

Abstract
We describe a collection of standardized image processing protocols for electron microscopy single-particle analysis using the XMIPP software package. These protocols allow performing the entire processing workflow starting from digitized micrographs up to the final refinement and evaluation of 3D models. A particular emphasis has been placed on the treatment of structurally heterogeneous data through maximum-likelihood refinements and self-organizing maps as well as the generation of initial 3D models for such data sets through random conical tilt reconstruction methods. All protocols presented have been implemented as stand-alone, executable python scripts, for which a dedicated graphical user interface has been developed. Thereby, they may provide novice users with a convenient tool to quickly obtain useful results with minimum efforts in learning about the details of this comprehensive package. Examples of applications are presented for a negative stain random conical tilt data set on the hexameric helicase G40P and for a structurally heterogeneous data set on 70S Escherichia coli ribosomes embedded in vitrified ice.
INTRODUCTION
Modern electron microscopes allow visualization of biological matter up to sub-nanometer resolutions1,2. In the single-particle approach, many images of assumedly identical copies of macromolecular complexes are combined to obtain 2D or 3D structural information. As the electron dose on the sample needs to be limited to avoid radiation damage, electron microscopy images typically present a very low signal-to-noise ratio, which is often between 0.3 and 0.1. These high levels of noise require robust image processing approaches. Consequently, the development of powerful image processing algorithms has gone hand in hand with the increasing success of the single-particle approach3,4. Besides, as many (often tens of thousands) experimental images need to be combined to eliminate the noise, electron microscopy image processing is computationally demanding, and the advances in this field have been tightly coupled to the availability of increasing computer power. Partly because of this dynamic character of the image processing field, to date, the experimentalist may choose between a large number of alternative data processing workflows, which have been implemented in many distinct computer programs. Besides a range of programs that allow one to perform specific data processing tasks, various packages for generalized single-particle analysis exist. A non-exhaustive list of these general packages includes SPIDER5, EMAN6, BSOFT7, IMAGIC8, SPARX9 and XMIPP10 (also see ref. 11 for an exhaustive review). In the following paragraphs, we will focus on the XMIPP package.
The software XMIPP was introduced over a decade ago12, and more recently, it was rewritten in an object-oriented approach, yielding a hierarchical organization of documented classes and programs10. The modular design of its functionalities aims to provide a convenient platform for rapid testing of new algorithms by software developers, although a large number of stand-alone programs offer a broad functionality to the user. For the more experienced user, the diversity of stand-alone XMIPP programs is a positive aspect, providing a high level of flexibility in devising optimal data processing strategies. Furthermore, the modularity of these programs allows changing from or to alternative packages at almost any point in the data processing workflow. For the inexperienced user, however, the multitude of programs may present a relatively steep learning curve. To overcome this problem, here we present an additional layer to the hierarchical structure of XMIPP, consisting of a collection of standardized protocols for XMIPP’s most popular functionalities. These protocols represent a major standardization of numerous existing scripts and recipes that circulated among the XMIPP user community, thereby representing years of experience by multiple researchers.
The diversity of the applications described in this article illustrates the comprehensiveness of the XMIPP package. Its usefulness is reflected by the numerous structural studies that employ XMIPP in their image processing analysis: examples that were published last year include studies on the 26S proteasome13, eukaryotic prefoldin14, bacterial photosynthetic core complex15, DNA transporter trwB16, bacteriophage T7 procapsids17, primosomal factor DnaB18, DNA repair complexes Ku70-Ku80 and DNA-PKcs19 and the cytoplasmic Syk kinase20. Naturally, the broad functionality of XMIPP by no means makes alternative packages redundant. For example, in XMIPP, the only way to generate 3D reconstructions ab initio from the data is by random conical tilt reconstruction, while other packages, like EMAN, SPIDER or IMAGIC, contain complementary functionalities to perform ab initio reconstructions using common lines in the so-called angular reconstitution approach21. Furthermore, XMIPP does not contain any automated particle selection algorithm, such that particle selection is restricted to the rather time-consuming process of manual picking. Finally, refinement of a 3D reference map may be performed in many different ways, and the optimal choice will often depend on the data at hand. In general, as each of the available software packages in the field has its own strengths, the more experienced experimentalist typically combines distinct functionalities from a range of different packages in designing his or her optimal data processing strategy.
To aid the user in designing his or her optimal data processing strategy, Figure 1 illustrates how each of the standardized protocols fits into a generalized processing workflow. Starting from digitized micrographs in TIFF format, the user may preprocess these (convert them to RAW format, downsample and estimate contrast transfer function parameters), launch a graphical program for interactive particle picking (both for single micrographs and tilt pairs) and preprocess the individual particles (windowing, background normalization and CTF phase correction). This procedure results in a list of extracted particles in SPIDER single-file format. Alternatively, the user may pick and extract his or her particles in an alternative package (e.g., EMAN or SPIDER), convert them to single-file SPIDER format and enter the XMIPP workflow at this stage. The user may then opt for 2D or 3D analysis of the data. In some 2D cases, where point symmetry plays a key role in distinguishing particles, image classification may be performed in a highly efficient manner based on differences in rotational symmetry only. If this is the case, the user may employ a specific protocol for classification of rotational spectra22 based on quantitative self-organizing maps (kerdenSOM)23. In the more general case, reference-free 2D image alignment and classification may be obtained through 2D maximum-likelihood multireference refinement (also known asML2D classification)24. Then, for the most challenging 2D cases, the classes thus obtained may be further subdivided using kerden-SOM classification. For 3D analysis, an initial reference map may be obtained from tilted micrograph pairs. Alignment and classification of the untilted images would typically be obtained through ML2D classification, and subsequent random conical tilt reconstruction may yield 3D maps for each of the classes obtained25. Once an initial 3D reference is available, projection data sets coming from a mixture of different conformations may be classified using 3D maximum likelihood multireference refinement (ML3D classification). This technique may yield structurally homogeneous subsets, even without knowing beforehand what kind of structural variability is present in the data26,27. Subsequent refinement of structurally homogeneous sets may then be performed by either one of two distinct refinement protocols. The first one is based on standard projection matching28,29, complemented with a realignment of each of the classes at every iteration. This realignment step follows the spirit of procedures implemented in EMAN6 and may serve to eliminate bias from an incorrect starting model. The second refinement protocol is based on a combination of multiresolution wavelet refinement30 and continuous angular assignments31 and allows correction of CTF amplitudes through iterative data refinement32. Both refinement protocols implement 3D reconstruction using the algebraic reconstruction technique (ART) with blobs, which may provide specific advantages over alternative reconstruction methods for small and very noisy data sets or uneven angular distributions33,34. The preferred choice between both refinement protocols depends on the case at hand. The projection matching protocol is relatively fast if intermediate resolutions are to be obtained and may start from worse reference maps than the multiresolution protocol. The latter, however, is not limited by the use of a discretely sampled reference projection library and may thus yield more accurate results and converge faster in higher-resolution refinements.
