

Abstract
Cellular uptake of therapeutic oligonucleotides and subsequent intracellular trafficking to their target sites represents the major technical hurdle for the biological effectiveness of these potential drugs. Accordingly, laboratories worldwide focus on the development of suitable delivery systems. Among the different available non-viral systems like cationic polymers, cationic liposomes and polymeric nanoparticles, cell-penetrating peptides (CPPs) represent an attractive concept to bypass the problem of poor membrane permeability of these charged macromolecules. While uptake per se in most cases does not represent the main obstacle of nucleic acid delivery in vitro, it becomes increasingly apparent that intracellular trafficking is the bottleneck. As a consequence, in order to optimize a given delivery system, a side-by-side analysis of nucleic acid cargo internalized and the corresponding biological effect is required to determine the overall efficacy. In this review, we will concentrate on peptide-mediated delivery of siRNAs and steric block oligonucleotides and discuss different methods for quantitative assessment of the amount of cargo taken up and how to correlate those numbers with biological effects by applying easy to handle reporter systems. To illustrate current limitations of non-viral nucleic acid delivery systems, we present own data as an example and discuss options of how to enhance trafficking of molecules entrapped in cellular compartments.
Key Words: Cell-Penetrating Peptides (CPPs), nucleic acid delivery, endocytosis, fluorescence microscopy, RNAi, siRNA, steric block, splice correction.
INTRODUCTION
Oligonucleotide-based strategies which can be used to modulate a vast variety of cellular functions represent a promising alternative to conventional therapies (for a review see: [1,2]). Among the different oligonucleotides with therapeutic potential are aptamers, transcription factor-binding decoy oligonucleotides, ribozymes, triplex-forming oligonucleotides (TFO), immunostimulatory CpG motifs, antisense oligonucleotides, small interfering RNAs (siRNAs) and antagomirs. Nowadays, these potential macromolecular drugs are generally either relatively easily derived by rational design (e.g. antisense or siRNA) or straightforward selection processes (e.g. aptamers). One of their main advantages over protein- or peptide-based approaches comprises the high specificity for their target while being non-immunogenic. However, despite these advances, a major impediment to the development of nucleic acid-based strategies for treatment and prevention of diseases is the relatively inefficient means to effectively deliver these macromolecules into the desired target cells. Although viral vectors have been widely used to transfer genetic material into cells [3,4], they bear an inherent risk for the patient to encounter severe immunological responses or even develop cancer [5-8]. As a result of these problems much attention has been paid in recent years to the development of non-viral delivery systems. This conception includes an assortment of fairly unrelated approaches yielding various degrees of enhanced cellular uptake of nucleic acids. Currently, liposomes and cationic polymers are used as a standard tool to transfect cells in vitro. However, these procedures are characterized by a significant lack of efficiency accompanied by a high level of toxicity rendering them mostly inadequate for in vivo applications. In this context cell-penetrating peptides (see below) represent an interesting alternative as they generally are less toxic than liposomes or cationic polymers. Moreover, they are commonly better suited to transfer cargo into different cell types like non-adherent cells and primary cells, which are hard to transfect using commercially available standard protocols. The most advanced approaches in the field, which are not subject of the present article, are complex carrier systems combining vantages of assorted strategies to generate nanoparticles with better defined properties aimed towards enhanced uptake as well as intracellular trafficking in combination with cell-specific functionalities. For example, there are attempts to combine peptides with cationic liposomes [9-16] or polyethyleneimine (PEI) [17]. Other strategies are aimed towards the synthesis of high or low molecular weight branched polymers and/or peptides [18-22] or dendrimers [23,24]. Even more complex systems are particularly promising with respect to in vivo delivery [25-29].
In this review we will report about particular aspects of non-viral oligonucleotide delivery in vitro, pinpointing the current limitations, and provide quantitative means for determining where the bottlenecks of such strategies at present are. The focus of this article is recent progress in the field of peptide-mediated cellular delivery of siRNA and steric block oligonucleotides in cell tissue culture as a starting point for further developments illustrated by own experimental data. Our intention is not to provide the reader with easy solutions on how to solve the existing problems encountered with such approaches but give some hints where to start optimizing a particular approach.
CELL-PENETRATING PEPTIDES
The idea of using peptides as carriers goes back some twenty years when it was discovered that the HIV-1 transactivating protein Tat is taken up by mammalian cells [30,31]. A few years later, the Antennapedia homeodomain of Drosophila melanogaster was shown to act similarly [32]. Later on, it could be shown that peptides derived from Tat and Antennapedia as well as other proteins are capable of transporting macromolecular cargo molecules into cells [33-35]. Based on such promising results, a rapidly expanding field focusing on the so-called cell-penetrating peptides (CPPs), also referred to as protein transduction domains (PTD), began to develop. Since the first reports about Tat, a large number of naturally occurring as well as engineered CPPs have been discovered [36-42]. Table 1 gives an overview of selected “classical” CPPs. Generally, CPPs are short polycationic sequences of less than 30 amino acids that are able to translocate different cargoes (e.g. nucleic acids, peptides and even entire proteins) into cells. The only common characteristic of these peptides appears to be that they are amphipathic and net positively charged at physiological pH. Frequently the cargo is covalently attached to the CPP which can be achieved by expression as a fusion construct or by chemical coupling (for a review see: [43]). In particular cases, cargo and carrier bind each other non-covalently through mainly ionic interactions [40,44,45].
Table 1.
Sequences of Selected “Classical” CPPs
| Peptide | Sequence | References |
|---|---|---|
| Tat48-60 | GRKKRRQRRRPPQ | [48] |
| penetratin (Antp43-58) | RQIKIWFQNRRMKWKK | [203] |
| transportan | GWTLNSAGYLLGKINLKALAALAKKIL | [204] |
| TP10 | AGYLLGKINLKALAALAKKIL | [205] |
| Oligoarginine (R8) | RRRRRRRR | [50] |
| MAP | KLALKLALKALKAALKLA | [163] |
| MPG | GALFLGFLGAAGSTMGAWSQPKKKRKV | [47] |
| MPGα | GALFLAFLAAALSLMGLWSQPKKKRKV | [69] |
Despite the widespread interest in peptide carriers, the mechanisms underlying the cellular translocation of CPPs are poorly understood. Early work relied upon fluorescence imaging or flow cytometry analysis of chemically fixed cells to examine intracellular localization of fluorescently labeled peptides in the absence or presence of cargo. According to these experiments peptides appeared to be internalized very rapidly within minutes even at 4 °C. From such observations it was concluded that CPPs penetrate cell membranes by an energy-independent mechanism [46-50]. Although it had been reported quite early on that certain fixation procedures may cause artefacts leading to an overestimation of the cellular uptake rates [51-53] the dimension of this problem was not commonly recognized until a side by side comparison of fixed and living cells was published [54]. Based on these findings, many groups re-examined their data. However, despite considerable technical improvements, there are still puzzling controversial results concerning the exact mechanism of CPP uptake. Though in most cases endocytosis has been suggested to be the main route of internalization (Fig. (1A)), substantial difficulties are encountered identifying the exact pathway ([42,55] and references therein). Prior to endocytosis CPPs interact electrostatically with the extracellular matrix of the cell surface mostly through binding to negatively charged glycosaminoglycans, i.e. heparan sulfate proteoglycans [56-59]. Recent studies indicate that the uptake mechanism of CPPs can be influenced by the attachment of cargos. For example, Richard et al. [54,60] reported a colocalization of Tat48-59 with markers of clathrin-mediated endocytosis, whereas Fittipaldi et al. [61] found a caveolae/lipid raft-dependent process for a Tat-GFP fusion protein and Wadia et al. [62] described a macropinocytotic uptake pathway for a fusion construct of Tat peptide with Cre recombinase. In summary, the precise mechanism of internalization remains elusive and strongly depends on the properties of both CPP and cargo as well as on the transfection conditions and the cell lines used [63-68].
Fig. (1).
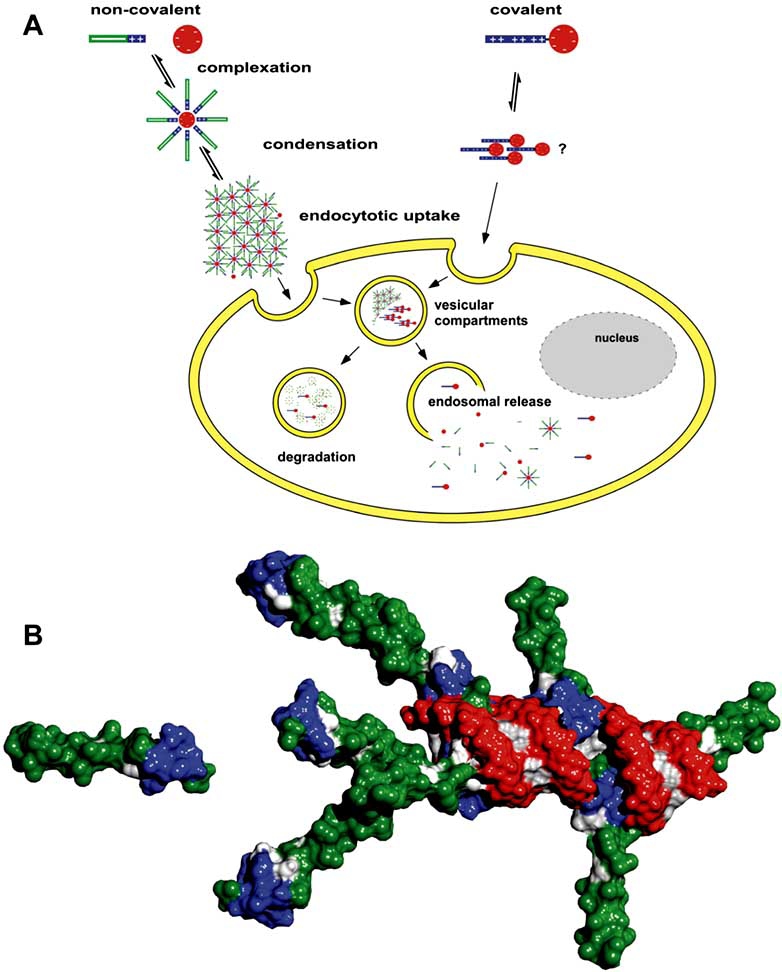
Simplistic scheme of peptide-based nucleic acid delivery systems (A). Interaction of CPP and cargo is either achieved by covalent attachment or by non-covalent complexation through mainly ionic interactions. In case of non-covalent complex formation, a further assembly of cargo/carrier complexes occurs, leading to the formation of large nanoparticles (confer Fig. (2)). In case of covalently joined molecules a similar scenario is less likely, yet cannot be excluded. Prior to the translocation process the particles attach to the cell surface by ionic interactions of positively charged CPP residues with negatively charged membrane components. Subsequently, complexes are taken up via an endocytotic pathway. Although less likely, direct penetration cannot be excluded and may occur simultaneously. Once inside the cell, the cargo has to escape from vesicular compartments, otherwise it eventually gets degraded in the lysosome. Red: negative charges, blue: positive charges, green: hydrophobic domains.
Three-dimensional model of MPGα/siRNA interactions (B). The model was generated by iterative rigid body docking cycles of siRNA (PDB 1R9F) and peptide using the program Hex 4.2 [201]. The PDB file of MPGα was generated with the program ICM (Molsoft LLC) taking into consideration different secondary structure predictions and energy minimization protocols. Out of many docking solutions particular ones were picked for illustration purposes using the program Chimera [202]. The phosphate backbone of the siRNA is shown in red, the nucleobases in light gray. Aliphatic, aromatic and hydrophobic residues of the peptide are shown in green, positive charged residues in blue and the remaining amino acids in gray. It is assumed that formation of larger particles is driven by hydrophobic peptide/peptide interactions generating free positive charges where other siRNA molecules can interact. This eventually drives complex formation in a sandwich or mesh like assembly reaction. In principle such a scenario holds true for any given nucleic acid cargo.
As opposed to the majority of CPP applications reported, which rely on covalent linkage of carrier and cargo, limiting their general use considerably as a new construct has to be generated as well as tested for any given nucleic acid cargo, we will focus in this article on a peptide termed MPGα which forms highly stable non-covalent complexes with nucleic acids (Fig. (1)). The peptide is a derivative of the original MPG peptide described by Morris and coworkers [47] and differs by five amino acids in the hydrophobic part. These changes result in an alteration of the overall structure of the peptide towards a higher tendency of adopting a helical conformation [69]. Accordingly, the two peptides behave differently with respect to their interaction with artificial lipids as well as Xenopus oocytes [70,71] and most probably, their exact mechanism of uptake is not the same. Besides ionic interactions responsible for the initial peptide/nucleic acid complex formation, hydrophobic peptide/peptide interactions drive the maturation of large nanoparticles in a sandwich-like assembly reaction (Fig. (1B) and Fig. (2)).
Fig. (2).
