experimental conditions (fig.1a). Pre-absorption with 25µg of their respective antigen peptides drastically abolished the detection of targeted G proteins by Western blotting in mouse brain homogenate (fig.1b). No immunoreactive band was observable in brain extracts from μOR−/− mice in presence of anti-μOR antibody (fig.1b). Elution with the highly acetic antigen elution buffer yelded μOR with an apparent molecular weight of 53kD (fig.1b). Elution with a neutral pH predominantly yielded μOR with an apparent molecular weight of 67kD (fig.1b).
Figure 1.
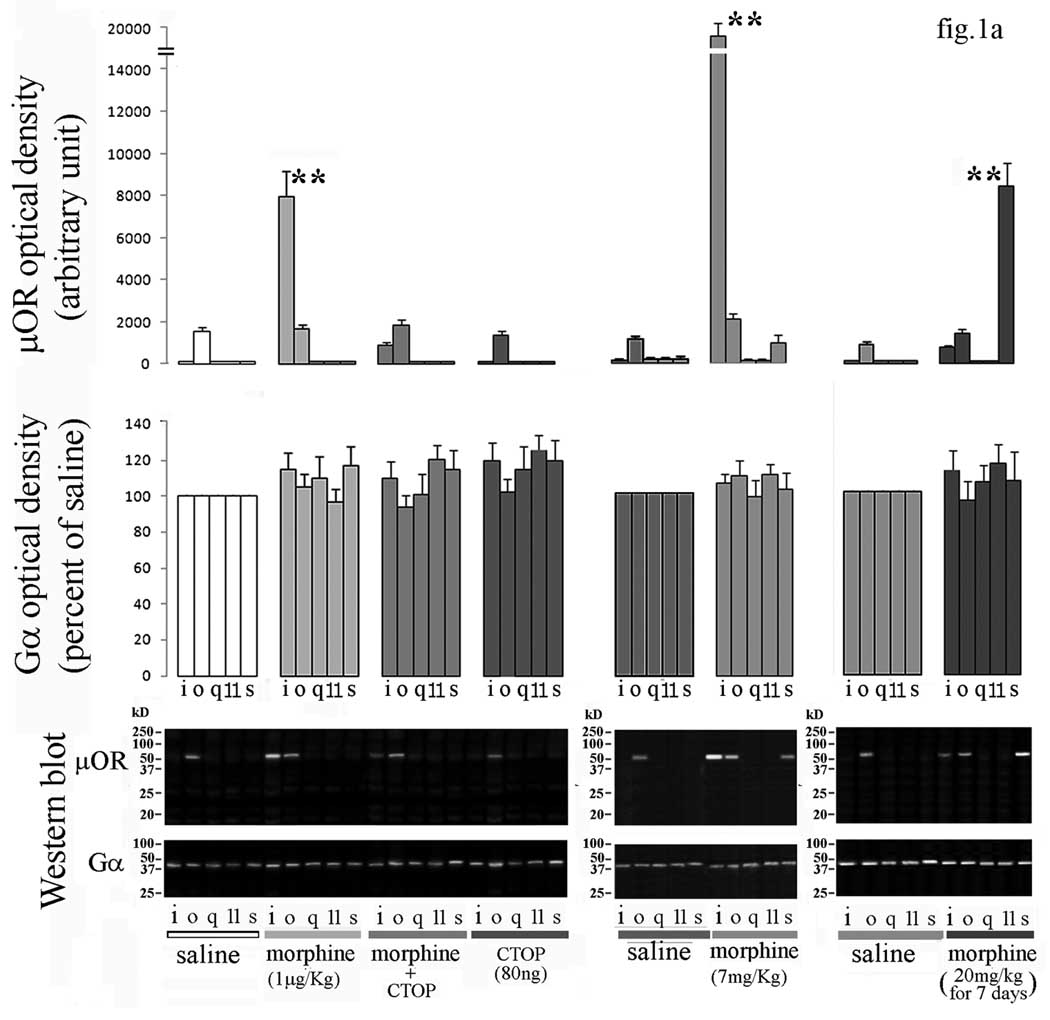

(a) G protein-μOR coupling in PAG from mice treated with morphine- A representative Western blots of the presence of μOR protein in immunoprecipitates of i, o, q, 11, and s subunits of Gα protein in PAG from mice submitted to acute or chronic morphine treatments in presence or absence of CTOP is shown at the bottom of the figure. The blots stripped and reprobed with antibodies against the above G proteins, are shown for the different treatments. Band optical density for Gα protein subunits after different treatments is represented at the middle of the figure. Each bar represents the mean density of each Gα subunit obtained from three independent experiments and expressed as percent of corresponding saline. Statistics were applied to the raw data prior to transformation to percent. Mean value of μOR density detected in immunoprecipitates of considered Gα subunits is represented at the top of the figure. Single values were normalized to surrounding background and expressed as arbitrary units. **=α<0.01 significant difference in comparison with corresponding saline value. Vertical lines represent S.E.M.