Figure 1.

A generalized XMIPP processing workflow. The protocols developed may be divided in data preprocessing, 2D processing and 3D processing (light-blue boxes). Computationally demanding protocols that allow multiprocessor computing (via message-passing interface, MPI) are shown in orange; all other protocols are shown in yellow.
The protocols described below have been implemented as stand-alone executable python scripts, each with a header that defines its corresponding parameters. The user may modify these parameters and execute the script either through a graphical user interface that was specifically developed for this purpose (see documentation at http://xmipp.cnb.csic.es/twiki/bin/view/Xmipp) or from the command line by editing the header in a standard text editor. As the output of one script can be used as the input for another, these scripts may guide the user through the generalized image processing workflow. Furthermore, they provide functionalities for standardized logging and visualization of the results. Although these scripts are primarily aimed to aid the inexperienced user, more expert users may also benefit from the standardized working environment that they provide, facilitating the exchange of intermediate results with alternative packages or other users, and improving repeatability of the experiments through the comprehensive logging functionalities.
In the following paragraphs, we will enumerate the distinct XMIPP commands that constitute the standardized protocols. Although, in principle, the user may subsequently execute each of these instructions from the command line, we recommend using the implemented python scripts instead. For the sake of clarity, non-XMIPP commands contained in these scripts are not shown here. These commands are a convenient but not essential part of the scripts, as they take care of the standardized directory structures and the logging functionalities. The XMIPP instructions alone should suffice to understand the essence of each protocol, and thus to provide it with an adequate set of parameters. (Also note that detailed help pages for each XMIPP program are available at http://xmipp.cnb.csic.es/twiki/bin/view/Xmipp/ListOfPrograms.) Thereby, a novice user should be able to obtain useful results with only minimal efforts in learning about the XMIPP package.
MATERIALS
EQUIPMENT SETUP
A computer with a Unix/Linux operating system, or preferably a multi-processor cluster for the more computationally intensive protocols (i.e., ML2D or ML3D classification, and projection matching or multi-resolution refinement)
XMIPP installation (version 2.0 or later; available from http://xmipp.cnb.csic.es)
XMIPP installation depends on the external Qt3, TIFF and message-passing interface libraries, which may be obtained from http://trolltech.com, http://www.libtiff.org, and http://www-unix.mcs.anl.gov/mpi/mpich1/, respectively. Most Unix/Linux operating systems provide these libraries in their standard distributions.
Python installation (version 2.3 or newer; available from http://www.python.org)
For the graphical-user interface: python-tk installation (installed by default with most python distributions, and also available from http://www.python.org)
Digitized micrographs in TIFF (Spider, MRC or RAW) format, or alternatively, single particle images in single-file SPIDER format5
PROCEDURE
Preprocessing
-
1| Preprocess micrographs (Steps 1–4). Convert each of the digitized micrographs (e.g., mic0001.tif) from TIFF to RAW format:
xmipp_convert_tiff2raw mic0001.tif mic0001.raw
? TROUBLESHOOTING
-
2| If the micrograph was digitized with a pixel size smaller than that needed in the processing of the individual particles, then perform a downsampling step (e.g., decrease the pixel size by a factor of 2):
xmipp_micrograph_downsample –i mic0001.raw –o down2_mic0001.raw
\–output_bits 32 –Xstep 2 –kernel rectangle 2 2
-
3| Estimate the contrast transfer function (CTF, i.e., the Fourier transform of the microscope’s point spread function) of the micrograph.
xmipp_ctf_estimate_from_micrograph –i mic00001_input.param
where parameter file mic00001_input.param describes the experiment (voltage in kV, spherical aberration in mm and sampling rate in Å pixel−1) and contains parameters for the CTF estimation algorithm: minimum and maximum frequency to be used in pixel−1 (i.e., normalized frequencies between 0 and 0.5), whether to use periodogram averaging and/or averaging over the entire micrograph and the size in pixels of the pieces of the micrograph used. Its format is:
image= down2_mic0001.raw
voltage= 200
spherical_aberration= 2.26
sampling_rate= 2.8
min_freq= 0.05
max_freq= 0.35
periodogram= yes
micrograph_averaging= yes
N_horizontal= 512
? TROUBLESHOOTING
-
4| Visualize the CTF of each micrograph and discard those micrographs that are of insufficient quality for further processing.
xmipp_show –img mic00001_Periodogramavg.ctfmodel_halfplane
▴ CRITICAL STEP Micrograph selection may strongly affect the outcome of all subsequent data processing steps. The CTFs of good micrographs typically have multiple concentric rings, extending from the image center toward its edges. Bad micrographs may lack any rings or only have very few rings that hardly extend from the image center. Other reasons to discard micrographs may be the presence of strongly asymmetric rings (astigmatism) or rings that fade in a particular direction (drift). Some examples that illustrate the micrograph selection based on their CTFs are shown in Figure 2a–c, and for further details, refer to ref. 35.
-
5| Manual particle selection. For each of the selected micrographs in Step 4, manually select the individual particles. This option can be carried out using option A or B, depending on whether it concerns single micrographs or tilt pairs, respectively.
-
Single micrographs
-
Use the following command:
-
xmipp_micrograph_mark –i down2_mic0001.raw
This command will launch an overview window of the entire micrograph and a zoom window showing part of it. Identify particles by clicking the left-mouse button in the zoom window and move the magnified area in the zoom window by clicking the left-mouse button in the overview window. Save the identified particle coordinates by typing CTRL-S or by clicking ‘Save Coordinates’ in the ‘File Menu’.
-
-
-
Tilt pairs
-
Use the following command:
xmipp_micrograph_mark –i down2_mic0001.raw –tilted mic0002.raw
This command will launch one overview window containing both micrographs and two zoom windows (one showing part of the untilted and one showing part of the tilted micrograph). Identify particle pairs by clicking the left-mouse button in the zoom windows of the untilted and the tilted micrographs. Save the particle coordinates in both windows (see option A), and save the calculated transformation between the two micrographs by clicking ‘Save angles’ in the ‘File Menu’ of the untilted micrograph. Note that once the correct transformation has been found, the program accurately predicts the position of the tilted particle after identifying the untilted one.
▴ CRITICAL STEP Note that some micrographs that were not discarded in Step 4 may be discarded at this stage, as a too high particle density or a strong heterogeneity in the particle population may hinder the selection process. The process of micrograph selection based on the particle density and heterogeneity is illustrated in Figure 2d–f.
▴ CRITICAL STEP For many specimens, in particular for particles embedded in vitrified ice and of relatively small size (100–500 kDa), particle selection has proved to be a major obstacle for single particle analysis. For such cases, a human expert typically performs much better than automated procedures. (For a recent overview of the state of the art in automated particle selection, the reader is referred to a dedicated special issue in the Journal of Structural Biology36.) Thereby, a careful interactive selection of the particles, although being time consuming and dependent on the experience of the user, will generally facilitate subsequent image processing steps and will typically yield better results (e.g., in terms of the resolution of the 3D reconstructions obtained in Steps 30–36 or 37–47).