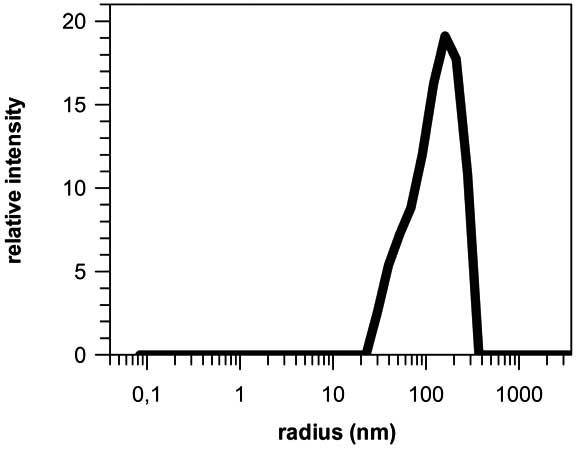
Dynamic light scattering measurements of MPGα/siRNA complexes. Experiments were performed with 5 µM peptide and 0.5 µM siRNA in a Tris-buffered (10 mM, pH 7.5) aqueous solution at 20°C using a DynaPro MS/X device (Protein Solutions, UK). Depending on buffer composition (e.g. ionic strength), complexes which range in size between 20 and 300 nm with an average of about 200 nm are observed.
MECHANISM OF RNA INTERFERENCE
In recent years, RNA interference (RNAi) has gained a lot of interest as a tool for functional genomics studies and probably equally important as a promising therapeutic approach for the treatment of various diseases [72,73]. RNAi is a highly evolutionally conserved and specific process of post-transcriptional gene silencing (PTGS) by which double stranded RNA (dsRNA), when introduced into a cell, causes sequence-specific degradation of homologous mRNA sequences [74,75]. Mechanistically the process can be divided into two steps. An initiator step where dsRNA is cleaved by dicer, a member of the RNase III family, into 21-25 nt long small interfering RNA (siRNA) fragments [76]. In a consecutive step, these fragments are transferred to RISC (RNA-induced silencing complex) where one of the strands, the so called guide strand, serves as a molecular template to recognize homologous mRNA that is cleaved by Argonaute [77,78], a protein component of RISC. Once the guide strand is bound to RISC this complex can undergo many rounds of mRNA binding and cleavage ([79], Fig. (3A)). To circumvent application of long double stranded RNAs, which inevitably trigger an interferon response, it is sufficient to extracellularly supply 21 nt long dsRNAs [80,81]. Alternatively, siRNAs can be expressed endogenously using DNA vectors which code for short hairpin (sh) RNAs [82-84]. These shRNAs are than cleaved by dicer to siRNAs. Short hairpin RNA constructs have advantages over siRNA because the effects of these constructs can lead to a more stable and long-term result. On the other hand, besides the fact that shRNAs might interfere with the microRNA pathway [85,86], this strategy requires a gene therapy approach in the long run [87]. For this reason we will not cover shRNAs.
Fig. (3).

Mechanistic principles of RNAi (A) and splice correction (B). Details are given in the text.
LITERATURE OVERVIEW: CPP-MEDIATED siRNA DELIVERY
As described above, siRNAs represent a valuable tool to inhibit the expression of a target gene in a sequence-specific manner. In the following section, selected examples of CPP-mediated siRNA delivery will be presented which are summarized in Table 2. Only a few studies describe the covalent attachment of nucleic acid cargo and peptide carrier (confer Table 2). In one approach, simple mixing of siRNA targeted against GFP or CDK9 and Tat peptide did not generate any measurable RNAi effect whereas cross-linked siRNA-Tat47-57 led to a significant down-regulation of the target proteins. However, high concentrations of siRNA (about 300 nM) had to be used [88]. Both LF- and Tat47-57-mediated transfections resulted in a perinuclear localization of siRNA. In contrast, fluorescently labeled Tat47-57 without cargo was mainly found in the nucleolus, suggesting that interactions with RISC influence subcellular localization. In another approach, significant uptake of siRNAs targeted against luciferase or GFP could be observed after disulfide coupling the 5’-end of the sense strand to penetratin or transportan [89]. Compared to LF2000, slightly higher levels of transfection were achieved. Interestingly, after LF2000-mediated transfection, basal luciferase activity returned to normal levels one day earlier than after CPP-mediated transfection although the same concentration of siRNA was applied. A remarkably strong RNAi effect in hard to transfect primary neuronal cells was reported by Davidson et al. [90]. Here, siRNAs directed against several endogenous proteins were coupled to penetratin via a disulfide bond. The observed down regulation of the target proteins after peptide-mediated siRNA delivery was found to be far more effective compared to LF2000. This was in part attributed to the toxicity of the lipids. As one of the first groups to report on Tat48-60- or penetratin-mediated siRNA delivery in vivo, Moschos et al. showed, that intratracheal administration of the conjugates did not lead to any intensification of the knockdown of the target gene p38 mitogen-activated protein kinase in mouse lungs in comparison to unmodified non-formulated siRNA [91]. Strikingly, it was found that the peptides alone triggered a detectable decrease in target gene expression and that the penetratin-conjugate induced elevated levels of the immune markers IFN-α , TNF-α , and IL-12p40 in lung tissue.
Table 2.
Examples for Peptide-Mediated Delivery of siRNA
| CPP/Delivery System | Mode of Linkage* | Target | Cell Line | Ref. |
|---|---|---|---|---|
| Tat47-57, Tat-derived oligocarbamate) | c | EGFP, CDK9 | HeLa | [88] |
| penetratin, transportan | c | luciferase, GFP | Cos-7, C166, EOMA, CHO-AA8 | [89] |
| penetratin | c | Cu-ZN SOD-1, Caspase-3/-8/-9 | primary rat hippocampal or sympathetic neurons | [90] |
| Tat48–60, penetratin | c | p38 MAP kinase | L929 (mouse fibroblasts), mouse lung (intratracheal) | [91] |
| MPG, MPGΔNLS | n-c | luciferase, GAPDH | HeLa, Cos-7, HS-68 | [94] |
| H3K8b, H3K8b(+RGD) | n-c | β-Gal, luciferase | SVR-bag4, MDA-MB-435, C6 | [21] |
| RVG-9R | n-c | GFP, Cu-ZN SOD-1, JEV | HeLa, Neuro2a, mouse brain (intravenously) | [95] |
| POD | n-c | EGFP | HER 911 | [96] |
| Chol-R9 | n-c | VEGF | CT-26, mouse (intratumoral) | [97] |
| EB1, MPGΔNLS,bPrPp | n-c | luciferase | HeLa, HepG2 | [101] |
| TatU1A | n-c | EGFP, EGFR | CHO, A431, | [104] |
| MPGα | n-c | luciferase | HeLa, ECV 304 | [55] |
| rCPP | n-c | EGFP, EF1A | HeLa, H1299 | [29] |
| stearyl-R8 | n-c | EGFP, MAP2B | primary rat hippocampal neurons | [206] |
| R8-MEND (siRNA/stearyl-R8 core) | n-c | luciferase | HeLa | [207] |
c = covalent / n-c = non-covalent.
Besides technical difficulties arising from the syntheses of conjugates consisting of short cationic or hydrophobic peptides and highly negatively charged siRNAs, Dowdy and his group [92] present a rather critical point of view referring to previous studies with CPP-siRNA-conjugates. They claim that the successful delivery described therein is solely the result of excess free peptide, which leads to additional complexation, and thereby cellular import of the siRNA. This is in accordance with Turner et al. [93], who were the first to observe that careful purification of CPP-antisense-conjugates abrogates their biological effect. Among other things, this might be the reason why most of the studies reporting on successful peptide-mediated delivery of siRNAs use a non-covalent complexation approach (confer Table 2).
In 2003, Simeoni et al. [94] were the first who non-covalently complexed siRNA with the peptide MPG. At a 1:10 ratio of negative nucleic acid to positive peptide charges a decrease in luciferase activity of about 80 % was detectable in HeLa or Cos-7 cells. This effect was further enhanced to about 90 % down-regulation by a mutation in the NLS sequence of the carrier peptide (MPGΔNLS), presumably due to an increased delivery to the cytoplasm, where RISC is localized. Recently, Veldhoen et al. [55] used a derivative of the MPG peptide for the delivery of siRNA, which will be described in the chapter “MPGα-mediated delivery of siRNA and steric block oligonucleotides”. Leng et al. [21] presented promising results with a prospect for cell-specific siRNA delivery. Different versions of a branched histidine/lysine-polymer (H3K8b) yielded up to 80 % knockdown of the target gene in several cell types. Structure-function studies revealed an important role of the composition of the histidine-rich domain as well as its position within the peptide and the branches for siRNA delivery, whereas size and surface charge did not have any effect. Furthermore, the toxicity was much lower than for the commercial cationic lipids Oligofectamine and LF2000. Finally, the attachment of the tripeptide RGD, an integrin-ligand, slightly enhanced siRNA delivery and turned this carrier into a cell-specific system. A similar concept has very recently been used by Kumar et al. [95] for a specific delivery approach into the brain. A peptide derived from rabies virus glycoprotein (RVG) interacts specifically with the nicotinic acetylcholine receptor (AchR) on neuronal cells to enable viral entry. The authors could show that the biotinylated form of the 29-amino-acid peptide (YTIWMPENPRPGTPCDIFTNSRGKRASNG) was taken up by neuronal cells. In order to transport nucleic acids with this vehicle, R9 was conjugated to RVG peptide. Systemic treatment of mice with siRNA in a non-covalent complex with this modified peptide promoted a highly specific cellular import of siRNA only into cells expressing AchR. Even more important, an antiviral siRNA treatment resulted in successful protection of mice against encephalitis caused by Japanese encephalitis virus (JEV). This is the first study to report on a non-toxic method to deliver siRNA across the blood brain barrier which could help to circumvent dangerous and ineffective injections into the brain. To date it presents one of the most promising tissue-specific delivery approaches which might be expandable to other in vivo applications.
Along these lines, most studies today are performed with the aim of CPP-mediated siRNA delivery in vivo. Although many of them are already showing promising results, e.g. concerning tumor-targeting and ocular delivery [96-98], this is beyond the scope of this review and will be discussed elsewhere in this issue [99,100].
With the aim to increase the endosomal escape of siRNAs after peptide-mediated delivery, Lundberg et al. [101] rationally modified penetratin to form a CPP (termed EB1) with improved endosomolytic properties. They achieved a pH-dependent conformational change of the peptide to a higher degree of helicity by the replacement of two basic amino acids with histidines and the N-terminal addition of six amino acids. In this study, several CPPs were compared in a non-covalent approach by measuring the overall cellular uptake via fluorescence and biological effect of siRNA targeted to luciferase mRNA. Penetratin- as well as TP10-mediated transfection did not lead to any silencing of luciferase gene expression, despite high amounts of intracellular siRNA [101] and in contrast to previous reports using siRNA-penetratin-conjugates [90] or TP10/DNA-complexes [102]. EB1-mediated delivery of 100 nM siRNA led to approximately 50 % reduction of luciferase activity. This silencing effect was slightly better than for bPrPp and in the same range as for MPGΔNLS, but still not as pronounced as for LF2000-mediated transfection of 100 nM siRNA. As it was described earlier, that addition of a pH-sensitive peptide derived from hemagglutinin (HA2) can promote endosomal escape [62], the authors linked HA2 to penetratin [101]. It turned out that although HA2-penetratin improved the silencing effect when coincubated with penetratin, EB1 was more potent than this combination of peptides. Together with confocal microscopy studies the authors concluded that the lack of biological effect after penetratin-mediated siRNA delivery is due to a lack of endosomal escape and that EB1 has a superior endosomolytic activity in comparison to HA2-penetratin.
Endoh et al. [103,104] very recently presented an innovative strategy, called CLIP-RNAi (i.e. CPP-linked RBP-mediated RNA internalization and photo-induced RNAi) combining delivery of a specific RNA sequence with enhanced photoinduced release of RNA from endosomes. This goal was accomplished by fusing the U1A RNA-binding domain (RBD) to the Tat peptide and extending the siRNA with a short stretch of nucleotides specifically recognized by this RBD. These complexes were efficiently internalized but exhibited a punctuate cytoplasmic localization pattern, indicative of endosomal entrapment. However, photostimulation of a fluorophore attached to the peptide led to a redistribution of complex into the cytosol followed by efficient RNAi-mediated gene silencing.