(b) Specificity assay of anti-G protein and anti-μOR antibodies- Homogenates from whole mouse brain were submitted to Western blotting after incubation with the proper antibody in presence or absence of respective immunogen sequence in excess as shown in the figure. Western blotting was performed on extracts of brain tissue from normal (+/+) and μOR knockout (−/−) mice with anti-μOR antibody. Molecular weight of μOR immunoprecipitated by the anti-μOR antibody after elution with an acidic (lane 1) versus a neutral (lane 2) buffer is reported in the last column. —=not preabsorbed; ----=preabsorbed.
Effect of M119 compound in morphine induced hyperalgesia
Morphine hyperalgesia induced by i.p. 1 µg/Kg dose in the mouse hot plate test was completely prevented by intra-PAG pre-treatment with M119 at 5–40 ng/ mouse (fig.2a). This effect appeared to be dose dependent. The M119 compound, when administered alone at the same doses, did not induce any significant change in licking latency (fig.2b).
Figure 2.

Effect of M119 coadministration on morphine induced hyperalgesic response-Licking latencies were measured before and after (15, 30, 45 and 60 min) i.p. morphine administration (1µg/Kg) in presence or absence of M119 intra-PAG coadministration at different doses (a). Licking latencies measured after M119 intra-PAG administration to mice are represented in (b). Each value represents the mean ± S.E.M. of licking latencies. Vertical bars represent S.E.M. ; **=α<0.01 significant difference in comparison with corresponding basal value. MF= morphine. The number of animals used for each experimental condition is reported at the top of control bars.
Rota-rod test and spontaneous activity meter
The spontaneous motility as well as inspection activity of mice were unmodified by pre-treatment with drugs in comparison with control group (fig. 3a). The number of falls from the rotating rod evaluated before and 15, 30 and 45 min after the beginning of the rota-rod test showed the lack of any impairment in motor coordination of mice submitted to the above treatments in comparison with control group (fig. 3b).
Figure 3.

(a) Morphine and M119 administration do not induce any significant difference with respect to saline or vehicle on inspection activity and spontaneous mobility evaluated in the mouse hole board test.
(b) Lack of effect of morphine and M119 administration on motor coordination evaluated in the mouse rota rod test. Vertical lines represent S.E.M.
Gβγ co-immunoprecipitation with PLCβ
In order to test the hypothesis that Gβγ associates with PLCβ in mouse PAG, anti-Gβ immune complexes were isolated from PAG of mice 15 min following 1 µg/Kg morphine administration and probed for PLCβ1–4 immunoreactivity. We found that Gβ immunoprecipitates with PLCβ3 whereas immunoreactivity associated with anti-PLCβ1, β2 and β4 was not different from the saline background (fig.4a). Pre-absorption with 25µg of their respective antigen peptides drastically abolished the detection of PLCβ1–4 and Gβ by Western blotting in mouse brain homogenate (fig.4b). Anti-Gβ immune complexes were isolated from PAG of mice 15 min following low dose morphine administration and assayed for associated PLC activity. These results demonstrate that Gβ associated with PLCβ3 in low dose morphine administration and this was catalytically active (fig.5). PLC activity could not be detected in anti-Gβ immune complexes isolated from PAG of mice previously administered with low morphine dose in presence of M119 or CTOP at the higher effective doses (fig.5).