-
-
-
6| Preprocess particles (Steps 6–9). For each of the selected micrographs in Step 4, extract the particles as individually windowed images from the micrographs. This option can be performed using option A or B, depending whether it concerns single micrographs or tilt pairs, respectively. Both options will generate images of 64 × 64 pixels ( -Xdim). For an improved CTF phase correction (Step 8), extract particles with twice the final desired size (e.g., 128 × 128).
-
Single micrographs
-
Use the following command:
xmipp_micrograph_scissor -i down2_mic0001.raw –pos \
down2_mic0001.raw.Common.pos -root mic0001_ -Xdim 64
-
-
Tilt pairs
-
Use the following command:
xmipp_micrograph_scissor -i down2_mic0001.raw –tilted \
down2_mic0002.raw -root mic0001_ -root_tilted mic0002_ -Xdim 64
-
-
-
7| Normalize the images to have zero mean and a standard deviation of unity for the background pixels.
xmipp_normalize -i down2_mic0001.raw.sel -background circle 30 -method \
Ramp -remove_black_dust -remove_white_dust
where -background circle 30defines the background pixels as those outside a circle of radius 30 pixels, and -method Ramp, -remove_black_dust and -remove_white_dust are optional flags to correct for ramping backgrounds and/or white or black outlier pixels (possibly dust particles).
-
8| Correct the CTF phases in the extracted particles (and window the particles to the final desired size if they were extracted with a larger size in Step 6).
xmipp_ctf_correct_phase –ctfdat down2_mic0001.ctfdat
xmipp_window -i down2_mic0001.raw.sel -size 64
where down2_mic0001.ctfdat is a two-column text file with the filenames of the individual images in the first column and the filename of their corresponding CTF parameter file in the second column.
-
9| Finally, create a single selection file containing all particles, sort them based on general statistics to identify outliers and display the sorted list.
xmipp_selfile_create ‘*.xmp’ > images.sel
xmipp_sort_by_statistics –i images.sel –o sorted_images
xmipp_show –sel sorted_images.sel
Figure 2.

Micrograph selection. This is based on (a–c) CTFs and on (d–f) particle appearance. (a) A suitable CTF has several rotationally symmetric rings. CTFs should be discarded if they present drift, that is, (b) fading in a particular direction, or astigmatism, that is, (c) rotationally asymmetric. Furthermore, suitable micrographs should present a (d) homogenous population of well-separated particles. (e) Micrographs should be discarded if the particle density is too high, that is, the particles are so close to each other that they almost overlap, or (f) if the particles are very heterogeneous in size or appearance, indicating aggregation or other forms of particle instability. Panels d–f are on the same scale, and the scale bar in panel d represents 50 nm.
2D analysis
-
10| Rotational spectra classification (Steps 10–14). Perform a 2D alignment for all particles in selection file images.sel.
xmipp_average –i images.sel
xmipp_align2d -i images.sel -ref images.med.xmp -Ri 3 -Ro 25 -iter 4
This will perform four iterations ( -iter) of a quick 2D alignment protocol, where the rotational alignments are performed using only pixels between 3 and 25 from the image center (parameters –Ri and –Ro).
-
11| Find the center of symmetry in the average of the aligned images.
xmipp_find_center2d -i images.med.xmp -x0 32 -y0 32 -r1 3 -r2 25 -low 27 -high 30
where –x0 and –y0 are half the image X and Y dimensions, -r1 and -r2 define which pixels to take into account (as in Step 10) and –low and –high are the parameters of a raised cosine filter, which are usually set to –r2 + 2 and –r2 + 5, respectively.
-
12| Calculate the rotational spectra for all individual particles.
xmipp_make_spectra -i images.sel -o images.sim -x0 31.0 -y0 30.625 -r1 3 -r2 25
where –x0 and –y0 are the coordinates of the center of symmetry as obtained in Step 11, and –r1 and –r2 define which pixels to take into account as in Steps 10 and 11.
-
13| Calculate a self-organizing map of all rotational spectra.
xmipp_classify_kerdensom -i images.sim -o kerd -xdim 7 -ydim 7 -reg0 1000 -reg1 200 -steps 5
where –xdim and –ydim define the X and Y dimensions of the output map and -reg0, –reg1 and –steps define the annealing procedure of the regularization parameters.
▴ CRITICAL STEP The algorithm proceeds from an initially high value of the regularization parameter ( -reg0) to a lower value ( -reg1) in a user-defined number of steps ( -steps). Too high regularization values result in too smooth output maps that do not explain the variance in the data, whereas too low values yield maps that are not organized. Typically, one repeats this calculation multiple times with varying annealing parameters to optimize the output map.
-
14| Inspect the self-organizing map and identify distinct classes. (These classes may be further aligned and classified using Steps 15–20.)
xmipp_show -spectsom kerd -din images.sim &
Select those nodes in the SOM that represent distinct classes by double-clicking the left-mouse button and save the corresponding particles in different selection files.
▴ CRITICAL STEP SOMs act as a summary of the structural variability in the data, providing the user with a convenient tool to interactively select different classes from large amounts of data. The philosophy behind this approach is that an expert user will typically perform better than automated procedures in deciding on which structural differences and how many distinct classes are present in the data. The self-organizing map algorithm outputs a 2D map of so-called code vectors that represent the distribution of the variability in the data. The organization of the map is reflected in the fact that similar code vectors are close to each other, whereas different code vectors tend to be separated. Thereby, the user may identify different regions of the map to correspond to distinct classes. Figure 3 shows an example of interactive class selection in a SOM. For a more detailed description on this topic, refer to the works by Pascual-Montano et al.23,37,38.
? TROUBLESHOOTING
-
15| ML2D classification (Steps 15–16). Perform a maximum-likelihood multireference refinement of all particles contained in selection file images.sel.
xmipp_ml_align2d –i images.sel –nref 5 -mirror –fast –o ml2d
This will align the particles and simultaneously classify them in five groups ( -nref). Optional parameters -mirror and –fast indicate that the mirrored version of each image is to be included in the alignment and that the fast version of the algorithm39 is to be used, respectively.
! CAUTION If this step is executed for Random Conical Tilt reconstruction (see Steps 21–24), do not include the mirror operation.
-
16| Visualize the classes and class averages of the multireference refinement. (For the most challenging cases, one may further subdivide each of the classes obtained using self-organizing maps, as explained in Steps 17–20.)
xmipp_show –sel ml2d.sel ml2d_ref00???.sel &
-
17| KerdenSOM classification (Steps 17–20). Store the optimal alignment parameters of the multireference refinement (Step 15) in the image headers.
xmipp_header_assign -i ml2d.doc -mirror
-
18| Graphically design a mask that defines the region of interest in the average image of all particles contained in selection file ml2d_ref00001.sel.
xmipp_mask_design -i ml2d_ref00001.sel –save_as mask.msk
▴ CRITICAL STEP Designing an optimal mask is important, as it serves to select those pixels of interest and to reduce the influence of noise. To do so, use the pop-up menu under the right-mouse key to select the shape of the mask that best includes the region of interest but minimizes the number of background pixels (e.g., a circle, ellipse, square, etc.). Move the center of this mask using the arrow keys and change its size using the CTRL key in combination with the arrow keys.