MECHANISMS OF ALTERNATIVE SPLICING
Human pre-mRNAs contain on average eight expressed sequences (exons) with an average length of 150 nt and up to 60 intervening sequences (introns) which can vary in length between 35 and 10,000 nt, therefore comprising up to 90 % of each transcriptional unit. In the nucleus, ribonucleoprotein complexes called spliceosomes recognize exon-intron boundaries and catalyze the precise removal of introns and subsequent joining of exons in a process called RNA splicing [105-107]. Additionally, each primary transcript can yield different mature RNAs through alternative splicing, thereby expanding the information content and versatility of the transcriptome, e.g. through the production of protein isoforms. A recent study of 10,000 human genes revealed, that at least 70 % of all multi-exon genes are alternatively spliced [108]. There are several different types of alternative splicing, amongst others affecting transcription start sites, splice sites, polyadenylation sites or even whole introns and exons. Disruptions of these intricate splicing patterns are tightly coupled with human pathophysiology, either as a determinant or a direct cause of disease or as a modifier of disease susceptibility and severity [109]. Among these diseases are β-thalassemia, cystic fibrosis, muscular dystrophies, Frasier syndrome, certain kinds of dementia and cancer. A more detailed description of the underlying mechanisms is beyond the focus of this article and can be found in a number of reviews [110-113]. López-Bigas et al. [114] proposed that 60 % of mutations that cause disease lead to splicing defects rather than changes in the amino acid sequence. Two common forms of mutations are depicted in Fig. (3B). On the one hand, a mutation in the splice donor can favor recognition of a cryptic splice donor and result in a mutant mRNA containing additional intronic sequences (part I). On the other hand, a mutation in the splice acceptor can lead to skipping of a whole exon and result in a shortened mRNA (part II). Both scenarios have been used in the context of antisense oligonucleotide-mediated approaches targeting alternative splicing. The use of antisense oligonucleotides interacting with mRNA to affect protein production goes back some 20 years [115,116]. Since then, three principle mechanisms have been exploited for this purpose (for a review see: [117,118]): (I) the oligonucleotide/RNA duplex forms a substrate for endogenous RNase H, leading to mRNA cleavage; (II) the oligonucleotide/RNA duplex prevents the productive assembly of the ribosomal complex or arrests a ribosomal complex already engaged in translation, in both cases affecting protein biosynthesis; (III) the oligonucleotide/RNA duplex alters pre-mRNA splicing in the nucleus. The following section will focus on the last approach with the aim to treat splicing disorders and give examples of possible applications for CPPs in this context.
LITERATURE OVERVIEW: CPP-MEDIATED STERIC BLOCK OLIGONUCLEOTIDE DELIVERY
Different forms of human β-thalassemia are caused by mutations within in the β-globin intron 2, which activate cryptic splice sites and thus lead to the formation of non-functional transcripts (Fig. (3B part I)). Those aberrantly used sites can be blocked by antisense steric block oligonucleotides, which leads to the synthesis of functional protein [119,120]. Kole and his group adopted this principle for the development of a splice correction assay [121]. In this model system, a firefly luciferase construct leads to the synthesis of inactive enzyme because the reporter gene pre-mRNA is interrupted by the human β-globin intron 2 containing an aberrant splice site. Upon binding of a steric block oligonucleotide, correct splicing is restored which in turn yields a functional luciferase protein. To achieve this, the oligonucleotide has to be delivered to the nucleus. Furthermore, only oligonucleotides that don’t activate RNase H are applicable [2], e.g. phosphorodiamidate morpholino oligomers (PMO, [122]), locked nucleic acids (LNA, [123]), peptide nucleic acids (PNA, [124]) or 2’-O-methyl-modified oligonucleotides (OMe). In addition to their inability to activate RNase H, most of these modifications confer higher affinity to the target RNA and increased resistance against enzymatic degradation than unmodified versions. Compared to RNAi-based model systems, this assay is less susceptible to side effects like cytotoxicity or off-target effects because the reporter gene activity is turned up rather than turned down. The splice correction assay has been successfully applied for the analysis of several carrier systems [24,93,125-135]. In the following section, selected examples will be presented, which are summarized in Table 3. In contrast to many non-covalent CPP-mediated siRNA delivery approaches, efficient splice correction was only achieved with conjugates of peptide and steric block oligonucleotide. Astriab-Fisher et al. [128] described delivery of OMe RNA phosphorothioate oligonucleotides linked via a disulfide bridge to Tat peptide and penetratin. A few hours after transfection, the CPP-oligonucleotide conjugates were detected both in cytoplasmic vesicles and in the nucleus and caused a dose-dependent increase in luciferase activity. These findings are in contrast to results of Turner et al. [93], who could not find a biological effect for several CPP-oligonucleotide conjugates in a HeLa cell assay for Tat-mediated transactivation of the HIV-1 long terminal repeat. The authors observed vesicular uptake but no nuclear import for their highly pure conjugates. Interestingly, the rate of uptake could be enhanced by addition of free CPP to the conjugates, though still no biological activity was detected. Based on these findings, Turner et al. [93] concluded that these free CPPs form complexes with CPP-cargo conjugates, which play a significant role in the uptake process. This is in accordance with observations by Meade et al. [92] for the uptake of CPP-siRNA conjugates described above. Moulton et al. [136] achieved correction of missplicing at low micromolar concentrations of a R9F2-PMO conjugate but not with complexes of peptide and PMO. The steric block activity of the R9F2-PMO conjugates could be further increased with longer spacers whereas variations in the conjugation chemistry did not result in any differences. Furthermore, transfection rates were higher than for conjugates with Tat peptide, penetratin or a Tat peptide analogue. Using the HIV-1 transactivation assay mentioned above, Turner et al. [137] could show that most CPP-oligo-nucleotide conjugates attained biologic activity only through co-administration of the endosomolytic substance chloroquine. Fluorescence microscopy analyses revealed that this treatment released fluorescently labeled conjugates from endosomal compartments into the nucleus. Besides the addition of chloroquine, different endosome disrupting strategies have been evaluated using the splice correction assay, for example co-treatment with endosome-disruptive peptides [129] or photochemical internalization [138] (see chapter “Strategies to enhance endosomal escape”). However, the most promising results have been achieved with two newly developed derivatives of classical CPPs (reviewed in [130]). The modification of oligoarginines with non-natural, uncharged amino acids [139] led, amongst others, to the peptide (R-Ahx-R)4, in which Ahx represents a six-atom aminohexanoic acid spacer. Abes et al. demonstrated that in contrast to Tat or oligoargine, PMO-conjugates of this peptide led to dose-dependent splice correction at low micromolar concentrations in the absence of endosomolytic agents. The underlying mechanism for this superior activity is not clear yet, as the uptake of (R-Ahx-R)4 constructs was less efficient than the uptake of Tat or oligoarginine constructs and also involved endocytotic routes [126]. The second peptide is a derivative of penetratin, to which six arginine residues were added at the N-terminus (R6Pen). R6Pen-PNA conjugates were shown to promote efficient splice correction at low concentrations and in the absence of endosomolytic agents [127]. Again, uptake of R6Pen-conjugates seemed to involve endocytosis and there was hardly any difference in splice correcting activity regardless of the nature of the linker used for conjugation, e.g. a stable thioether versus a reducible disulfide linker [130].
Table 3.
Examples for Peptide-Mediated Delivery of Steric Block Oligonucleotides
| Oligonucleotide Cargo | CPP/Delivery System | Application | Ref. |
|---|---|---|---|
| 2’-OMe phosphorothioate RNA | Tat49-60, penetratin | splice correction | [128] |
| RNA analogues | Tat48-58, penetratin, R6-penetratin, transportan, R9, R9F2 and further peptides | HIV-1 transactivation | [93] |
| PMO | R9F2, Tat peptide, penetratin | splice correction | [136] |
| PNA | Tat48-58, penetratin, transportan analogues, R9F2, R6-penetratin | HIV-1 transactivation | [137] |
| PMO | (R-Ahx-R)4 | splice correction | [126] |
| PNA | R6-penetratin | splice correction | [127] |
| PMO | (R-Ahx-R)4AhxB | exon skipping | [152] |
| PMO | R8-derivatives containing non-α amino acids | exon skipping | [153] |
| PNA | Tat, MSP, AAV6, AAV8 | exon skipping | [154] |
Part II of Fig. (3B) illustrates a phenomenon that represents a strategy for the treatment of Duchenne muscular dystrophy (DMD). DMD is a severe progressive neuromuscular disorder caused by several different mutations in the dystrophin gene that abolish the production of functional protein [140]. Depending on the location of the mutation, the corresponding exon is skipped by covering the responsible splice sites with steric block oligonucleotides. This allows the transcription of internally deleted, but largely functional, dystrophin proteins and converts a severe DMD into a milder Becker muscular dystrophy phenotype. A more detailed description of this approach and its application in a number of animal models can be found in several excellent recent reviews [141,145]. Successful systemic delivery of splice switching oligonucleotides with or without chemical modifications (PMO, LNA, OMe) has been accomplished via injection of naked nucleic acids [146,147], with the help of viral vectors [148], through re-implantation of ex vivo manipulated stem cells [149] or in combination with CPPs [150-154]. For the latter purpose, several studies were carried out with novel derivatives of arginine-rich peptides containing different numbers of non-α amino acids, e.g. aminohexanoic acid and/or β-alanine. These CPP-PMO conjugates showed higher serum stability, less endosomal trapping and led to efficient exon skipping in myoblasts and mice at lower dosages than the splice switching PMO alone [152,153]. Yin et al. [154] used a PNA-modified splice-switching oligonucleotide conjugated to Tat, a muscle specific peptide (MSP) or different functional domains of the adenovirus capsid protein VP1 (AAV6, AAV8) and examined exon skipping efficiency in vitro and in vivo. Surprisingly, both after transfection and intramuscular injection, the activity of these PNA-peptide conjugates was not significantly better than that achieved by naked neutral PNA, presumably due to endosomal trapping.
STRATEGIES FOR QUANTIFICATION
Intracellular trafficking represents one of the major limitations of current non-viral nucleic acid delivery approaches [155]. In other words a large percentage of intracellular cargo molecules are entrapped in vesicular compartments and thus will not trigger the desired effect. Moreover, degradation or retrograde transport might further reduce the number of active molecules. So in order to determine the overall efficacy of a given delivery approach it is essential to know the numbers of intact cargo molecules inside the cell along with the minimal numbers of molecules required to cause a particular effect. Based on such information one can easily calculate the percentage of bioactive molecules. In the following chapter we will briefly describe selected examples of variable suitability for a quantitative determination of nucleic acids in a cellular context.
In principle, either the peptide or the cargo can be labeled by a reporter group, e.g. a radioisotope [156] or a fluorophore [157]. Fluorescent peptides or cargos have been quantitatively evaluated by FACS [54] or fluorescence correlation microscopy (FCS) [158] or FRET [159]. In all cases, it is crucial to distinguish between internalized and membrane-associated signals. For this purpose, a simple wash step with just buffer is not sufficient to completely remove membrane-bound peptide/cargo-complexes [54,55]. Extracellularly bound complexes can be efficiently removed for example by enzymatic digestion with trypsin [160], acid wash [161] or heparin treatment [55,162]. Alternatively, discrimination between intra- and extracellular material is possible through chemical modification of extracellular components [163] or fluorescence quenching [164].
Having established that only intracellular signals are taken into account, it is still challenging to distinguish between intact and degraded forms of peptide or cargo. In two studies, a fluorescence-based quantification method was combined with either HPLC analysis [163] or “cell activity by capillary electrophoresis” [165] to verify the integrity of cargo and carrier.
Recently, a technique to measure cellular uptake of CPPs by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) was reported by the group of Burlina [166-168]. This quantification is based on the addition of an internal standard, i.e. a peptide with a stable isotope label. The method has been used to determine the amount and stability of intact internalized peptides, e.g. Penetratin, R9 and several novel CPPs, and can also be used for the quantification of peptidic cargoes, e.g. an inhibitor of protein kinase C [166].
Varga et al. [169-171] developed an “integrative systems” approach, combining quantitative experiments and computational modeling studies of vector uptake and trafficking kinetics, with the aim to take multiple potentially rate-limiting cellular and molecular processes into account. By applying their mathematical model to plasmid delivery with either Lipofectamine, several PEI-based vector formulations or an adenoviral vector, they could successfully predict experimentally observed effects and identify endosomal escape as the most important rate-limiting intracellular barrier for non-viral vectors. Recently, Zhou et al. [172] applied a similar strategy for the characterization of a novel lipopolymer (WLSP). This carrier shows an increased rate of endosomal escape compared to conventional PEI-based carriers.
With the aim to quantify rhodamine-labeled plasmid DNA in cellular compartments while avoiding problems arising from subcellular fractionation, like recovery and leakage, Akita et al. [173] developed a novel quantitative strategy, the confocal image-assisted three-dimensionally integrated quantification (CIDIQ) method. To distinguish endosomes/lysosomes and the nucleus from the cytosol, they were stained with LysoSensor DND-189 and Hoechst 33258, respectively, and sequential Z-series images were captured by CLSM. By applying this quantification method, the authors could show that due to a rapid endosomal escape, Lipofectamine Plus delivered more plasmid into the nucleus than R8 or stearylated R8. Hama et al. [174] used the same method in combination with TaqMan PCR to evaluate the uptake and intracellular distribution of plasmid DNA after delivery with viral as well as non-viral vectors. Due to superior cell surface binding, the efficiency of cellular uptake was significantly higher for Lipofectamine Plus than for adenovirus whereas intracellular trafficking, i.e. endosomal escape and nuclear transfer, were essentially the same. However, to achieve comparable transgene expression, 8000-fold higher intranuclear plasmid numbers were required in case of Lipofectamine Plus. This finding suggests a difference in nuclear transcription efficiency after non-viral delivery.