Figure 4.


(a) Representative blot of co-immunoprecipitation of Gβ proteins with PLCβ1–4 in PAG from low dose morphine administered mice is shown in figure. The blots stripped and reprobed with Gβ antibody are shown at the bottom of the figure. Band optical density for Gβ protein subunits after saline or morphine is represented in the middle of the figure. Each bar represents the mean density of Gβ subunit obtained from three independent experiments and expressed as percent of corresponding saline. Statistics was applied to the raw data prior to transformation to percent. Mean value of PLCβ subunit density detected in immunoprecipitates of Gβ is represented at the top of the figure. Single values were normalized to surrounding background and expressed as arbitrary units. **=α<0.01 significant difference in comparison with corresponding saline value. Vertical lines represent S.E.M.
(b) Specificity assay of anti-PLCβ1–4 and anti-Gβ protein antibodies- Western blot of whole mouse brain tissue preincubated with the proper antibody in presence or absence of respective immunogen sequence in excess are shown in figure.
Figure 5.

PLC activity present in anti-Gβ immunoprecipitates from PAG of mice previously administered with morphine (1 µg/Kg) in presence or absence of CTOP (80ng) or M119(40ng). Each bar represents PLC activity obtained from three independent experiments under different treatment conditions. Immune complex PLC activity was measured and expressed as pmol Ins(1,4,5)P3 product formed/min/mg protein. Vertical lines represent S.E.M.;**=α<0.01.
Histology
Histology confirmed that 95% mice used in the experiments had cannula placement within the PAG. Only data from mice in which the cannula was correctly placed within the PAG were considered.
DISCUSSION
μOR-G protein coupling after morphine administration to mice
Although changes in μOR signaling have been previously demonstrated in the excitatory effects of opiates throughout opioid tolerance and dependence, alterations in μOR signaling that mediate excitatory effects in an in vivo treatment acute paradigm have not been yet studied. To determine whether alterations in μOR-G protein coupling occur at μOR-expressing CNS tissue after morphine low dose systemic administration, we isolated the PAG from mice receiving systemic morphine at a dose which produces acute thermal hyperalgesia (Crain and Shen, 2001, Galeotti et al, 2006; Esmaeili-Mahani et al, 2008). PAG is an important site of opioid analgesia (Yaksh et al, 1976) and tolerance to the antinociceptive effects of both systemic and locally applied morphine (Siuciak and Advocat 1987; Lane et al, 2004). In mouse PAG, μOR activation hyperpolarizes most neurons via activation of G-protein-gated inwardly rectifying potassium channels (Vaughan et al, 2003), and also inhibits GABA release from nerve terminals (Hack et al, 2003), consistent with the disinhibitory mechanisms proposed to be responsible for PAG-mediated opioid analgesia. Otherwise, opioid effect may extend beyond inhibition in the PAG. In brain slices, the excitatory action of NMDA on PAG neurons is potentiated by a μOR agonist at low nanomolar concentration (Kow et al, 2002). Exposure of PAG neurons to selective μOR antagonist CTOP completely reversed the morphine low dose induced acute hyperalgesic response showing that μOR activation in PAG is necessary to the excitatory response (Ghelardini et al, 2008). Under non-denaturing conditions that keep μOR-G protein complexes intact, specific Gα proteins together with their coupled receptors were immunoprecipitated with selective anti-Gα antibodies from solubilized synaptic membranes obtained from the PAG under both basal and morphine-stimulated conditions. In our experiments, μOR coupled exclusively to Go in PAG from control mice. When mice were administered with systemic low morphine dose, a pronounced coupling of μOR to α subunit of Gi protein emerged in PAG area; this coupling was markedly decreased from that in the morphine-treated animals in presence of selective μOR antagonist CTOP administered at the PAG site showing that low dose morphine effect is triggered by μOR activated Gi protein at supraspinal level. Morphine administration to mice at analgesic dose induced a dramatic increase in μOR coupling to α subunit of Gi and a small, nonsignificant increase in μOR coupling to Go and Gs with respect to control, in agreement with previous results (Sánchez-Blázquez et al, 2001; Askari et al, 2008). When mice were submitted to chronic morphine administration, the coupling of μOR to Gs protein emerged in PAG whereas Gi protein-μOR coupling dramatically decreased as previously obtained by different authors (Crain and Shen, 1998; Wang et al, 2005). In the classic G protein signaling cascade, Gβγ subunits are released as a complex from the α subunit after activation of an associated receptor at the cell surface (Smrcka, 2008). Bonacci et al. identified a compound, M119, that bound to Gβγ subunit and selectively inhibited Gβγ downstream signaling from the Gβγ subunit (Bonacci et al, 2006; Mathews et al, 2008). In our experiments, we used a hot plate test for evaluating thermal nociception in mice. Intra-PAG co-administration of M119 at the higher dose caused complete prevention of low dose morphine-induced acute thermal hyperalgesia demonstrating that thermal hyperalgesia is dependent on Gβγ at the supraspinal level.