-
19| Calculate a self-organizing map for the selected class (see also Step 13).
xmipp_convert_img2data -i ml2d_ref00001.sel -mask mask.msk -o data.dat
xmipp_classify_kerdensom -i data.dat -o som -xdim 7 -ydim 5 -reg0 1000 -reg1 200 -steps 5
xmipp_convert_data2img -i som.cod -mask mask.msk
-
20| Visualize the self-organizing map and identify distinct classes (see also Step 14).
xmipp_show -som som &
? TROUBLESHOOTING
Figure 3.

Example of class selection from a self-organizing map of rotational spectra. The user interactively identifies distinct classes, each of which may comprehend several (neighboring) code vectors. In this example, two classes were identified, one with sixfold symmetry and one with threefold symmetry. Code vectors were identified as belonging to the sixfold symmetric class when they contained a single peak at the sixfold harmonic. Code vectors were identified to belong to the threefold symmetric class when they contained the largest peak at the threefold harmonic and a secondary peak at the sixfold harmonic. The graphical interface of the xmipp_show program (Steps 14 and 20) allows calculating averages for all experimental measurements corresponding to the selected classes (insets).
3D processing
-
21| Random conical tilt (Steps 21–24). After performing a maximum-likelihood multireference refinement (Step 15), visualize the resulting class averages, and decide for which classes to perform an RCT reconstruction.
xmipp_show –sel ml2d.sel &
For each of the selected classes, perform Steps 22–24. In the following paragraphs, we will use the first class (with corresponding selection file ml2d_ref00001.sel and class average ml2d_ref00001.xmp).
-
22| To correctly set the image headers and to allow non-integer shifts, perform a realignment of the untilted images.
xmipp_align2d -i ml2d_ref00001.sel -ref ml2d_ref00001.xmp –iter 2
-
23| Transfer the in-plane rotation angles of the untilted images to their corresponding tilted pairs and center the tilted images.
xmipp_align_tilt_pairs -u ml2d_ref00001.sel –t ml2d_ref00001_tilted.sel
where ml2d_ref00001_tilted.sel is a selection file containing the tilted pair of each of the images in ml2d_ref00001.sel (in the same order).
? TROUBLESHOOTING
-
24| Perform a 3D reconstruction with the tilted images. This option can be performed using the faster option A or the potentially more accurate option B.
-
Faster
-
Use the following command:
xmipp_reconstruct_wbp –i ml2d_ref00001_tilted.sel –o wbp_ml2d_ref00001_tilted.vol
-
-
Potentially more accurate
-
Use the following command:
xmipp_reconstruct_art –i ml2d_ref00001_tilted.sel –o art_ml2d_ref00001_tilted –l 0.01
-
▴ CRITICAL STEP The quality of the ART reconstruction may depend strongly on the value of the relaxation parameter used ( -l). Typically, one performs multiple reconstructions varying this parameter to reach optimal results. As a rule of thumb, at higher levels of noise, larger and higher numbers of images require lower relaxation factors40.
? TROUBLESHOOTING
-
-
25| ML3D classification (Steps 25–29). If the gray scale of your reference map is not on the correct absolute scale, correct it using a single cycle of projection matching and weighted back-projection reconstruction with all particles (contained in selection file images.sel), using the map to be corrected ( reference.vol) as reference. Otherwise, proceed to Step 26.
xmipp_angular_projection_matching –i images.sel –o correct –vol reference.vol \
–dont_modify_header –output_refs
xmipp_angular_class_average –i correct.doc –lib correct_lib.doc –o correct
xmipp_reconstruct_wbp –i correct_classes.sel –o reference.vol \
-use_each_image –weight
▴ CRITICAL STEP The probability functions in maximum-likelihood refinement are based on squared differences between projections of the reference map(s) and the experimental images. Therefore, it is crucial to provide a reference map that has the correct absolute grayscale. Any map reconstructed in XMIPP is guaranteed to have the correct grayscale, but maps coming from alternative packages (like EMAN or SPIDER) may have to be corrected first.
-
26| Perform a low-pass filtering of the reference volume.
xmipp_fourier_filter -i reference.vol -o filtered_reference.vol -sampling 2.8 -low_pass 50
where –sampling defines the pixel size in Å and –low_pass defines the resolution of the low-pass filter in Å.
▴ CRITICAL STEP Low-pass filtering of the reference map to generate the unbiased seeds (i.e., the initial reference maps for the ML3D classification in Step 29) has proven to be crucial for optimal convergence of the ML3D classification protocol. Typically, to prevent bias of high-resolution features in the initial reference, one aims to low-pass filter as much as possible (i.e., still allowing correct convergence).
-
27| Divide the input data set into random subsets for the generation of a user-defined number of unbiased seeds (initial reference maps) for the ML3D classification run.
xmipp_selfile_split –i images.sel –o images_split –n 3
where –n defines the number of random subsets to be created.
-
28| For each of the generated random subsets (e.g., the one in selection file images_split_1.sel), perform a single iteration of 3D maximum-likelihood refinement.
xmipp_ml_refine3d -i images_split_1.sel -o seeds_split_1 -vol \
filtered_reference.vol -iter 1
? TROUBLESHOOTING
-
29| Perform ML3D classification of the entire input data set, using the unbiased seeds as initial references.
xmipp_selfile_create ‘seeds_split_*it00001.vol’ > seeds.sel
xmipp_ml_refine3d -i images.sel -o ml3d -vol seeds.sel -iter 25
? TROUBLESHOOTING
-
30| Projection matching refinement (Steps 30–36). Mask the user-supplied initial reference map with a user-supplied mask:
xmipp_mask –i reference.vol -o masked.vol -mask user_suplied.mask
▴ CRITICAL STEP Do not mask the reference map too tightly. Ideally, one would mask away all surrounding background noise without altering the volume of interest.
-
31| Perform a single iteration of projection matching of all images contained in the selfile images.sel against the masked reference map:
xmipp_angular_projection_matching -i images.sel -vol masked.vol -o proj_match -sam 10 \
-output_refs
xmipp_angular_class_average –i proj_match.doc –lib proj_match_lib.doc –o proj_match
▴ CRITICAL STEP -sam 10 provides the angular sampling in degrees. The value of this parameter should be inversely proportional to the particle size and data quality. Typically, the value for the angular sampling is decreased during the various iterations of this protocol.