In another approach, Jiang et al. [175] extended the 3’ end of the sense strand of a siRNA with a nuclease-resistant DNA hairpin to obtain a so-called “crook” siRNA. This modification had no effect on RNAi-mediated reporter gene inhibition and served as a primer for a filling-in reaction followed by PCR. Parameters were chosen so that the initial rate of template amplification correlates with the initial concentration of the “crook” siRNA. Under these conditions, quantification of attomolar siRNA levels per cell was possible after liposomal transfections with Oligofectamine.
A highly sensitive method was developed by Overhoff et al. [176] for the detection of siRNA after phosphorothioate-stimulated uptake [177] and adapted for the quantification of siRNA or steric block oligonucleotides after non-covalent peptide-mediated delivery ([55] and Laufer et al., manuscript in preparation, see chapter “MPGα -mediated delivery of siRNA and steric block oligonucleotides”). This so-called liquid hybridization assay is based on the extraction of total cellular RNA and the subsequent hybridization in solution of a radioactively labeled probe which is complementary to the oligonucleotide to be detected. Finally, following PAGE analysis, absolute amounts of internalized oligonucleotide can be quantified with high accuracy down to ~10 molecules per cell using internal standards [55]. In addition to this outstanding sensitivity, no amplification step is needed and only intact oligonucleotides are taken into account.
MPGα -MEDIATED DELIVERY OF siRNA AND STERIC BLOCK OLIGONUCLEOTIDES – AN EXAMPLE OF A QUANTITATIVE EVALUATION
Considering the multitude of available CPPs, nucleic acid cargos and cellular as well as animal model systems, a comparison of different delivery strategies seems nearly impossible. In this context, we have for the first time undertaken a detailed side by side comparison of two different model systems using the peptide MPGα as delivery agent. In the following paragraph we present own experimental data to exemplarily illustrate particular aspects regarding current limitations of peptide-based delivery systems. In contrast to many other CPP procedures, which rely on covalent linkage of carrier and cargo, the peptide MPGα forms highly stable non-covalent complexes with nucleic acids, displaying binding constants in the low nanomolar range ([55], A. Trampe, unpublished data). The high flexibility of this non-covalent approach can be exploited to easily transport a wide variety of nucleic acid cargos without having to synthesize a new construct for each oligonucleotide. We have used MPGα for the delivery of siRNAs in the context of an RNAi-based reporter system and for the delivery of steric block oligonucleotides in the context of the splice correction assay described above [121]. After MPGα -mediated transfection of a luciferase-targeted siRNA, we observed strong inhibition of reporter gene readout with an IC50 in the subnanomolar range [55]. After MPGα -mediated transfection of a luciferase-targeted steric block oligonucleotide (further on also referred to as ON-705), we observed a moderate up-regulation of reporter gene readout, representative of low splice correcting activity (Laufer et al., manuscript in preparation). One possible explanation for the different degree of reporter gene regulation could be the different intracellular target sites, i.e. the cytoplasm for siRNAs and the nucleus for splice correction oligonucleotides. To attain more information about the subcellular localization of MPGα -oligonucleotide complexes, we performed confocal laser scanning as well as conventional fluorescence microscopy studies with fluorescently labeled siRNAs or steric block oligonucleotides. In both cases, a punctuate non-homogenous distribution of the nucleic acids inside the cells was observed. This pattern is indicative of an accumulation of nucleic acids in endocytotic vesicles, which was verified by coincubation with LysoSensor (Fig. (4A)). A quantitative computational analysis of fluorescence microscopy data yielded an average of approximately 50 % colocalization between endosomes and siRNA (Fig. (4B)). In contrast to earlier assumptions that CPPs directly traverse the lipid bilayer, it has commonly become accepted that for most peptide-cargo combinations endocytosis plays a major role in cellular uptake. As described above and in the chapter “Strategies to enhance endosomal escape” below, administration of endosome disruptive substances like chloroquine can greatly increase endosomal release of trapped nucleic acids. Chloroquine is a weak base, non-charged at neutral pH but charged at pH 5.5 [178]. It is able to pass easily through membranes in its uncharged form, but becomes protonated and accumulates within acidic vesicles in its positively charged, membrane-impermeable form. Although its exact mode of action has not yet been resolved, it is generally accepted that chloroquine works via prevention of endosome acidification which in turn increases the residence time of cargo within the endosomes eventually resulting in a higher probability of transfer to the cytoplasm. Fluorescence microscopy analyses in the presence of 100 µM chloroquine yielded two quite contrary outcomes. While the localization of siRNA did not change after addition of chloroquine (Fig. (4C)), for the steric block oligonucleotide the picture changed completely (Fig. (4D)). In the latter case, a diffuse fluorescence all over the cytoplasm with an accumulation of ON-705 in the nucleus could be observed. Nonetheless, considerable amounts of nucleic acid molecules were still visible as a punctual pattern, which indicates that the chloroquine treatment liberates only a certain fraction. On the whole, these qualitative observations are in full agreement with the observed biologic effects in the absence or presence of chloroquine (Fig. (5A)). For MPGα -mediated transfection of siRNA, even under conditions where the amount of bio-available siRNA was severely limited, only a minor increase in RNAi (ca. 30 %) was measurable. For MPGα -mediated transfection of steric block oligonucleotide, on the other hand, a dramatic increase of reporter gene up-regulation by a factor of 50 - 100 was observed. However, in both cases, the overall uptake did not change upon incubation with chloroquine (Fig. (5B)), which proves that the endosomolytic substance does not interfere with uptake but leads to a re-distribution of internalized nucleic acids. The underlying mechanism for the different effects triggered by chloroquine in case of peptide/siRNA and peptide/steric block oligonucleotide complexes remains unclear. Though, this is a good example that the cargo can substantially affect the properties and thereby intracellular trafficking of a particular carrier system.
Fig. (4).

Confocal (A) and semiconfocal (C and D) microscopic analyses of intracellular RNA localization following peptide-mediated transfection. (A) Living ECV304 cells were transfected for 4 h with 4.2 µM MPGα and 10 nM Cy3-labeled siRNA. Subsequently cells were incubated with 5 µM LysoSensor (Invitrogen). (I) Overlay of the phase contrast image with all fluorescence channels, (II) Cy3-channel (siRNA - red), (III) LysoSensor channel (green) and (IV) overlay of red and green channel. Yellow dots indicate colocalization. (B) Quantitative analysis of siRNA/LysoSensor colocalization using the program ImageJ. For each data point 5 cells were analyzed and averaged. (C) Living ECV-GFP-Nuc cells [55] were transfected for 4 h with 4.2 µM MPGα and 10 nM Cy3-labeled siRNA in the absence and presence of 100 µM chloroquine (CQ).The left images show the red (siRNA) channels followed by an overlay of the red and green (nuclei) channels. The right panel shows plots of the intensity profiles for the green channel and the Cy3-channel along the line indicated in the figure to the left. (D) Living HeLa Luc/705 cells were transfected for 4 h with 2.5 µM MPGα and 278 nM Cy3-labeled ON-705 PTO in the absence and presence of 100 µM chloroquine (CQ). The very left images show an overlay of the phase contrast with all fluorescence channels followed by the red (steric block) and an overlay of the red and blue (nuclei) channels. The right panel shows plots of the intensity profiles for the DAPI-channel (blue) and the Cy3-channel (red) along the line indicated in the figure to the left. Cell nuclei are either stained via GFP-Nuc (green) or with DAPI (blue).
Fig. (5).
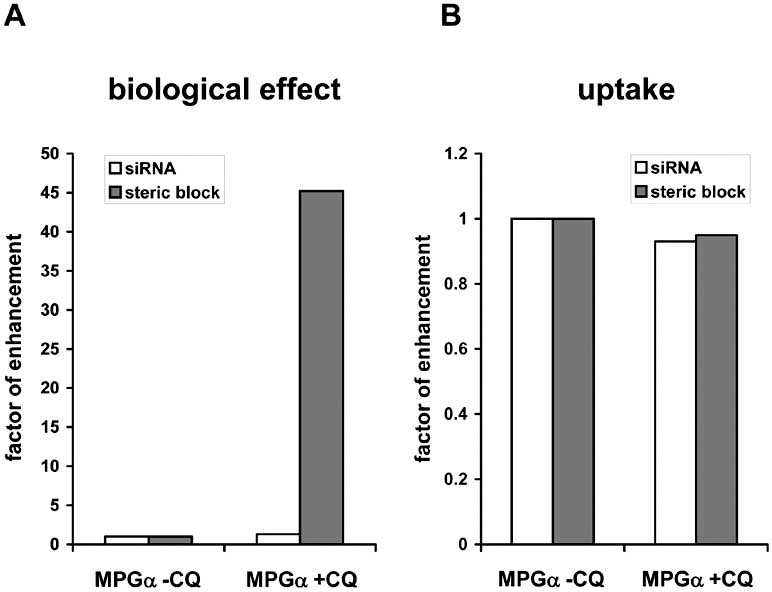
Influence of chloroquine on MPGα -mediated delivery of siRNA and steric block oligonucleotide. Cells were transfected with complexes of 2.1 µM MPGα and 1 nM siRNA or 2.5 µM MPGα and 278 nM steric block oligonucleotide either in the presence or absence of 100 µM chloroquine (CQ). 24 h later, RNA interference and splice correction were measured via reporter gene activity (A) and uptake was determined using the liquid hybridization protocol [55] (B). The factor of enhancement was calculated from three independent experiments based on the situation without CQ.
Ultimately, to assess the overall efficacy of this carrier system, we were interested to elucidate which percentage of molecules taken up after MPGα -mediated delivery is biologically active. To derive such information, the exact intracellular amount of intact oligonucleotide, the corresponding reporter signal and the minimal number of molecules necessary to trigger a specific degree of reporter gene modulation have to be known. For the quantification of internalized cargo, we adapted a highly sensitive method first described by Overhoff et al. [176], enabling us to detect intracellular oligonucleotide amounts down to ≥ 10 copies per cell [55]. The method is based on the liquid hybridization of a radioactively labeled probe with the corresponding oligonucleotide in cellular lysates. In this context it should be noted that a stringent heparin wash following the transfection procedure is crucial to avoid an overestimation of intracellular nucleic acid molecules due to complexes attached to the outside of the cell membrane [54,55]. In order to correlate the numbers derived from the quantification experiments with the minimal number of molecules essential to trigger the observed effect, an independent assay had to be established. The gold standard in this case is microinjection as this technique enables one to deliver definite amounts of nucleic acids with a high degree of bioavailability into the cytoplasm or the nucleus of a mammalian cell along with a low toxicity profile and great accuracy. Considering that after cytoplasmic microinjection only 12 siRNA molecules are sufficient for half-maximal inhibition of reporter gene expression, one can estimate that of the 10,000 molecules measured after MPGα -mediated transfection, only ca. 0.1 % are biologically active (Table 4). For the splice correction assay, the numbers are different in terms of absolute numbers but the outcome remains the same. Compared to the 300,000 molecules sufficient for maximal splice correction after nuclear microinjection, of the 70,000,000 molecules required following peptide-mediated delivery only ca. 0.5 % are biologically active (Table 5). In both cases >> 99 % of internalized oligonucleotides are most likely retained in endosomes and subsequently degraded in lysosomes after peptide-mediated delivery. Frankly, this is a sobering result and puts in numbers how much room for improvement there actually is. Though it certainly is not legitimate to generalize these findings, there are countless reports in the literature suggesting similar limitations for the majority of non-viral strategies.
Table 4.
Summary of Quantitative Studies Concerning Peptide-Mediated siRNA Uptake
| Mode of Delivery | Molecules Per Cell for Half-Maximal Inhibition |
|---|---|
| LF2000-mediated transfection | ~300 |
| MPGα-mediated transfection | ~10,000 |
| cytoplasmic microinjection | ~12 |
Transfections were performed with 2.1 µM MPGα and 1 nM siRNA or 10 µg/ml LF2000 and 0.02 nM siRNA, i.e. in the range of the IC50 value [55]. Quantification was performed after 24 h according to the liquid hybridization protocol [55]. Molecules per cell were calculated based on the cell number seeded for transfection. For microinjection experiments, molecules per cell were calculated on the basis of the injection volume.
Table 5.
Summary of Quantitative Studies Concerning Peptide-Mediated Steric Block Oligonucleotide Uptake
| Mode of Delivery | Molecules Per Cell for Maximal Splice Correction |
|---|---|
| LF2000-mediated transfection | ~7,000,000 |
| MPGα -mediated transfection | ~70,000,000 |
| nuclear microinjection | ~300,000 |
Transfections were performed with 2.5 µM MPGα or 7 µg/ml LF2000 and 278 nM steric block oligonucleotide, respectively, to achieve maximal splice correction. Quantification was performed after 24 h according to the liquid hybridization protocol ([55] and Laufer et al., manuscript in preparation). Molecules per cell were calculated based on the cell number seeded for transfection. For microinjection experiments, molecules per cell were calculated on the basis of the injection volume.