Gβγ-PLCβ interaction in low dose morphine induced hyperalgesia
Opioids are known to generate different neurochemical adaptations as inhibition of adenylyl cyclase (Childers, 1991), activation of inwardly rectifying K+ channels (North et al, 1987), and inhibition of voltage-activated calcium channels (Schroeder et al, 1991). Additionally, there is growing evidence that modulation of phosphoinositide-specific PLC, and consequently altered formation of inositol 1,4,5trisphosphate/diacylglycerol/Ca2+ signaling, plays a key role in mediating excitatory opioid effects (Smith et al, 1999). PLC is one of only two signaling effector enzymes, the other being adenylyl cyclase, whose activity is directly modulated by opioids and several physiological studies have implicated PLC-linked pathways in a diverse range of opioid-modulated events as in vivo pain regulation (Galeotti et al, 2006; Bonacci et al, 2006; Esmaeili-Mahani et al, 2008, Mathews et al, 2008) and opioid tolerance (Smith et al, 1999). Among the large PLC family, the PLCβ3 isoform was localized in regions of the brain important for nociceptive transmission, including PAG, and was activated in this region after morphine administration to mice at a hyperalgesic dose (Bianchi et al, 2009). Galeotti et al (2006) recently demonstrated that low dose morphine induced hyperalgesic effect is mediated by the activation of PLCβ3. Studies in vitro showed that G protein mediation, pertussis toxin-insensitive (via the α subunit of Gq) or sensitive (via the Gβγ subunit of Gi/Go) is a prerequisite for receptor activation of PLCβ isoforms (Wu et al, 1992; Smrcka and Sternweis, 1993). In order to test the hypothesis that Gβγ associates with PLCβ subfamily in PAG after low morphine dose administration, anti Gβ-immune-complex were isolated from PAG area and probed for anti-PLCβ immunoreactivity after saline or low dose morphine administration. Our results show that, at 15 min after morphine administration which corresponds at the time of the maximum hyperalgesic effect, PLCβ3 was associated with Gβ and appeared catalytically active; this effect was reversed by supraspinal administration of the Gβγ blocker, M119. When μOR was blocked at supraspinal site by CTOP, phospholipase C activity remained unmodified showing that Gβγ-dependent PLC activation is dependent on μOR.