-
32| To remove model bias from the refinement procedure, perform a 2D realignment for the images assigned to each reference projection direction (e.g., the first projection directions, with selection file proj_match_ref00001.sel and corresponding average proj_match_ref00001.xmp):
xmipp_align2d –i proj_match_ref0001.sel –ref proj_match_ref00001.xmp –iter 4
-
33| Perform a 3D reconstruction with the aligned images. This option can be performed using the faster option A or the potentially more accurate option B. If the projection directions are distributed evenly over the entire projection space, option A is recommended. Otherwise, one would typically choose option A during the initial stages of refinement and option B in the last iteration.
-
Faster
-
Use the following command:
xmipp_selfile_create ‘proj_match_ref?????.med.xmp’ > averages.sel
xmipp_reconstruct_wbp –i averages.sel -o reconstructed.vol –weight -use_each_image
-
-
Potentially more accurate
-
Use the following command:
xmipp_selfile_create ‘proj_match_ref?????.med.xmp’ > averages.sel
-
xmipp_reconstruct_art –i averages.sel -o reconstructed –WLS –l 0.2
▴ CRITICAL STEP The relaxation factor of the ART algorithm ( -l) is a critical parameter (see Step 24).
? TROUBLESHOOTING
-
-
-
34| Estimate the resolution limit of the reconstructed volume by calculating the Fourier Shell Correlation (FSC)41:
xmipp_selfile_split –i averages.sel -o split -n 2
xmipp_reconstruct_wbp -i split_1.sel -o split_1.vol -weight -use_each_image
xmipp_reconstruct_wbp -i split_2.sel -o split_2.vol -weight -use_each_image
xmipp_resolution_fsc -i split_1.vol -ref split_2.vol
This step will provide a text file called split_1.vol.frc with the FSC at each resolution shell. For filtration purposes, we estimate the resolution limit of the current model as the shell where the FSC drops below 0.5 (see Step 35).
-
35| Filter the reconstructed volume at its estimated resolution limit plus a user-defined constant.
xmipp_fourier_filter –i reconstructed.vol -o filtered.vol –low_pass 0.25
where parameter -low_pass defines the high-resolution limit of the low-pass filter in pixel−1.
▴ CRITICAL STEP The FSC=0.5 criterion typically underestimates the effective resolution. Therefore, we recommend adding a small constant in the range of 0.1–0.2 pixel−1 to the value determined in Step 34.
-
36| Repeat Steps 30–35, using the filtered volume obtained from Step 35 as the reference volume in Step 30. Iterate until the resolution does not improve anymore.
? TROUBLESHOOTING
-
37| Multi-resolution refinement (Steps 37–47). The wavelet-based angular assignment in this protocol (Step 40) requires images of size 2n × 2n pixels, where n is an integer (e.g., 64 × 64 or 128 × 128). If this is not the case, rescale the images in selection file images.sel and the initial reference map (otherwise, proceed to Step 38):
xmipp_scale –i images.sel –xdim 128
xmipp_scale –i reference.vol –xdim 128
where ‘ –xdim 128’ is the rescaled image size, chosen using the smallest n for which 2n is larger than the current image size.
-
38| Precenter the images:
xmipp_average –i images.sel
xmipp_align2d –i images.sel –ref images.med.xmp –iter 4 –only_trans
-
39| Choose the image size to be used in the multiresolution approach, and prepare the data for the angular assignment and reconstruction steps:
xmipp_scale_pyramid –i images.sel –reduce –levels 1 –oext pyr
xmipp_scale_pyramid –i reference.vol –reduce –levels 1
xmipp_normalize –i images_pyr.sel –background radius 60
where –levels 1 defines the scale reduction factor of a pyramid-type scaling42. The downscaled images will have a size of times their original size, where p is the pyramid scale reduction factor. That is, a value of 0 yields images of the original image size, a value of 1 yields images with half the original size, and so on; -background circle 60 defines the background pixels as those outside a circle of radius 60 pixels in the downscaled images. Note that the latter value will depend on the pyramid scale reduction factor used and 60 would be a reasonable value for images of size 128.
▴ CRITICAL STEP One typically performs this protocol in a multiresolution manner to achieve a larger radius of convergence and increased robustness to noise. Use downscaled images (i.e., with a pyramid scale reduction factor larger than zero, but typically not leading to images smaller than 32 × 32 pixels) during the initial iterations of this protocol. Increase the image size during the refinement process, reaching the original size in the final iterations.
-
40| Perform a discrete angular assignment:
xmipp_angular_discrete assign –i images_pyr.sel –ref reference.vol –proj_step 5 \
–psi_step 5 –oang disc_angles.doc
where –proj_step 5 –psi_step 5 defines the angular sampling rate.
▴ CRITICAL STEP The angular step should be decreased as iterations advance. We recommend starting with an angular step of 8 degrees and gradually reducing it to 3 degrees.
-
41| Perform a continuous angular assignment to refine the discrete assignments:
xmipp_angular_continuous_assign –i images_pyr.sel –ref reference.vol –oang cont_angles.doc
▴ CRITICAL STEP The continuous assignment should not be used in the early iterations if the reference volume does not have enough resolution (generally speaking, less than 30 Å). Otherwise, the continuous assignment may lead the optimization too soon to a local minimum. It is recommended to apply the continuous angular assignment only after seven or eight iterations with the discrete angular assignment.
-
42| Correct the images for the effects of the CTF amplitude. (This step is optional.)
xmipp_ctf_correct_idr -vol reference.vol -ctfdat all_images.ctfdat
Here, all_image.ctfdat is a two-column text file as explained in Step 8.
? TROUBLESHOOTING
-
43| Mask the images for 3D reconstruction.
xmipp_selfile_copy images_pyr.sel images_recons
xmipp_mask -i images_recons.sel -mask raised_cosine -60 -64
where the raised cosine is a circular mask that drops continuously between radii of 60 and 64 pixels. Choose the radii of the mask so that its maximum still fits into the image (in this example, the image size was 128).
-
44| Perform a 3D reconstruction. This option can be performed using the faster option A or the potentially more accurate option B. Typically, one would choose option A during the initial stages of refinement, and one would use option B for the last iteration.
-
Faster
-
Use the following command:
xmipp_header_assign -i cont_angles.doc -o images_recons.sel -force
xmipp_reconstruct_wbp –i images_recons.sel -o reconstructed.vol
-
-
Potentially more accurate
-
Use the following command:
xmipp_header_assign -i cont_angles.doc -o images_recons.sel -force
-
xmipp_reconstruct_art –i images_recons.sel –o reconstructed –l 0.01
▴ CRITICAL STEP The relaxation factor of the ART algorithm ( -l) is a critical parameter (see Step 24).