According to actual conceptions, siRNA enters a multiple-turnover pathway with one siRNA molecule capable of RISC-mediated cleavage of 50 or more mRNA molecules [79]. Even though the fate of steric block oligonucleotides is not really clear, it can be assumed that per splicing event one molecule is used up and translated into a functional mRNA molecule (e.g. single-turnover pathway). As a result the number of molecules needed to trigger an apparent effect is much higher in the splice correction assay compared to the RNAi-based reporter system (confer Table 4 and Table 5). On the other hand, being a single-turnover pathway, the splice correction assay should be much more sensitive to even minor changes in intracellular steric block oligonucleotide concentrations whereas the catalytic nature of the multiple-turnover RNAi mechanism might mask such small variations. This is in accordance with the data described above.
Taken together, based on the results presented as well as unpublished data (Laufer et al., manuscript in preparation), the splice correction assay appears to be the superior tool for a quantitative assessment of nucleic acid delivery strategies.
STRATEGIES TO ENHANCE ENDOSOMAL ESCAPE
As outlined above, endosomal release is one of the major rate-limiting steps for cellular delivery of macromolecules via cationic lipids, polyplexes and especially CPPs. In the following chapter we will present some examples of how to increase endosomal release.
Endosome-disrupting substances, like chloroquine, calcium or sucrose, were used to significantly enhance the activity of antisense PNA oligonucleotides conjugated to Tat, oligoarginines or oligolysines [125,179,180]. This effect did not result from increased uptake, but rather improved bioavailability in the cytoplasm or nucleus after endosomal escape. Takeuchi et al. [181] showed that by incubation of the target cells with pyrenebutyrate, delivery of arginine-rich peptides could be shifted from endocytic uptake to direct membrane translocation, yielding a rapid distribution of the peptide throughout the cytoplasm, even at 4 °C. Pyrenebutyrate acts as a counteranion and, by interacting with the positively charged peptide, increases the overall hydrophobicity, thereby facilitating a direct translocation through the lipid bilayer, as earlier shown with artificial membranes [182]. This method, which works only in the absence of a medium or serum, was successfully applied for administration of a fluorescent protein and an apoptosis-inducing peptide into dividing as well as non-dividing cells [181]. However, in general the strategies described above are not feasible for in vivo applications, due to high cytotoxicity or other undesirable secondary effects.
The imidazole group of histidine (His) can absorb protons in the acidic environment of the endosome, leading to osmotic swelling, membrane disruption and eventually nucleic acid escape. Accordingly, Lo et al. [183] modified Tat, which can bind and condense DNA through ionic interactions but has no acidic residues that can promote endosomal release, with different numbers of His residues. Highest reporter gene expression could be achieved after plasmid delivery with a Tat peptide covalently fused to 10 His residues (Tat-10H). Insertion of two additional cysteine residues into Tat-10H further enhanced stability of peptide/DNA complexes and transgene expression through formation of interpeptide disulfide bonds. Youngblood et al. [184] evaluated the influence of the stability of arginine-rich peptide PMO conjugates on cellular uptake and antisense activity. They could show that the stability is affected by the amino acid composition and the type of linkage to the cargo. Moreover, they found that degraded fragments could not escape anymore from endosomal or lysosomal compartments.
Another concept makes use of photosensitive substances, which induce the release of macromolecules from vesicles by light exposure. This so-called photochemical internalization (PCI) has, in the past, been used for intracellular delivery of a large variety of macromolecules (reviewed in [185]) and, more recently, for the endosomal release of nucleic acids after delivery mediated by liposomes [186,187], polyplexes [188] or CPPs [138,189]. Shiraishi et al. [138] investigated the biological activity of PNAs conjugated either to Tat, R7 or KLA-peptide in combination with a PCI treatment. Depending on the peptide, nuclear as well as cytosolic antisense effects could be enhanced by up to two orders of magnitude. Similar results were presented by Folini et al. [189] for a PNA targeting human telomerase reverse transcriptase conjugated to Tat. In both studies, lower nucleic acid doses were sufficient, thereby reducing the probability of off-target effects. In light of encouraging data from ongoing anticancer clinical trials employing photodynamic therapy [190,191], an in vivo application of PCI seems feasible and will be discussed in more detail by Oliveira et al. [192] in this issue. Furthermore, target specificity could be increased by local illumination of cells or tissue.
Viral fusion proteins drive the fusion process between the viral membrane and the endosomal host cell membrane in a pH-dependent manner, which is required to translocate the viral genome into the cytoplasm after receptor-mediated endocytosis. Fusogenic peptides, usually hydrophobic, rich in glycine residues and found at the amino terminus of these proteins, were shown to have membrane perturbing and lipid mixing activities [193]. Many well studied representatives of this group are derived from the fusion sequence of influenza virus HA or HIV-1 gp41 [194] and have been used to improve the transfection efficiency of non-viral delivery systems [195].
Addition of the influenza-derived dimeric peptide diINF-7 to LF2000/siRNA complexes had no effect on the particle size of ca. 120 nm, but significantly improved gene silencing activity of siRNAs targeting the epidermal growth factor receptor or the K-ras oncogene [196]. Similar results were obtained for plasmid delivery through addition of a fusogenic peptide derived from herpes simplex virus glycoprotein H to Lipofectamine/DNA complexes [197]. To the same end, Futaki et al. [198] used the peptide GALA, which was specially designed to mimic the function of viral fusion sequences, together with various commercially available cationic liposomes. Although they could not detect significant differences in cellular localization, plasmid transfection efficiency was increased and liposomal dosage could be reduced.
PEI covalently modified with the HIV-1 gp41-derived peptide HGP led to a 38-fold increase of gene expression after plasmid DNA delivery and also enhanced siRNA-mediated knockdown of GAPDH by approximately 2-fold [199]. 30 % of cells incubated with PEI-HGP polyplexes showed not only the punctuate plasmid DNA staining observed with PEI polyplexes alone, but also a diffuse fluorescence throughout the cell, indicative of endosomal release of vectors. This would explain the observed increase in transfection efficiency, since the overall uptake was unaffected.
Wadia et al. [62] were the first to use the influenza hemagglutinin-derived fusogenic peptide HA2 in combination with a CPP. Delivery of a Tat-HA2 conjugate with increasing concentrations of a Tat-Cre conjugate enhanced reporter protein activity, presumably through an increased release from macropinosomes. The same fusogenic peptide, linked to a polyarginine-p53 conjugate, promoted release of p53 from macropinosomes and subsequent translocation to the nucleus, accompanied by an enhanced anti-cancer effect [200].
CONCLUSIONS AND FUTURE PROSPECTS
The development of delivery systems for therapeutic oligonucleotides is a fast growing field. Owing to the enormous potential of short nucleic acids as alternative drugs such a growth is not unexpected. Besides viral vectors there is a highly diverse and constantly increasing number of non-viral systems evolving. However, despite considerable progress achieved in recent years, even the most advanced systems either lack the efficiencies required for downstream drug development or do show a substantial degree of toxicity or both. Of the many factors which limit their use, cellular uptake of the cargo/carrier complexes and subsequent intracellular trafficking to reach the target site are the most important. In addition to such essential considerations there are various additional parameters to be taken into account like serum stability, pharmacokinetic features and tissue barriers as well as target cell specificity.
Now the question arises where to start optimizing a given delivery system. Currently, there is a clear trend towards in vivo testing. In principle such a development is a step in the right direction since many of the experimental data derived from artificial cell tissue culture systems with established cell lines are not applicable to the in vivo situation. On the other hand, it is questionable to what extent such animal experiments will eventually pay off as long as important fundamental problems remain largely unsolved. As outlined above, our quantitative studies along with microscopic analyses of siRNAs and steric block oligonucleotides using either a peptide or a commercially availably cationic lipid as carrier clearly show that less than 0.1% - 5% of molecules taken up are involved in a biological response, i.e. RNAi-mediated down regulation or splice correction-mediated up regulation of reporter gene activity. Evidently, the vast majority of internalized cargo never reaches the target. This implies that uptake per se is not the limiting factor here. Although there are no such detailed quantitative numbers available in the literature for other systems, there are countless reports showing that to various degrees this applies to almost any non-viral delivery approach currently available. Taken together, if one would succeed to optimize intracellular trafficking this holds the potential to boost overall efficacy by up to 3 orders of magnitude. Moreover, it is reasonable to assume that such fundamental cellular restrictions can be adequately investigated in tissue culture without the use of animal models. So it might be worthwhile to reconsider the concept of maybe premature in vivo testing by moving backwards one step and first optimizing the systems with regard to intracellular limitations before dealing with the next level of complexity. Accordingly, in vitro model systems like the ones described above for siRNAs or steric block oligonucleotides are valuable tools to study particular aspects of nucleic acid delivery. However, currently available data are based on studies using a variety of different cell lines and techniques, which renders a direct comparison of different delivery approaches impossible. In this context it would be highly desirable to introduce standardized protocols for in vitro testing (e.g. the splice correction system developed by Kole and coworkers [121]) together with methods for detailed quantitative analyses (e.g. the liquid hybridization protocol [55,176]). This would facilitate a direct quantitative comparison of exceedingly diverse approaches on at least the cellular level.
One reason for the problem with intracellular trafficking of oligonucleotides arises from the mode carrier/cargo complexes are taken up by cells. Today it is well established that the majority of these complexes are taken up via endosomal pathways and therefore end up in vesicular compartments from which they have to escape in order to reach their target. Although there are many attempts reported to trigger endosomal escape by various strategies, they either proved to be toxic or did not achieve sustained success. Additionally, there might be a further reason for the encountered difficulties to overcome these intracellular barriers. It is not unreasonable to speculate that during evolution cells might have evolved mechanisms to avoid large amounts of foreign nucleic acids freely floating in the cytoplasm and thus safely contain them in vesicular compartments where they are eventually degraded. Alternatively they might be exported by retrograde transport. Co-evoluting viruses evidently have developed strategies to circumvent such defense mechanisms. So it might be somewhat naive to expect that a rather simple man-made carrier system is capable to efficiently overcome such an intrinsic barrier. Current developments towards more complex and elaborate carrier systems take into account such considerations. In any case it appears there is no simple solution for this problem.
In conclusion, despite significant progress in the field of nucleic acid delivery in vitro as well as in vivo, there still is a long way to go before this will become a standard procedure in the clinic. Certain problems like endosomal escape are known for more than twenty years and still far from being resolved. In order to develop new strategies, more information about intracellular processes involved in nucleic acid trafficking is needed. Moreover, it would be desirable if the field would move from a qualitative description towards a quantitative evaluation preferentially using standardized model systems. This would allow for comparison of different approaches with one another. While animal studies are inevitable in the long run, there still is a lot of room for improvement on the cellular level. So it might be worthwhile to fathom how far we can push the different systems on this level.
ACKNOWLEDGEMENTS
We apologize to those authors whose work was not cited directly owing to space limitations. We thank Alexander Trampe for microscopic images, Anna Lena Recke, Sandra Veldhoen, Georg Sczakiel and members of his group for fruitful discussions, Silke Degen for critically reading the manuscript and our co-workers in the EC project ’Peptides in Drug Delivery’, Annette G. Beck-Sickinger, Ernest Giralt, Fréderic Heitz, Christian LeGrimellec and Hans-Peter Merkle for constructive collaborations. T.R. acknowledges funding by EC-grants QLK2-2001-01451 and LSHG-CT-2003-503480.