Conclusion
Collectively, our data support that hyperalgesic doses of morphine exposure induces a Gβγ-dependent stimulation of PLCβ3 triggered from Gi through its coupling to μOR at supraspinal level. Previous findings demonstrated that the α subunit of the Gi triggered the analgesic effect at higher morphine doses (Sanchez-Blazquez et al, 2001), suggesting a bimodal opioid receptor induced activation of the same protein. A similar pattern was previously proved in smooth muscle where the βγ subunit of the Gi protein has been showed to activate PLC signaling whereas the α subunit inhibits adenylyl cyclase activity (Murthy and Makhlouf, 1996). The β3 isoform of PLC appears to be implicated also in analgesic morphine effects. When an acute analgesic morphine dose is administered to PLCβ3 knockout or downregulated mice, a potentiation in the analgesic effect was obtained by different authors (Xie et al,1999; Galeotti et al, 2006; Bonacci et al, 2006). Gβγ blocker M119 coadministration resulted in a dramatic increase in acute thermal analgesic potency of morphine (Mathews et al, 2008). This led to the proposal that μOR activation by morphine might trigger separate stimulatory and inhibitory effects linked to different effector systems. Assuming that the overall pharmacological effect of morphine is equal to the sum of these two processes, the high dose morphine inhibitory analgesic system would not be opposed by the excitatory nociceptive component when the PLCβ3-dependent stimulatory pathway is switched off. It has been reported that systemic injection of morphine produces a rebound hyperalgesia after the antinociceptive effect was terminated (Ossipov et al, 2005). The residual low concentration of morphine that remains after cessation of its administration might elicit the stimulatory withdrawal hyperalgesia which was shown both after single or chronic morphine administration when nociceptive facilitatory systems are not overwhelmed by the opponent antinociceptive inhibitory systems. The blockage of withdrawal hyperalgesia by naloxone confirmed the involvement of opioid receptors in the phenomen supporting that withdrawal hyperalgesia is a direct effect of a residual, low concentration of morphine. A large dose of intraoperative opioids before the onset of noxious stimuli, that is preemptive analgesia, could lead to the development of abnormal pain sensitivity postoperatively (Guignard et al, 2000). The patients treated intraoperatively with opioids reported more postoperative pain than the matched non-opioid control subjects (Guignard et al, 2000). Prolonged hyperalgesia following short term morphine exposure, when the opiate concentration is expected to be as low as after low dose administration, may explain why the results of clinical studies of preemptive analgesia as a means of reducing postsurgical pain have been sometimes disappointing. Increasing the opioid dose to restore the analgesic effect may not always be the answer to morphine decreased efficacy. Knowledge of the molecular mechanism of possible excitatory action of opiates may allow the development of new chemical approaches that can prevent these effects as well as change the way in which these drugs are used clinically. Selectively inhibiting excitatory signaling represents a novel approach to target opioid-induced abnormal pain sensitivity confirming the potential use of M119 in clinical management of pain.
Acknowledgements
This work was supported by grants from MiUR and NIH GM 081772.
Abbreviations
- (Gi)
G inhibitory
- (μOR)
μ-opioid receptors
- (PLC)
phospholipase C
- (CTOP)
D-Phe–Cys–Tyr–D-Trp–Orn–Thr–Pen–Thr–NH2
- (morphine)
morphine HCl
- (M119)
2-(3,4,5-trihydroxy-6-oxoxanthen-9-yl)cyclohexane-1-carboxylic acid
- (PAG)
periaqueductal gray
REFERENCES