? TROUBLESHOOTING
-
-
-
45| Calculate the resolution of the current model. This step can be done by Fourier Shell Correlation (as in Step 34) or based on its 3D spectral signal-to-noise ratio43. For the latter option, make a reconstruction without masking the experimental images and a reconstruction from pure noise images. Except for the absence of mask, these reconstructions must be performed in exactly the same way as in Step 43.
xmipp_header_assign –i cont_angles.doc \ -o images_pyr.sel -force
xmipp_reconstruct_art –i images_pyr.sel \ –o signal –l 0.001
xmipp_selfile_copy images_pyr.sel noise \ xmipp_mask -i noise.sel -mask circular 128
xmipp_add_noise -i noise.sel -gaussian 1
xmipp_reconstruct_art –i noise.sel –o noise \ –l 0.001
xmipp_ssnr –S signal.vol –N noise.vol \ –selS images_pyr.sel –selN noise.sel
–sampling_rate 2 –o reconstructed.vol.ssnr
The output text file reconstructed.vol.ssnr contains the estimated spectral signal-to-noise ratio and is explained in more detail in the XMIPP manual pages. The parameter –sampling_rate is the pixel size (in Å) in the (possibly) reduced-size images.
-
46| Optionally, one may post-process the reconstructed volume by masking, low-pass filtering and positioning its center of mass at the origin.
xmipp_mask –i reconstructed.vol \
–mask raised_cosine -60 -64
xmipp_fourier_filter –i reconstructed.vol \
–low_pass 0.3
xmipp_find_center3d –i reconstructed.vol \
–center_volume
xmipp_mask –i reconstructed.vol \
–mask user_provided_mask.vol
Parameter -low_pass for the fourier_filter program defines the resolution of the current model as determined in Step 45 (in pixel−1).
-
47| Repeat Steps 39–46, using the post-processed map from Step 46 as the reference in Step 39. Iterate until the resolution (as calculated in Step 45) does not improve anymore. If the next iteration uses a larger image size than the current one, rescale the reference volume correspondingly:
xmipp_scale_pyramid –i reconstructed.vol –expand –levels 1
cp reconstructed.vol reference.vol
? TROUBLESHOOTING
• TIMING
Unless otherwise mentioned, computing times were measured using a single 1 GHz Alpha processor.
Steps 1–4, preprocess micrographs: 15 min per micrograph for a single Kodak SO-163 plate digitized in a Zeiss-Intergraph scanner at a pixel size of 7 μm and with a downsample step of 3
Step 5, manual particle selection: 0.5–1 h per micrograph, but strongly dependent on the sample and on the experience of the user
Steps 6–9, preprocess particles: 1–2 min per micrograph
Steps 10–14, rotational spectra classification: 1 h for 14,000 images of 80 × 80 pixels
Steps 15–16, ML2D classification: 8 h for 14,000 images of 80 × 80 pixels using three references and using sixty four 2.2-GHz BladeCenter JS20 processors in parallel
Steps 17–20, KerdenSOM classification: 9 h for 14,000 images of 80 × 80 pixels and a 12×6 output map
Steps 21–24, random conical tilt: 10 min for 1,000 images of 80 × 80 pixels
Steps 25–29, ML3D classification: 72 h for 20,000 images of 64 × 64 pixels using four references and using sixty four 2.2-GHz BladeCenter JS20 processors in parallel
Steps 30–36, projection matching refinement: 24 h for eight iterations with 16,588 images of 128 × 128 pixels with angular sampling intervals down to 3 degrees and using sixty four 2.2-GHz BladeCenter JS20 processors in parallel.
Steps 37–47, multiresolution refinement: 60 h for ten iterations with 16,588 images of 128 × 128 pixels with discrete angular sampling intervals down to 3 degrees and using sixty four 2.2-GHz BladeCenter JS20 processors in parallel.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
TABLE 1.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| All | XMIPP executables are not found | XMIPP (version 2.0 or higher) was not installed correctly | Install XMIPP-2.0 or ask your system manager to do so |
| All | Python gives the following error message—ImportError: No module named Tkinter | Python-tk was not installed | Install python-tk or ask your system manager to do so |
| All | The graphical XMIPP programs do not work | XMIPP did not find the qt3 or qt3-devel libraries | Install qt3 and qt3-devel or ask your system manager to do so |
| All | The parallelized XMIPP programs do not work | XMIPP did not find the MPI libraries | Install MPI or ask your system manager to do so |
| 1 | TIFF to RAW conversion fails | Unrecognized TIFF format | Different scanners may produce different TIFF formats. If XMIPP does not recognize your scanned micrographs, use scanner-specific software to convert to MRC or Spider format, and use the programs xmipp_convert_ raw22spi and/or xmipp_convert_-spi22ccp4 to convert these to RAW format |
| TIFF to RAW conversion fails | XMIPP did not find the TIFF libraries | Make sure you have the TIFF libraries, and if not, install them, or ask your system manager to do this | |
| 3 | The estimated CTF does not fit the experimental power spectral density | The CTF estimation algorithm has converged to a local optimum | Adjust parameters min_freq and max_freq to those frequencies where Thon rings are visible, or guide the CTF estimation algorithm through the user-interactive, graphical mode: xmipp_micrograph_mark –i mic00001.raw –ctf mic00001_input.param |
| 14 and 20 | The SOM looks too smooth | The regularization factors ( -reg0 and –reg1) are too high | Rerun the SOM calculation with lower regularization factors |
| The SOM does not look organized | The regularization factors ( -reg0 and –reg1) are too low | Rerun the SOM calculation with higher regularization factors | |
| 23 | The tilted particles are not well centered | The implemented centering algorithm may not be suited for your particles | Rerun xmipp_align_tilt_pairs with any of the following options: -force_x_zero, -skip_stretching or –skip_centering |
| 24, 33 and 44 | The ART reconstruction looks very blurred | The relaxation parameter ( -l) may be too low for the number of cycles through the data performed | Rerun xmipp_reconstruct_art with a higher relaxation parameter ( -l) or perform more cycles through the data |
| The ART reconstruction looks very noisy | The relaxation parameter ( -l) may be too high | Rerun xmipp_reconstruct_art with a lower relaxation parameter ( -l) | |
| 28 and 29 | The resulting maps are streaked in one direction | The reference map may not be on the correct grayscale | Perform Step 25 |
| 29 | The resulting maps are very noisy and do not explain any structural heterogeneity in the data | The initial reference map may have contained too many high frequencies, directing the ML3D classification toward a local minimum | Apply a lower-resolution low-pass filter to the initial map (Step 26) |
| 36 and 47 | The refinement process yields noisy maps that resemble the initial reference too much | The initial reference may be outside the radius of convergence of the refinement | Try to generate a different initial reference map |
| 42 | The refinement process yields maps of bad quality | The CTF amplitude correction algorithm will not converge if the images have not been phase corrected previously | Perform a CTF phase correction on the images (Step 8) |
ANTICIPATED RESULTS
In this section, we present the results of applying the protocols presented to two experimental data sets. The first one is a negative stain data set on the hexameric helicase G40P. These data, and the corresponding results previously obtained, are described in detail by Núñez-Ramirez et al.44. These data and the results obtained with the protocols presented here are available for testing and may be downloaded from: http://xmipp.cnb.csic.es/twiki/bin/view/Xmipp/Protocols. The second set is structurally heterogeneous data on 70S E. coli ribosome particles embedded in vitrified ice. These data and the classification results previously obtained are described in detail by Scheres et al.26. As in a similar experiment reported before27, we used a randomly selected subset of 20,000 projections of the originally much larger data set.