ABBREVIATIONS
- AchR
= Acetylcholine receptor
- bPrPp
= Bovine prion protein derived peptide
- CLSM
= Confocal laser scanning microscopy
- CPP
= Cell-penetrating peptide
- CQ
= Chloroquine
- DMD
= Duchenne muscular dystrophy
- FACS
= Fluorescence assisted cell sorting
- FCS
= Fluorescence correlation spectroscopy
- GFP
= Green fluorescent protein
- gp41
= Glycoprotein 41
- HA
= Hemagglutinin
- HIV
= Human immunodeficiency virus
- IL
= Interleukin
- IFN
= Interferon
- JEV
= Japanese encephalitis virus
- LF
= Lipofectamine™
- LF2000
= Lipofectamine™ 2000
- MALDI-TOF MS
= Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry
- MAP
= Model amphipathic peptide
- MEND
= Multifunctional envelope-type nano device
- NLS
= Nuclear localization sequence
- OMe
2’-O-Methyl
- PCI
= Photochemical internalization
- PEG
= Polyethylene glycol
- PEI
= Polyethyleneimine
- PMO
= Phosphorodiamidate morpholino oligomer
- PNA
= Peptide nucleic acid
- POD
= Peptide for ocular delivery
- PTD
= Protein transduction domains
- PTGS
= Post-transcriptional gene silencing
- RBD
= RNA-binding domain
- RNAi
= RNA interference
- RVG
= Rabies virus glycoprotein
- siRNA
= Small interfering RNA
- shRNA
= Short hairpin RNA
- STR-R8
= Stearyl-R8
- TFO
= Triplex forming oligonucleotide
- TNF
= Tumour necrosis factor
- TP10
= Transportan 10
REFERENCES
- 1.Opalinska JB, Gewirtz AM. Nucleic-acid therapeutics: basic principles and recent applications. Nat Rev Drug Discov. 2002;1:503–14. doi: 10.1038/nrd837. [DOI] [PubMed] [Google Scholar]
- 2.Eckstein F. The versatility of oligonucleotides as potential therapeutics. Expert Opin Biol Ther. 2007;7:1021–34. doi: 10.1517/14712598.7.7.1021. [DOI] [PubMed] [Google Scholar]
- 3.Kootstra NA, Verma IM. Gene therapy with viral vectors. Annu Rev Pharmacol Toxicol. 2003;43:413–39. doi: 10.1146/annurev.pharmtox.43.100901.140257. [DOI] [PubMed] [Google Scholar]
- 4.Verma IM, Weitzman MD. Gene therapy: twenty-first century medicine. Annu Rev Biochem. 2005;74:711–38. doi: 10.1146/annurev.biochem.74.050304.091637. [DOI] [PubMed] [Google Scholar]
- 5.Raper SE, Yudkoff M, Chirmule N, Gao GP, Nunes F, Haskal ZJ, et al. A pilot study of in vivo liver-directed gene transfer with an adenoviral vector in partial ornithine transcarbamylase deficiency. Hum Gene Ther. 2002;13:163–75. doi: 10.1089/10430340152712719. [DOI] [PubMed] [Google Scholar]
- 6.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–9. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 7.Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP, et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. 2003;80:148–58. doi: 10.1016/j.ymgme.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 8.Check E. Gene therapy put on hold as third child develops cancer. Nature. 2005;433:561. doi: 10.1038/433561a. [DOI] [PubMed] [Google Scholar]
- 9.Torchilin VP, Rammohan R, Weissig V, Levchenko TS. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc Natl Acad Sci USA. 2001;98:8786–91. doi: 10.1073/pnas.151247498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Preuss M, Tecle M, Shah I, Matthews DA, Miller AD. Comparison between the interactions of adenovirus-derived peptides with plasmid DNA and their role in gene delivery mediated by liposome-peptide-DNA virus-like nanoparticles. Org Biomol Chem. 2003;1:2430–8. doi: 10.1039/b302361c. [DOI] [PubMed] [Google Scholar]
- 11.Read ML, Bremner KH, Oupicky D, Green NK, Searle PF, Seymour LW. Vectors based on reducible polycations facilitate intracellular release of nucleic acids. J Gene Med. 2003;5:232–45. doi: 10.1002/jgm.331. [DOI] [PubMed] [Google Scholar]
- 12.Torchilin VP, Levchenko TS, Rammohan R, Volodina N, Papahadjopoulos-Sternberg B, D'Souza GG. Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome-DNA complexes. Proc Natl Acad Sci USA. 2003;100:1972–7. doi: 10.1073/pnas.0435906100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hyndman L, Lemoine JL, Huang L, Porteous DJ, Boyd AC, Nan X. HIV-1 Tat protein transduction domain peptide facilitates gene transfer in combination with cationic liposomes. J Control Release. 2004;99:435–44. doi: 10.1016/j.jconrel.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 14.Futaki S, Masui Y, Nakase I, Sugiura Y, Nakamura T, Kogure K, et al. Unique features of a pH-sensitive fusogenic peptide that improves the transfection efficiency of cationic liposomes. J Gene Med. 2005;7:1450–8. doi: 10.1002/jgm.796. [DOI] [PubMed] [Google Scholar]
- 15.Read ML, Singh S, Ahmed Z, Stevenson M, Briggs SS, Oupicky D, et al. A versatile reducible polycation-based system for efficient delivery of a broad range of nucleic acids. Nucleic Acids Res. 2005;33:e86. doi: 10.1093/nar/gni085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torchilin VP. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev. 2008;60:548–58. doi: 10.1016/j.addr.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 17.Kilk K, El-Andaloussi S, Järver P, Meikas A, Valkna A, Bartfai T, et al. Evaluation of transportan 10 in PEI mediated plasmid delivery assay. J Control Release. 2005;103:511–23. doi: 10.1016/j.jconrel.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 18.Midoux P, Monsigny M. Efficient gene transfer by histidylated polylysine/pDNA complexes. Bioconjug Chem. 1999;10:406–11. doi: 10.1021/bc9801070. [DOI] [PubMed] [Google Scholar]
- 19.Chen QR, Zhang L, Stass SA, Mixson AJ. Branched co-polymers of histidine and lysine are efficient carriers of plasmids. Nucleic Acids Res. 2001;29:1334–40. doi: 10.1093/nar/29.6.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ritter W, Plank C, Lausier J, Rudolph C, Zink D, Reinhardt D, et al. A novel transfecting peptide comprising a tetrameric nuclear localization sequence. J Mol Med. 2003;81:708–17. doi: 10.1007/s00109-003-0483-2. [DOI] [PubMed] [Google Scholar]
- 21.Leng Q, Scaria P, Zhu J, Ambulos N, Campbell P, Mixson AJ. Highly branched HK peptides are effective carriers of siRNA. J Gene Med. 2005;7:977–86. doi: 10.1002/jgm.748. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z, Li M, Cui D, Fei J. Macro-branched cell-penetrating peptide design for gene delivery. J Control Release. 2005;102:699–710. doi: 10.1016/j.jconrel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 23.Bayele HK, Ramaswamy C, Wilderspin AF, Srai KS, Toth I, Florence AT. Protein transduction by lipidic peptide dendrimers. J Pharm Sci. 2006;95:1227–37. doi: 10.1002/jps.20606. [DOI] [PubMed] [Google Scholar]
- 24.Kang H, Delong R, Fisher MH, Juliano RL. Tat-Conjugated PAMAM Dendrimers as Delivery Agents for Antisense and siRNA Oligonucleotides. Pharm Res. 2005;22:2099–106. doi: 10.1007/s11095-005-8330-5. [DOI] [PubMed] [Google Scholar]
- 25.Kale AA, Torchilin VP. Design, synthesis, and characterization of pH-sensitive PEG-PE conjugates for stimuli-sensitive pharmaceutical nanocarriers: the effect of substitutes at the hydrazone linkage on the ph stability of PEG-PE conjugates. Bioconjug Chem. 2007;18:363–70. doi: 10.1021/bc060228x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kale AA, Torchilin VP. "Smart" drug carriers: PEGylated TATp-modified pH-sensitive liposomes. J Liposome Res. 2007;17:197–203. doi: 10.1080/08982100701525035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khalil IA, Kogure K, Futaki S, Hama S, Akita H, Ueno M, et al. Octaarginine-modified multifunctional envelope-type nanoparticles for gene delivery. Gene Ther. 2007;14:682–9. doi: 10.1038/sj.gt.3302910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soundara Manickam D, Oupický D. Multiblock reducible copolypeptides containing histidine-rich and nuclear localization sequences for gene delivery. Bioconjug Chem. 2006;17:1395–403. doi: 10.1021/bc060104k. [DOI] [PubMed] [Google Scholar]
- 29.Rahbek UL, Howard KA, Oupický D, Manickam DS, Dong M, Nielsen AF, et al. Intracellular siRNA and precursor miRNA trafficking using bioresponsive copolypeptides. J Gene Med. 2008;10:81–93. doi: 10.1002/jgm.1120. [DOI] [PubMed] [Google Scholar]
- 30.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–93. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 31.Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 1988;55:1179–88. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- 32.Joliot AH, Triller A, Volovitch M, Pernelle C, Prochiantz A. alpha-2,8-Polysialic acid is the neuronal surface receptor of antennapedia homeobox peptide. New Biol. 1991;3:1121–34. [PubMed] [Google Scholar]
- 33.Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, et al. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci USA. 1994;91:664–8. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allinquant B, Hantraye P, Mailleux P, Moya K, Bouillot C, Prochiantz A. Downregulation of amyloid precursor protein inhibits neurite outgrowth in vitro. J Cell Biol. 1995;128:919–27. doi: 10.1083/jcb.128.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–72. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 36.Lindgren M, Hällbrink M, Prochiantz A, Langel Ü. Cell-penetrating peptides. Trends Pharmacol Sci. 2000;21:99–103. doi: 10.1016/s0165-6147(00)01447-4. [DOI] [PubMed] [Google Scholar]
- 37.Zorko M, Langel Ü. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv Drug Deliv Rev. 2005;57:529–45. doi: 10.1016/j.addr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 38.Langel Üe. Handbook of Cell-Penetrating Peptides. Boca Raton: CRC Press; 2006. [Google Scholar]
- 39.Patel LN, Zaro JL, Shen WC. Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharm Res. 2007;24:1977–92. doi: 10.1007/s11095-007-9303-7. [DOI] [PubMed] [Google Scholar]
- 40.Morris MC, Deshayes S, Heitz F, Divita G. Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol Cell. 2008;100:201–17. doi: 10.1042/BC20070116. [DOI] [PubMed] [Google Scholar]
- 41.Foged C, Nielsen HM. Cell-penetrating peptides for drug delivery across membrane barriers. Expert Opin Drug Deliv. 2008;5:105–17. doi: 10.1517/17425247.5.1.105. [DOI] [PubMed] [Google Scholar]
- 42.Veldhoen S, Laufer SD, Restle T. Recent Developments in Peptide-based Nucleic Acid Delivery. Int J Mol Sci. 2008;9:1276–320. doi: 10.3390/ijms9071276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zatsepin TS, Turner JJ, Oretskaya TS, Gait MJ. Conjugates of oligonucleotides and analogues with cell penetrating peptides as gene silencing agents. Curr Pharm Des. 2005;11:3639–54. doi: 10.2174/138161205774580769. [DOI] [PubMed] [Google Scholar]
- 44.Deshayes S, Morris M, Heitz F, Divita G. Delivery of proteins and nucleic acids using a non-covalent peptide-based strategy. Adv Drug Deliv Rev. 2008;60:537–47. doi: 10.1016/j.addr.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 45.Crombez L, Morris MC, Deshayes S, Divita G. Peptide-based Nanoparticle for ex vivo and in vivo drug delivery. Curr Pharm Des. doi: 10.2174/138161208786898842. In press. [DOI] [PubMed] [Google Scholar]
- 46.Derossi D, Calvet S, Trembleau A, Brunissen A, Chassaing G, Prochiantz A. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J Biol Chem. 1996;271:18188–93. doi: 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- 47.Morris MC, Vidal P, Chaloin L, Heitz F, Divita G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997;25:2730–6. doi: 10.1093/nar/25.14.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–7. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 49.Schwarze SR, Dowdy SF. In vivo protein transduction: intracellular delivery of biologically active proteins, compounds and DNA. Trends Pharmacol Sci. 2000;21:45–8. doi: 10.1016/s0165-6147(99)01429-7. [DOI] [PubMed] [Google Scholar]
- 50.Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, et al. Arginine-rich peptides. An abundant source of membranepermeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276:5836–40. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 51.Pichon C, Monsigny M, Roche AC. Intracellular localization of oligonucleotides: influence of fixative protocols. Antisense Nucleic Acid Drug Dev. 1999;9:89–93. doi: 10.1089/oli.1.1999.9.89. [DOI] [PubMed] [Google Scholar]
- 52.Lundberg M, Johansson M. Is VP22 nuclear homing an artifact? Nat Biotechnol. 2001;19:713–4. doi: 10.1038/90741. [DOI] [PubMed] [Google Scholar]
- 53.Lundberg M, Johansson M. Positively charged DNA-binding proteins cause apparent cell membrane translocation. Biochem Biophys Res Commun. 2002;291:367–71. doi: 10.1006/bbrc.2002.6450. [DOI] [PubMed] [Google Scholar]