- Abbott FV, Franklin KB, Connell B. The stress of a novel environment reduces formalin pain: possible role of serotonin. Eur J Pharmacol. 1986;126:141–144. doi: 10.1016/0014-2999(86)90750-8. [DOI] [PubMed] [Google Scholar]
- Allan AM, Weeber EJ, Savage DD, Caldwell KK. Effects of prenatal ethanol exposure on phospholipase C-beta 1 and phospholipase A2 in hippocampus and medial frontal cortex of adult rat offspring. Alcohol Clin Exp Res. 1997;21:1534–1541. [PubMed] [Google Scholar]
- Askari N, Mahboudi F, Haeri-Rohani A, Kazemi B, Sarrami R, Edalat R, Ahmadiani A. Effects of single administration of morphine on G-protein mRNA level in the presence and absence of inflammation in the rat spinal cord. Scand J Immunol. 2008;67:47–52. doi: 10.1111/j.1365-3083.2007.02043.x. [DOI] [PubMed] [Google Scholar]
- Bianchi E, Lehmann D, Vivoli E, Norcini M, Ghelardini C. Involvement of PLC-{beta}3 in the effect of morphine on memory retrieval in passive avoidance task. J Psychopharmacol. 2009 Mar 12; doi: 10.1177/0269881108102013. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Bonacci TM, Mathews JL, Yuan C, Lehmann DM, Malik S, Wu D, Font JL, Bidlack JM, Smrcka AV. Differential targeting of Gbetagamma-subunit signaling with small molecules. Science. 2006;312:443–446. doi: 10.1126/science.1120378. [DOI] [PubMed] [Google Scholar]
- Carvalho-Netto EF, Litvin Y, Nunes-de-Souza RL, Blanchard DC, Blanchard RJ. Effects of intra-PAG infusion of ovine CRF on defensive behaviors in Swiss-Webster mice. Behav Brain Res. 2007;176:222–229. doi: 10.1016/j.bbr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GG, Chalazonitis A, Shen KF, Crain SM. Inhibitor of cyclic AMP-dependent protein kinase blocks opioid-induced prolongation of the action potential of mouse sensory ganglion neurons in dissociated cell cultures. Brain Res. 1988;462:372–377. doi: 10.1016/0006-8993(88)90568-9. [DOI] [PubMed] [Google Scholar]
- Chen C, Xue JC, Zhu J, Chen YW, Kunapuli S, Kim de Riel J, Yu L, Liu-Chen LY. Characterization of irreversible binding of beta-funaltrexamine to the cloned rat mu opioid receptor. J Biol Chem. 1995;270:17866–17870. doi: 10.1074/jbc.270.30.17866. [DOI] [PubMed] [Google Scholar]
- Childers SR. Opioid receptor-coupled second messenger systems. Life Sci. 1991;48:1991–2003. doi: 10.1016/0024-3205(91)90154-4. [DOI] [PubMed] [Google Scholar]
- Crain SM, Shen KF. Opioids can evoke direct receptor-mediated excitatory effects on sensory neurons. Trends Pharmacol Sci. 1990;11:77–81. doi: 10.1016/0165-6147(90)90322-y. [DOI] [PubMed] [Google Scholar]
- Crain SM, Shen KF. After chronic opioid exposure sensory neurons become supersensitive to the excitatory effects of opioid agonists and antagonists as occurs after acute elevation of GM1 ganglioside. Brain Res. 1992;575:13–24. doi: 10.1016/0006-8993(92)90417-8. [DOI] [PubMed] [Google Scholar]
- Crain SM, Shen KF. Modulation of opioid analgesia, tolerance and dependence by Gs-coupled, GM1 ganglioside-regulated opioid receptor functions. Trends Pharmacol Sci. 1998;19:358–365. doi: 10.1016/s0165-6147(98)01241-3. [DOI] [PubMed] [Google Scholar]
- Crain SM, Shen KF. Acute thermal hyperalgesia elicited by low-dose morphine in normal mice is blocked by ultra-low-dose naltrexone, unmasking potent opioid analgesia. Brain Res. 2001;888:75–82. doi: 10.1016/s0006-8993(00)03010-9. [DOI] [PubMed] [Google Scholar]
- Esmaeili-Mahani S, Shimokawa N, Javan M, Maghsoudi N, Motamedi F, Koibuchi N, Ahmadiani A. Low-dose morphine induces hyperalgesia through activation of G alphas, protein kinase C, and L-type Ca 2+ channels in rats. J Neurosci Res. 2008;86:471–479. doi: 10.1002/jnr.21489. [DOI] [PubMed] [Google Scholar]