Figure 4 depicts the processing workflow used to classify and reconstruct two distinct conformations of the hexameric helicase G40P from pairs of tilted micrographs. Steps 1–9 were performed to yield 14,000 single-particle images. These particles were classified based on their rotational spectra (Steps 10–14). Two structurally distinct classes were selected, one containing 1,300 threefold symmetric particles and the other containing 800 sixfold symmetric ones. The images of both groups were then aligned using ML2D classification with a single reference (Steps 15–16), and subsequently, a random conical tilt reconstruction (Steps 21–24) was performed for each of them.
Figure 4.
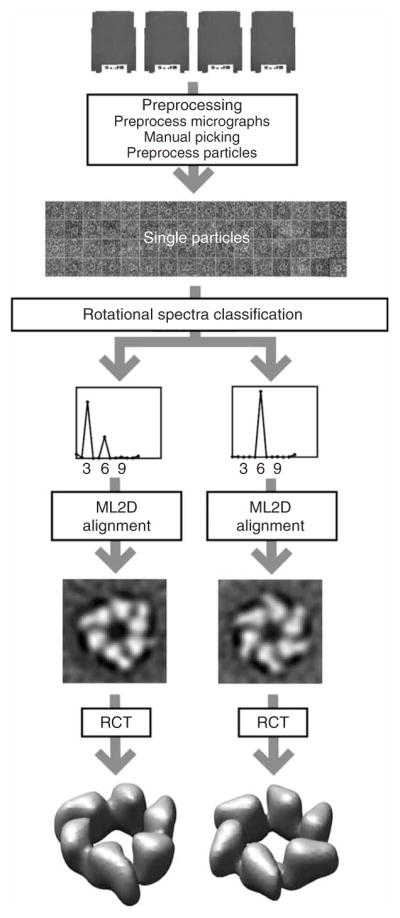
Anticipated results for the G40P case. Tilted pairs of digitized micrographs were processed to yield a data set of 14,000 particle pairs. The untilted particles were classified in threefold and sixfold symmetric particles based on their rotational spectra. For both classes, the untilted particles were aligned using maximum-likelihood refinement, and the tilted particles were used to calculate the corresponding random conical tilt reconstructions.
Figure 5 shows the processing workflow used to classify and refine a structurally heterogeneous data set containing 20,000 experimental projections of 70S E. coli ribosomes embedded in vitrified ice. ML3D classification (Steps 25–29) using four references served to identify two structurally distinct classes: 70S ribosomes without EF-G and with three tRNAs bound (classes 1–3) and 70S ribosomes in complex with EF-G and a single tRNA (class 4). Subsequent refinement of the subset containing 16,588 images corresponding to the ribosome complex without EFG using either projection matching (Steps 30–36) or multiresolution refinement (Steps 37–47) yielded 3D reconstructions up to 16 Å resolution in both cases (according to the FSC=0.5 criterium described in Step 34).
Figure 5.

Anticipated results for the ribosome case. A structurally heterogeneous data set of 70S E. coli ribosome particles in complex with or without EFG were classified using ML3D classification, and the corresponding class with particles lacking EFG was further refined using either projection matching or multiresolution refinement.
Acknowledgments
We thank Haixiao Gao and Joachim Frank for providing the ribosome data, and we thank the Barcelona Supercomputing Center (Centro Nacional de Supercomputación) for providing computer resources. This work was funded by the European Union (FP6-502828 and UE-512092), the US National Institutes of Health (HL740472), the Spanish Comisión Interministerial de Ciencia y Tecnología (BFU2004-00217), the Spanish Ministerio de Educación y Ciencias (CSD2006-0023, BIO2007-67150-C03-01 and -03), the Spanish Fondo de Investigación Sanitaria (04/0683) and the Comunidad de Madrid (S-GEN-0166-2006).
Footnotes
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Henderson R. Realizing the potential of electron cryo-microscopy. Q Rev Biophys. 2004;37:3–13. doi: 10.1017/s0033583504003920. [DOI] [PubMed] [Google Scholar]
- 2.Subramaniam S, Milne JLS. Three-dimensional electron microscopy at molecular resolution. Annu Rev Biophys Biomol Struct. 2004;33:141–155. doi: 10.1146/annurev.biophys.33.110502.140339. [DOI] [PubMed] [Google Scholar]
- 3.Carragher B, Smith PR. Special issue on Advances in Computational Image Processing for Microscopy. J Struct Biol. 1996;116:2–8. doi: 10.1006/jsbi.1996.0002. [DOI] [PubMed] [Google Scholar]
- 4.Chiu SLAW. Special Issue on Single Particle Processing. In: Ludtke SJ, Chiu W, editors. J Struct Biol. Vol. 173. 2001. [Google Scholar]
- 5.Frank J, et al. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 6.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 7.Heymann JB, Belnap DM. Bsoft: image processing and molecular modeling for electron microscopy. J Struct Biol. 2007;157:3–18. doi: 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 8.van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A new generation of the IMAGIC image processing system. J Struct Biol. 1996;116:17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 9.Hohn M, et al. SPARX, a new environment for Cryo-EM image processing. J Struct Biol. 2007;157:47–55. doi: 10.1016/j.jsb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Sorzano COS, et al. XMIPP: a new generation of an open-source image processing package for electron microscopy. J Struct Biol. 2004;148:194–204. doi: 10.1016/j.jsb.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Software tools for molecular microscopy. Wikipedia; 2007. http://en.wikipedia.org/wiki/Software_tools_for_molecular_microscopy. [Google Scholar]
- 12.Marabini R, et al. Xmipp: an image processing package for electron microscopy. J Struct Biol. 1996;116:237–240. doi: 10.1006/jsbi.1996.0036. [DOI] [PubMed] [Google Scholar]
- 13.Nickell S, et al. Automated cryoelectron microscopy of ‘single particles’ applied to the 26S proteasome. FEBS Lett. 2007;581:2751–2756. doi: 10.1016/j.febslet.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 14.Martin-Benito J, et al. Divergent substrate-binding mechanisms reveal an evolutionary specialization of eukaryotic prefoldin compared to its archaeal counterpart. Structure. 2007;15:101–110. doi: 10.1016/j.str.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 15.Busselez J, et al. Structural basis for the PufX-mediated dimerization of bacterial photosynthetic core complexes. Structure. 2007;15:1674–1683. doi: 10.1016/j.str.2007.09.026. [DOI] [PubMed] [Google Scholar]
- 16.Tato I, et al. The ATPase activity of the DNA transporter TrwB is modulated by protein TrwA: implications for a common assembly mechanism of DNA translocating motors. J Biol Chem. 2007;282:25569–25576. doi: 10.1074/jbc.M703464200. [DOI] [PubMed] [Google Scholar]