- 54.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, et al. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278:585–90. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 55.Veldhoen S, Laufer SD, Trampe A, Restle T. Cellular delivery of small interfering RNA by a non-covalently attached cell-penetrating peptide: quantitative analysis of uptake and biological effect. Nucleic Acids Res. 2006;34:6561–73. doi: 10.1093/nar/gkl941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Console S, Marty C, Garcia-Echeverria C, Schwendener R, Ballmer-Hofer K. Antennapedia and HIV transactivator of transcription (TAT) "protein transduction domains" promote endocytosis of high molecular weight cargo upon binding to cell surface glycosaminoglycans. J Biol Chem. 2003;278:35109–14. doi: 10.1074/jbc.M301726200. [DOI] [PubMed] [Google Scholar]
- 57.Tyagi M, Rusnati M, Presta M, Giacca M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J Biol Chem. 2001;276:3254–61. doi: 10.1074/jbc.M006701200. [DOI] [PubMed] [Google Scholar]
- 58.Rusnati M, Tulipano G, Spillmann D, Tanghetti E, Oreste P, Zoppetti G, et al. Multiple interactions of HIV-I Tat protein with size-defined heparin oligosaccharides. J Biol Chem. 1999;274:28198–205. doi: 10.1074/jbc.274.40.28198. [DOI] [PubMed] [Google Scholar]
- 59.Rusnati M, Coltrini D, Oreste P, Zoppetti G, Albini A, Noonan D, et al. Interaction of HIV-1 Tat protein with heparin. Role of the backbone structure, sulfation, and size. J Biol Chem. 1997;272:11313–20. doi: 10.1074/jbc.272.17.11313. [DOI] [PubMed] [Google Scholar]
- 60.Richard JP, Melikov K, Brooks H, Prevot P, Lebleu B, Chernomordik LV. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem. 2005;280:15300–6. doi: 10.1074/jbc.M401604200. [DOI] [PubMed] [Google Scholar]
- 61.Fittipaldi A, Ferrari A, Zoppe M, Arcangeli C, Pellegrini V, Beltram F, et al. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J Biol Chem. 2003;278:34141–9. doi: 10.1074/jbc.M303045200. [DOI] [PubMed] [Google Scholar]
- 62.Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10:310–5. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 63.Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J Am Chem Soc. 2004;126:9506–7. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- 64.Maiolo JR, Ferrer M, Ottinger EA. Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim Biophys Acta. 2005;1712:161–72. doi: 10.1016/j.bbamem.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 65.Mano M, Teodosio C, Paiva A, Simoes S, Pedroso de Lima MC. On the mechanisms of the internalization of S4(13)-PV cell-penetrating peptide. Biochem J. 2005;390:603–12. doi: 10.1042/BJ20050577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Coupade C, Fittipaldi A, Chagnas V, Michel M, Carlier S, Tasciotti E, et al. Novel human-derived cell-penetrating peptides for specific subcellular delivery of therapeutic biomolecules. Biochem J. 2005;390:407–18. doi: 10.1042/BJ20050401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.El-Andaloussi S, Johansson HJ, Holm T, Langel Ü. A novel cell-penetrating peptide, M918, for efficient delivery of proteins and peptide nucleic acids. Mol Ther. 2007;15:1820–6. doi: 10.1038/sj.mt.6300255. [DOI] [PubMed] [Google Scholar]
- 68.Edenhofer F. Protein transduction revisited: Novel insights into the mechanism underlying intracellular delivery of proteins. Curr Pharm Des. doi: 10.2174/138161208786898833. In press. [DOI] [PubMed] [Google Scholar]
- 69.Deshayes S, Plenat T, Aldrian-Herrada G, Divita G, Le Grimellec C, Heitz F. Primary amphipathic cell-penetrating peptides: structural requirements and interactions with model membranes. Biochemistry. 2004;43:7698–706. doi: 10.1021/bi049298m. [DOI] [PubMed] [Google Scholar]
- 70.Deshayes S, Gerbal-Chaloin S, Morris MC, Aldrian-Herrada G, Charnet P, Divita G, et al. On the mechanism of non-endosomial peptide-mediated cellular delivery of nucleic acids. Biochim Biophys Acta. 2004;1667:141–7. doi: 10.1016/j.bbamem.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 71.Deshayes S, Morris MC, Divita G, Heitz F. Interactions of amphipathic CPPs with model membranes. Biochim Biophys Acta. 2006;1758:328–35. doi: 10.1016/j.bbamem.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 72.Bumcrot D, Manoharan M, Koteliansky V, Sah DW. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol. 2006;2:711–9. doi: 10.1038/nchembio839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.de Fougerolles A, Vornlocher HP, Maraganore J, Lieberman J. Interfering with disease: a progress report on siRNA-based therapeutics. Nat Rev Drug Discov. 2007;6:443–53. doi: 10.1038/nrd2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–11. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 75.Rana TM. Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- 76.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–6. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 77.Hammond SM, Boettcher S, Caudy AA, Kobayashi R, Hannon GJ. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science. 2001;293:1146–50. doi: 10.1126/science.1064023. [DOI] [PubMed] [Google Scholar]
- 78.Hutvagner G, Simard MJ. Argonaute proteins: key players in RNA silencing. Nat Rev Mol Cell Biol. 2008;9:22–32. doi: 10.1038/nrm2321. [DOI] [PubMed] [Google Scholar]
- 79.Haley B, Zamore PD. Kinetic analysis of the RNAi enzyme complex. Nat Struct Mol Biol. 2004;11:599–606. doi: 10.1038/nsmb780. [DOI] [PubMed] [Google Scholar]
- 80.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 81.Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948–58. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu JY, DeRuiter SL, Turner DL. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci USA. 2002;99:6047–52. doi: 10.1073/pnas.092143499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leung RK, Whittaker PA. RNA interference: from gene silencing to gene-specific therapeutics. Pharmacol Ther. 2005;107:222–39. doi: 10.1016/j.pharmthera.2005.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–41. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 86.Snøve O Jr, Rossi JJ. Toxicity in mice expressing short hairpin RNAs gives new insight into RNAi. Genome Biol. 2006;7:231. doi: 10.1186/gb-2006-7-8-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Snøve O Jr, Rossi JJ. Expressing short hairpin RNAs in vivo. Nat Methods. 2006;3:689–95. doi: 10.1038/nmeth927. [DOI] [PubMed] [Google Scholar]
- 88.Chiu YL, Ali A, Chu CY, Cao H, Rana TM. Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells. Chem Biol. 2004;11:1165–75. doi: 10.1016/j.chembiol.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 89.Muratovska A, Eccles MR. Conjugate for efficient delivery of short interfering RNA (siRNA) into mammalian cells. FEBS Lett. 2004;558:63–8. doi: 10.1016/S0014-5793(03)01505-9. [DOI] [PubMed] [Google Scholar]
- 90.Davidson TJ, Harel S, Arboleda VA, Prunell GF, Shelanski ML, Greene LA, et al. Highly efficient small interfering RNA delivery to primary mammalian neurons induces MicroRNA-like effects before mRNA degradation. J Neurosci. 2004;24:10040–6. doi: 10.1523/JNEUROSCI.3643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moschos SA, Jones SW, Perry MM, Williams AE, Erjefalt JS, Turner JJ, et al. Lung delivery studies using siRNA conjugated to TAT(48-60) and penetratin reveal peptide induced reduction in gene expression and induction of innate immunity. Bioconjug Chem. 2007;18:1450–9. doi: 10.1021/bc070077d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Meade BR, Dowdy SF. Enhancing the cellular uptake of siRNA duplexes following noncovalent packaging with protein transduction domain peptides. Adv Drug Deliv Rev. 2007 doi: 10.1016/j.addr.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Turner JJ, Arzumanov AA, Gait MJ. Synthesis, cellular uptake and HIV-1 Tat-dependent trans-activation inhibition activity of oligonucleotide analogues disulphide-conjugated to cell-penetrating peptides. Nucleic Acids Res. 2005;33:27–42. doi: 10.1093/nar/gki142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Simeoni F, Morris MC, Heitz F, Divita G. Insight into the mechanism of the peptide-based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003;31:2717–24. doi: 10.1093/nar/gkg385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL, et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 96.Johnson LN, Cashman SM, Kumar-Singh R. Cell-penetrating peptide for enhanced delivery of nucleic acids and drugs to ocular tissues including retina and cornea. Mol Ther. 2008;16:107–14. doi: 10.1038/sj.mt.6300324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim WJ, Christensen LV, Jo S, Yockman JW, Jeong JH, Kim YH, et al. Cholesteryl oligoarginine delivering vascular endothelial growth factor siRNA effectively inhibits tumor growth in colon adenocarcinoma. Mol Ther. 2006;14:343–50. doi: 10.1016/j.ymthe.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 98.Moschos SA, Williams AE, Lindsay MA. Cell-penetrating-peptide-mediated siRNA lung delivery. Biochem Soc Trans. 2007;35:807–10. doi: 10.1042/BST0350807. [DOI] [PubMed] [Google Scholar]
- 99.Aigner A. Cellular delivery in vivo of siRNA-based therapeutics. Curr Pharm Des. doi: 10.2174/138161208786898815. In press. [DOI] [PubMed] [Google Scholar]
- 100.Moschos SA, Spinks K, Williams AE, Lindsay MA. Targeting the lung using siRNA and antisense based oligonucleotides. Curr Pharm Des. doi: 10.2174/138161208786898851. In press. [DOI] [PubMed] [Google Scholar]
- 101.Lundberg P, El-Andaloussi S, Sutlu T, Johansson H, Langel Ü. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007;21:2664–71. doi: 10.1096/fj.06-6502com. [DOI] [PubMed] [Google Scholar]
- 102.El-Andaloussi S, Johansson H, Magnusdottir A, Järver P, Lundberg P, Langel Ü. TP10, a delivery vector for decoy oligonucleotides targeting the Myc protein. J Control Release. 2005;110:189–201. doi: 10.1016/j.jconrel.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 103.Endoh T, Sisido M, Ohtsuki T. Photo inducible RNA interference using cell permeable protein carrier. Nucleic Acids Symp Ser (Oxf) 2007:127–8. doi: 10.1093/nass/nrm064. [DOI] [PubMed] [Google Scholar]
- 104.Endoh T, Sisido M, Ohtsuki T. Cellular siRNA delivery mediated by a cell-permeant RNA-binding protein and photoinduced RNA interference. Bioconjug Chem. 2008;19:1017–24. doi: 10.1021/bc800020n. [DOI] [PubMed] [Google Scholar]
- 105.Sharp PA. Split genes and RNA splicing. Cell. 1994;77:805–15. doi: 10.1016/0092-8674(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 106.Nilsen TW. The spliceosome: the most complex macromolecular machine in the cell? Bioessays. 2003;25:1147–9. doi: 10.1002/bies.10394. [DOI] [PubMed] [Google Scholar]
- 107.Matlin AJ, Clark F, Smith CW. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005;6:386–98. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- 108.Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, et al. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141–4. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- 109.Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749–61. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 110.Caceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet. 2002;18:186–93. doi: 10.1016/s0168-9525(01)02626-9. [DOI] [PubMed] [Google Scholar]
- 111.Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–37. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 112.Garcia-Blanco MA, Baraniak AP, Lasda EL. Alternative splicing in disease and therapy. Nat Biotechnol. 2004;22:535–46. doi: 10.1038/nbt964. [DOI] [PubMed] [Google Scholar]
- 113.Orengo JP, Cooper TA. Alternative splicing in disease. Adv Exp Med Biol. 2007;623:212–23. doi: 10.1007/978-0-387-77374-2_13. [DOI] [PubMed] [Google Scholar]
- 114.Lopez-Bigas N, Audit B, Ouzounis C, Parra G, Guigo R. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 2005;579:1900–3. doi: 10.1016/j.febslet.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 115.Stephenson ML, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci USA. 1978;75:285–8. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zamecnik PC, Stephenson ML. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci USA. 1978;75:280–4. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Crooke ST. Progress in antisense technology. Annu Rev Med. 2004;55:61–95. doi: 10.1146/annurev.med.55.091902.104408. [DOI] [PubMed] [Google Scholar]
- 118.Chan JH, Lim S, Wong WS. Antisense oligonucleotides: from design to therapeutic application. Clin Exp Pharmacol Physiol. 2006;33:533–40. doi: 10.1111/j.1440-1681.2006.04403.x. [DOI] [PubMed] [Google Scholar]