- Galeotti N, Stefano GB, Guarna M, Bianchi E, Ghelardini C. Signaling pathway of morphine induced acute thermal hyperalgesia in mice. Pain. 2006;123:294–305. doi: 10.1016/j.pain.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Ghelardini C, Galeotti N, Vivoli E, Norcini M, Zhu W, Stefano GB, Guarna M, Bianchi E. Molecular interaction in the mouse PAG between NMDA and opioid receptors in morphine-induced acute thermal nociception. J Neurochem. 2008;105:91–100. doi: 10.1111/j.1471-4159.2007.05117.x. [DOI] [PubMed] [Google Scholar]
- Gintzler AR, Chakrabarti S. Opioid tolerance and the emergence of new opioid receptor-coupled signaling. Mol Neurobiol. 2001;21:21–33. doi: 10.1385/MN:21:1-2:021. [DOI] [PubMed] [Google Scholar]
- Gray EG, Whittaker VP. The isolation of nerve endings from brain: an electron-microscopic study of cell fragments derived by homogenization and centrifugation. J Anat. 1962;96:79–88. [PMC free article] [PubMed] [Google Scholar]
- Guignard B, Bossard AE, Coste C, Sessler DI, Lebrault C, Alfonsi P, Fletcher D, Chauvin M. Acute opioid tolerance: intraoperative remifentanil increases postoperative pain and morphine requirement. Anesthesiology. 2000;93:409–417. doi: 10.1097/00000542-200008000-00019. [DOI] [PubMed] [Google Scholar]
- Hack SP, Vaughan CW, Christie MJ. Modulation of GABA release during morphine withdrawal in midbrain neurons in vitro. Neuropharmacology. 2003;45:575–584. doi: 10.1016/s0028-3908(03)00205-3. [DOI] [PubMed] [Google Scholar]
- Kayser V, Besson JM, Guilbaud G. Paradoxical hyperalgesic effect of exceedingly low doses of systemic morphine in an animal model of persistent pain (Freund's adjuvant-induced arthritic rats) Brain Res. 1987;414:155–157. doi: 10.1016/0006-8993(87)91338-2. [DOI] [PubMed] [Google Scholar]
- Kow LM, Commons KG, Ogawa S, Pfaff DW. Potentiation of the excitatory action of NMDA in ventrolateral periaqueductal gray by the mu-opioid receptor agonist, DAMGO. Brain Res. 2002;935:87–102. doi: 10.1016/s0006-8993(02)02532-5. [DOI] [PubMed] [Google Scholar]
- Lane DA, Tortorici V, Morgan MM. Behavioral and electrophysiological evidence for tolerance to continuous morphine administration into the ventrolateral periaqueductal gray. Neuroscience. 2004;125:63–69. doi: 10.1016/j.neuroscience.2004.01.023. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Mathews JL, Smrcka AV, Bidlack JM. A novel Gbetagamma-subunit inhibitor selectively modulates mu-opioid-dependent antinociception and attenuates acute morphine-induced antinociceptive tolerance and dependence. J Neurosci. 2008;28:12183–12189. doi: 10.1523/JNEUROSCI.2326-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy KS, Makhlouf GM. Opioid mu, delta, and kappa receptor-induced activation of phospholipase C-beta 3 and inhibition of adenylyl cyclase is mediated by Gi2 and G(o) in smooth muscle. Mol Pharmacol. 1996;50:870–877. [PubMed] [Google Scholar]
- North RA, Williams JT, Surprenant A, Christie MJ. Mu and delta receptors belong to a family of receptors that are coupled to potassium channels. Proc Natl Acad Sci U S A. 1987;84:5487–5491. doi: 10.1073/pnas.84.15.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono T, Inoue M, Rashid MH, Sumikawa K, Ueda H. Stimulation of peripheral nociceptor endings by low dose morphine and its signaling mechanism. Neurochem Int. 2002;41:399–407. doi: 10.1016/s0197-0186(02)00047-5. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Porreca F. Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers. 2005;80:319–324. doi: 10.1002/bip.20254. [DOI] [PubMed] [Google Scholar]
- Pan YX, Cheng J, Xu J, Rossi G, Jacobson E, Ryan-Moro J, Brooks AI, Dean GE, Standifer KM, Pasternak GW. Cloning and functional characterization through antisense mapping of a kappa 3-related opioid receptor. Mol Pharmacol. 1995;47:1180–1188. [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic Press; 2001. [Google Scholar]