- 17.Agirrezabala X, et al. Quasi-atomic model of bacteriophage t7 procapsid shell: insights into the structure and evolution of a basic fold. Structure. 2007;15:461–472. doi: 10.1016/j.str.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Nunez-Ramirez R, et al. Loading a ring: structure of the Bacillus subtilis DnaB protein, a co-loader of the replicative helicase. J Mol Biol. 2007;367:764–769. doi: 10.1016/j.jmb.2006.12.075. [DOI] [PubMed] [Google Scholar]
- 19.Rivera-Calzada A, Spagnolo L, Pearl LH, Llorca O. Structural model of full-length human Ku70-Ku80 heterodimer and its recognition of DNA and DNA-PKcs. EMBO Rep. 2007;8:56–62. doi: 10.1038/sj.embor.7400847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arias-Palomo E, Recuero-Checa MA, Bustelo XR, Llorca O. 3D structure of Syk kinase determined by single-particle electron microscopy. Biochim Biophys Acta. 2007;1774:1493–1499. doi: 10.1016/j.bbapap.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Heel M. Angular reconstitution: a posteriori assignment of projection directions for 3D reconstruction. Ultramicroscopy. 1987;21:111–123. doi: 10.1016/0304-3991(87)90078-7. [DOI] [PubMed] [Google Scholar]
- 22.Crowther RA, Amos LA. Harmonic analysis of electron microscope images with rotational symmetry. J Mol Biol. 1971;60:123–130. doi: 10.1016/0022-2836(71)90452-9. [DOI] [PubMed] [Google Scholar]
- 23.Pascual-Montano A, et al. A novel neural network technique for analysis and classification of EM single-particle images. J Struct Biol. 2001;133:233–245. doi: 10.1006/jsbi.2001.4369. [DOI] [PubMed] [Google Scholar]
- 24.Scheres SHW, et al. Maximum-likelihood multi-reference refinement for electron microscopy images. J Mol Biol. 2005;348:139–149. doi: 10.1016/j.jmb.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 25.Radermacher M, Wagenknecht T, Verschoor A, Frank J. Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli. J Microsc. 1987;146:113–136. doi: 10.1111/j.1365-2818.1987.tb01333.x. [DOI] [PubMed] [Google Scholar]
- 26.Scheres SHW, et al. Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization. Nat Methods. 2007;4:27–29. doi: 10.1038/nmeth992. [DOI] [PubMed] [Google Scholar]
- 27.Scheres SHW, et al. Modeling experimental image formation for likelihood-based classification of electron microscopy data. Structure. 2007;15:1167–1177. doi: 10.1016/j.str.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng RH, et al. Functional implications of quasi-equivalence in a T = 3 icosahedral animal virus established by cryo-electron microscopy and X-ray crystallography. Structure. 1994;2:271–282. doi: 10.1016/s0969-2126(00)00029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Penczek PA, Grassucci RA, Frank J. The ribosome at improved resolution: new techniques for merging and orientation refinement in 3D cryo-electron microscopy of biological particles. Ultramicroscopy. 1994;53:251–270. doi: 10.1016/0304-3991(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 30.Sorzano COS, et al. A multiresolution approach to orientation assignment in 3D electron microscopy of single particles. J Struct Biol. 2004;146:381–392. doi: 10.1016/j.jsb.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 31.Jonic S, et al. Spline-based image-to-volume registration for three-dimensional electron microscopy. Ultramicroscopy. 2005;103:303–317. doi: 10.1016/j.ultramic.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Sorzano COS, Marabini R, Herman GT, Censor Y, Carazo JM. Transfer function restoration in 3D electron microscopy via iterative data refinement. Phys Med Biol. 2004;49:509–522. doi: 10.1088/0031-9155/49/4/003. [DOI] [PubMed] [Google Scholar]
- 33.Marabini R, Herman GT, Carazo JM. 3D reconstruction in electron microscopy using ART with smooth spherically symmetric volume elements (blobs) Ultramicroscopy. 1998;72:53–65. doi: 10.1016/s0304-3991(97)00127-7. [DOI] [PubMed] [Google Scholar]
- 34.Sorzano COS, et al. The effect of overabundant projection directions on 3D reconstruction algorithms. J Struct Biol. 2001;133:108–118. doi: 10.1006/jsbi.2001.4338. [DOI] [PubMed] [Google Scholar]
- 35.Jonic S, Sorzano COS, Cottevieille M, Larquet E, Boisset N. A novel method for improvement of visualization of power spectra for sorting cryo-electron micrographs and their local areas. J Struct Biol. 2007;157:156–167. doi: 10.1016/j.jsb.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 36.Zhu Y, et al. Automatic particle selection: results of a comparative study. J Struct Biol. 2004;145:3–14. doi: 10.1016/j.jsb.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 37.Pascual A, Barcena M, Merelo JJ, Carazo JM. Mapping and fuzzy classification of macromolecular images using self-organizing neural networks. Ultramicroscopy. 2000;84:85–99. doi: 10.1016/s0304-3991(00)00022-x. [DOI] [PubMed] [Google Scholar]
- 38.Pascual-Montano A, Taylor KA, Winkler H, Pascual-Marqui RD, Carazo JM. Quantitative self-organizing maps for clustering electron tomograms. J Struct Biol. 2002;138:114–122. doi: 10.1016/s1047-8477(02)00008-4. [DOI] [PubMed] [Google Scholar]
- 39.Scheres SHW, Valle M, Carazo JM. Fast maximum-likelihood refinement of electron microscopy images. Bioinformatics. 2005;21(Suppl 2):ii243–ii244. doi: 10.1093/bioinformatics/bti1140. [DOI] [PubMed] [Google Scholar]
- 40.Sorzano COS, Marabini R, Herman GT, Carazo JM. Multiobjective algorithm parameter optimization using multivariate statistics in threedimensional electron microscopy reconstruction. Pattern Recognit. 2005;38:2587–2601. [Google Scholar]
- 41.Van Heel M. Similarity measures between images. Ultramicroscopy. 1987;21:95–99. [Google Scholar]
- 42.Unser M, Aldroubi A, Eden M. The L(2) polynomial spline pyramid. IEEE Trans Pattern Anal Mach Intell. 1993;15:364–379. [Google Scholar]
- 43.Unser M, et al. Spectral signal-to-noise ratio and resolution assessment of 3D reconstructions. J Struct Biol. 2005;149:243–255. doi: 10.1016/j.jsb.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nunez-Ramirez R, et al. Quaternary polymorphism of replicative helicase G40P: structural mapping and domain rearrangement. J Mol Biol. 2006;357:1063–1076. doi: 10.1016/j.jmb.2006.01.091. [DOI] [PubMed] [Google Scholar]