- Sánchez-Blázquez P, Gómez-Serranillos P, Garzón J. Agonists determine the pattern of G-protein activation in mu-opioid receptor-mediated supraspinal analgesia. Brain Res Bull. 2001;54:229–235. doi: 10.1016/s0361-9230(00)00448-2. [DOI] [PubMed] [Google Scholar]
- Schroeder JE, Fischbach PS, Zheng D, McCleskey EW. Activation of mu opioid receptors inhibits transient high- and low-threshold Ca2+ currents, but spares a sustained current. Neuron. 1991;6:13–20. doi: 10.1016/0896-6273(91)90117-i. [DOI] [PubMed] [Google Scholar]
- Shen KF, Crain SM. Dual opioid modulation of the action potential duration of mouse dorsal root ganglion neurons in culture. Brain Res. 1989;491:227–242. doi: 10.1016/0006-8993(89)90059-0. [DOI] [PubMed] [Google Scholar]
- Siuciak JA, Advokat C. Tolerance to morphine microinjections in the periaqueductal gray (PAG) induces tolerance to systemic, but not intrathecal morphine. Brain Res. 1987;424:311–319. doi: 10.1016/0006-8993(87)91476-4. [DOI] [PubMed] [Google Scholar]
- Smith FL, Lohmann AB, Dewey WL. Involvement of phospholipid signal transduction pathways in morphine tolerance in mice. Br J Pharmacol. 1999;128:220–226. doi: 10.1038/sj.bjp.0702771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrcka AV, Sternweis PC. Regulation of purified subtypes of phosphatidylinositol-specific phospholipase C beta by G protein alpha and beta gamma subunits. J Biol Chem. 1993;268:9667–9674. [PubMed] [Google Scholar]
- Smrcka AV. G protein βγ subunits: central mediators of G protein-coupled receptor signaling. Cell Mol Life Sci. 2008;65:2191–2214. doi: 10.1007/s00018-008-8006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl GR, Childers S, Pasternak G. An opiate-receptor gene family reunion. Trends Neurosci. 1994;17:89–93. doi: 10.1016/0166-2236(94)90110-4. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, Bagley EE, Drew GM, Schuller A, Pintar JE, Hack SP, Christie MJ. Cellular actions of opioids on periaqueductal grey neurons from C57B16/J mice and mutant mice lacking MOR-1. Br. J. Pharmacol. 2003;139:362–367. doi: 10.1038/sj.bjp.0705261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaught JL, Pelley K, Costa LG, Setler P, Enna SJ. A comparison of the antinociceptive responses to the GABA-receptor agonists THIP and baclofen. Neuropharmacology. 1985;24:211–216. doi: 10.1016/0028-3908(85)90076-0. [DOI] [PubMed] [Google Scholar]
- Wang L, Gintzler AR. Altered mu-opiate receptor-G protein signal transduction following chronic morphine exposure. J Neurochem. 1997;68:248–254. doi: 10.1046/j.1471-4159.1997.68010248.x. [DOI] [PubMed] [Google Scholar]
- Wang HY, Friedman E, Olmstead MC, Burns LH. Ultra-low-dose naloxone suppresses opioid tolerance, dependence and associated changes in mu opioid receptor–G protein coupling and Gβγ signaling. Neuroscience. 2005;135:247–261. doi: 10.1016/j.neuroscience.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Wu DQ, Lee CH, Rhee SG, Simon MI. Activation of phospholipase C by the alpha subunits of the Gq and G11 proteins in transfected Cos-7 cells. J Biol Chem. 1992;267:1811–1817. [PubMed] [Google Scholar]
- Xie W, Samoriski GM, McLaughlin JP, Romoser VA, Smrcka A, Hinkle PM, Bidlack JM, Gross RA, Jiang H, Wu D. Genetic alteration of phospholipase C beta3 expression modulates behavioral and cellular responses to mu opioids. Proc Natl Acad Sci U S A. 1999;96:10385–10390. doi: 10.1073/pnas.96.18.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Yeung JC, Rudy TA. Systematic examination in the rat of brain sites sensitive to the direct application of morphine: observation of differential effects within the periaqueductal grey. Brain Res. 1976;114:83–103. doi: 10.1016/0006-8993(76)91009-x. [DOI] [PubMed] [Google Scholar]