- 119.Lacerra G, Sierakowska H, Carestia C, Fucharoen S, Summerton J, Weller D, et al. Restoration of hemoglobin A synthesis in erythroid cells from peripheral blood of thalassemic patients. Proc Natl Acad Sci USA. 2000;97:9591–6. doi: 10.1073/pnas.97.17.9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sazani P, Kole R. Therapeutic potential of antisense oligonucleotides as modulators of alternative splicing. J Clin Invest. 2003;112:481–6. doi: 10.1172/JCI19547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kang SH, Cho MJ, Kole R. Up-regulation of luciferase gene expression with antisense oligonucleotides: implications and applications in functional assay development. Biochemistry. 1998;37:6235–9. doi: 10.1021/bi980300h. [DOI] [PubMed] [Google Scholar]
- 122.Summerton J, Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997;7:187–95. doi: 10.1089/oli.1.1997.7.187. [DOI] [PubMed] [Google Scholar]
- 123.Vester B, Wengel J. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry. 2004;43:13233–41. doi: 10.1021/bi0485732. [DOI] [PubMed] [Google Scholar]
- 124.Nielsen PE. PNA Technology. Mol Biotechnol. 2004;26:233–48. doi: 10.1385/MB:26:3:233. [DOI] [PubMed] [Google Scholar]
- 125.Abes S, Williams D, Prevot P, Thierry A, Gait MJ, Lebleu B. Endosome trapping limits the efficiency of splicing correction by PNA-oligolysine conjugates. J Control Release. 2006;110:595–604. doi: 10.1016/j.jconrel.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 126.Abes S, Moulton HM, Clair P, Prevot P, Youngblood DS, Wu RP, et al. Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents. J Control Release. 2006;116:304–13. doi: 10.1016/j.jconrel.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 127.Abes S, Turner JJ, Ivanova GD, Owen D, Williams D, Arzumanov A, et al. Efficient splicing correction by PNA conjugation to an R6-Penetratin delivery peptide. Nucleic Acids Res. 2007;35:4495–502. doi: 10.1093/nar/gkm418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Astriab-Fisher A, Sergueev D, Fisher M, Shaw BR, Juliano RL. Conjugates of antisense oligonucleotides with the Tat and antennapedia cell-penetrating peptides: effects on cellular uptake, binding to target sequences, and biologic actions. Pharm Res. 2002;19:744–54. doi: 10.1023/a:1016136328329. [DOI] [PubMed] [Google Scholar]
- 129.El-Andaloussi S, Johansson HJ, Lundberg P, Langel Ü. Induction of splice correction by cell-penetrating peptide nucleic acids. J Gene Med. 2006;8:1262–73. doi: 10.1002/jgm.950. [DOI] [PubMed] [Google Scholar]
- 130.Lebleu B, Moulton HM, Abes R, Ivanova GD, Abes S, Stein DA, et al. Cell penetrating peptide conjugates of steric block oligonucleotides. Adv Drug Deliv Rev. 2008;60:517–29. doi: 10.1016/j.addr.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rasmussen FW, Bendifallah N, Zachar V, Shiraishi T, Fink T, Ebbesen P, et al. Evaluation of transfection protocols for unmodified and modified peptide nucleic acid (PNA) oligomers. Oligonucleotides. 2006;16:43–57. doi: 10.1089/oli.2006.16.43. [DOI] [PubMed] [Google Scholar]
- 132.Shiraishi T, Bendifallah N, Nielsen PE. Cellular delivery of polyheteroaromate-peptide nucleic acid conjugates mediated by cationic lipids. Bioconjug Chem. 2006;17:189–94. doi: 10.1021/bc050246z. [DOI] [PubMed] [Google Scholar]
- 133.Thierry AR, Abes S, Resina S, Travo A, Richard JP, Prevot P, et al. Comparison of basic peptides- and lipid-based strategies for the delivery of splice correcting oligonucleotides. Biochim Biophys Acta. 2006;1758:364–74. doi: 10.1016/j.bbamem.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 134.Vacek M, Sazani P, Kole R. Antisense-mediated redirection of mRNA splicing. Cell Mol Life Sci. 2003;60:825–33. doi: 10.1007/s00018-003-3042-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wolf Y, Pritz S, Abes S, Bienert M, Lebleu B, Oehlke J. Structural requirements for cellular uptake and antisense activity of peptide nucleic acids conjugated with various peptides. Biochemistry. 2006;45:14944–54. doi: 10.1021/bi0606896. [DOI] [PubMed] [Google Scholar]
- 136.Moulton HM, Nelson MH, Hatlevig SA, Reddy MT, Iversen PL. Cellular uptake of antisense morpholino oligomers conjugated to arginine-rich peptides. Bioconjug Chem. 2004;15:290–9. doi: 10.1021/bc034221g. [DOI] [PubMed] [Google Scholar]
- 137.Turner JJ, Ivanova GD, Verbeure B, Williams D, Arzumanov AA, Abes S, et al. Cell-penetrating peptide conjugates of peptide nucleic acids (PNA) as inhibitors of HIV-1 Tat-dependent trans-activation in cells. Nucleic Acids Res. 2005;33:6837–49. doi: 10.1093/nar/gki991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shiraishi T, Nielsen PE. Photochemically enhanced cellular delivery of cell penetrating peptide-PNA conjugates. FEBS Lett. 2006;580:1451–6. doi: 10.1016/j.febslet.2006.01.077. [DOI] [PubMed] [Google Scholar]
- 139.Rothbard JB, Kreider E, VanDeusen CL, Wright L, Wylie BL, Wender PA. Arginine-rich molecular transporters for drug delivery: role of backbone spacing in cellular uptake. J Med Chem. 2002;45:3612–8. doi: 10.1021/jm0105676. [DOI] [PubMed] [Google Scholar]
- 140.Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–28. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 141.Wilton SD, Fletcher S. Modification of pre-mRNA processing: application to dystrophin expression. Curr Opin Mol Ther. 2006;8:130–5. [PubMed] [Google Scholar]
- 142.Aartsma-Rus A, van Ommen GJ. Antisense-mediated exon skipping: a versatile tool with therapeutic and research applications. RNA. 2007;13:1609–24. doi: 10.1261/rna.653607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bertoni C. Clinical approaches in the treatment of Duchenne muscular dystrophy (DMD) using oligonucleotides. Front Biosci. 2008;13:517–27. doi: 10.2741/2697. [DOI] [PubMed] [Google Scholar]
- 144.van Ommen GJ, van Deutekom J, Aartsma-Rus A. The therapeutic potential of antisense-mediated exon skipping. Curr Opin Mol Ther. 2008;10:140–9. [PubMed] [Google Scholar]
- 145.Yokota T, Pistilli E, Duddy W, Nagaraju K. Potential of oligonucleotide-mediated exon-skipping therapy for Duchenne muscular dystrophy. Expert Opin Biol Ther. 2007;7:831–42. doi: 10.1517/14712598.7.6.831. [DOI] [PubMed] [Google Scholar]
- 146.Alter J, Lou F, Rabinowitz A, Yin H, Rosenfeld J, Wilton SD, et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med. 2006;12:175–7. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- 147.Roberts J, Palma E, Sazani P, Orum H, Cho M, Kole R. Efficient and persistent splice switching by systemically delivered LNA oligonucleotides in mice. Mol Ther. 2006;14:471–5. doi: 10.1016/j.ymthe.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 148.Goyenvalle A, Vulin A, Fougerousse F, Leturcq F, Kaplan JC, Garcia L, et al. Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science. 2004;306:1796–9. doi: 10.1126/science.1104297. [DOI] [PubMed] [Google Scholar]
- 149.Benchaouir R, Meregalli M, Farini A, D'Antona G, Belicchi M, Goyenvalle A, et al. Restoration of human dystrophin following transplantation of exon-skipping-engineered DMD patient stem cells into dystrophic mice. Cell Stem Cell. 2007;1:646–57. doi: 10.1016/j.stem.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 150.Fletcher S, Honeyman K, Fall AM, Harding PL, Johnsen RD, Steinhaus JP, et al. Morpholino oligomer-mediated exon skipping averts the onset of dystrophic pathology in the mdx mouse. Mol Ther. 2007;15:1587–92. doi: 10.1038/sj.mt.6300245. [DOI] [PubMed] [Google Scholar]
- 151.McClorey G, Moulton HM, Iversen PL, Fletcher S, Wilton SD. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006;13:1373–81. doi: 10.1038/sj.gt.3302800. [DOI] [PubMed] [Google Scholar]
- 152.Moulton HM, Fletcher S, Neuman BW, McClorey G, Stein DA, Abes S, et al. Cell-penetrating peptide-morpholino conjugates alter pre-mRNA splicing of DMD (Duchenne muscular dystrophy) and inhibit murine coronavirus replication in vivo. Biochem Soc Trans. 2007;35:826–8. doi: 10.1042/BST0350826. [DOI] [PubMed] [Google Scholar]
- 153.Jearawiriyapaisarn N, Moulton HM, Buckley B, Roberts J, Sazani P, Fucharoen S, et al. Sustained Dystrophin Expression Induced by Peptide-conjugated Morpholino Oligomers in the Muscles of mdx Mice. Mol Ther. 2008 doi: 10.1038/mt.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yin H, Lu Q, Wood M. Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol Ther. 2008;16:38–45. doi: 10.1038/sj.mt.6300329. [DOI] [PubMed] [Google Scholar]
- 155.Mescalchin A, Detzer A, Wecke M, Overhoff M, Wünsche W, Sczakiel G. Cellular uptake and intracellular release are major obstacles to the therapeutic application of siRNA: novel options by phosphorothioate-stimulated delivery. Expert Opin Biol Ther. 2007;7:1531–8. doi: 10.1517/14712598.7.10.1531. [DOI] [PubMed] [Google Scholar]
- 156.Gammon ST, Villalobos VM, Prior JL, Sharma V, Piwnica-Worms D. Quantitative analysis of permeation peptide complexes labeled with Technetium-99m: chiral and sequence-specific effects on net cell uptake. Bioconjug Chem. 2003;14:368–76. doi: 10.1021/bc0256291. [DOI] [PubMed] [Google Scholar]
- 157.Zhang X, Wan L, Pooyan S, Su Y, Gardner CR, Leibowitz MJ, et al. Quantitative assessment of the cell penetrating properties of RI-Tat-9: evidence for a cell type-specific barrier at the plasma membrane of epithelial cells. Mol Pharm. 2004;1:145–55. doi: 10.1021/mp034014y. [DOI] [PubMed] [Google Scholar]
- 158.Fischer R, Waizenegger T, Kohler K, Brock R. A quantitative validation of fluorophore-labelled cell-permeable peptide conjugates: fluorophore and cargo dependence of import. Biochim Biophys Acta. 2002;1564:365–74. doi: 10.1016/s0005-2736(02)00471-6. [DOI] [PubMed] [Google Scholar]
- 159.Hällbrink M, Floren A, Elmquist A, Pooga M, Bartfai T, Langel U. Cargo delivery kinetics of cell-penetrating peptides. Biochim Biophys Acta. 2001;1515:101–9. doi: 10.1016/s0005-2736(01)00398-4. [DOI] [PubMed] [Google Scholar]
- 160.Lindgren M, Gallet X, Soomets U, Hallbrink M, Brakenhielm E, Pooga M, et al. Translocation properties of novel cell penetrating transportan and penetratin analogues. Bioconjug Chem. 2000;11:619–26. doi: 10.1021/bc990156s. [DOI] [PubMed] [Google Scholar]
- 161.Kameyama S, Horie M, Kikuchi T, Omura T, Tadokoro A, Takeuchi T, et al. Acid wash in determining cellular uptake of Fab/cell-permeating peptide conjugates. Biopolymers. 2007;88:98–107. doi: 10.1002/bip.20689. [DOI] [PubMed] [Google Scholar]
- 162.Kaplan IM, Wadia JS, Dowdy SF. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J Control Release. 2005;102:247–53. doi: 10.1016/j.jconrel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 163.Oehlke J, Scheller A, Wiesner B, Krause E, Beyermann M, Klauschenz E, et al. Cellular uptake of an alpha-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim Biophys Acta. 1998;1414:127–39. doi: 10.1016/s0005-2736(98)00161-8. [DOI] [PubMed] [Google Scholar]
- 164.Drin G, Mazel M, Clair P, Mathieu D, Kaczorek M, Temsamani J. Physico-chemical requirements for cellular uptake of pAntp peptide. Role of lipid-binding affinity. Eur J Biochem. 2001;268:1304–14. doi: 10.1046/j.1432-1327.2001.01997.x. [DOI] [PubMed] [Google Scholar]
- 165.Soughayer JS, Wang Y, Li H, Cheung SH, Rossi FM, Stanbridge EJ, et al. Characterization of TAT-mediated transport of detachable kinase substrates. Biochemistry. 2004;43:8528–40. doi: 10.1021/bi036296d. [DOI] [PubMed] [Google Scholar]
- 166.Aussedat B, Sagan S, Chassaing G, Bolbach G, Burlina F. Quantification of the efficiency of cargo delivery by peptidic and pseudo-peptidic Trojan carriers using MALDI-TOF mass spectrometry. Biochim Biophys Acta. 2006;1758:375–83. doi: 10.1016/j.bbamem.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 167.Burlina F, Sagan S, Bolbach G, Chassaing G. Quantification of the cellular uptake of cell-penetrating peptides by MALDI-TOF mass spectrometry. Angew Chem Int Ed Engl. 2005;44:4244–7. doi: 10.1002/anie.200500477. [DOI] [PubMed] [Google Scholar]
- 168.Burlina F, Sagan S, Bolbach G, Chassaing G. A direct approach to quantification of the cellular uptake of cell-penetrating peptides using MALDI-TOF mass spectrometry. Nat Protoc. 2006;1:200–5. doi: 10.1038/nprot.2006.30. [DOI] [PubMed] [Google Scholar]
- 169.Varga CM, Hong K, Lauffenburger DA. Quantitative analysis of synthetic gene delivery vector design properties. Mol Ther. 2001;4:438–46. doi: 10.1006/mthe.2001.0475. [DOI] [PubMed] [Google Scholar]
- 170.Varga CM, Tedford NC, Thomas M, Klibanov AM, Griffith LG, Lauffenburger DA. Quantitative comparison of polyethylenimine formulations and adenoviral vectors in terms of intracellular gene delivery processes. Gene Ther. 2005;12:1023–32. doi: 10.1038/sj.gt.3302495. [DOI] [PubMed] [Google Scholar]
- 171.Varga CM, Tedford NC, Thomas M, Klibanov AM, Griffith LG, Lauffenburger DA. Quantitative comparison of polyethylenimine formulations and adenoviral vectors in terms of intracellular gene delivery processes. Gene Ther. 2005;12:1023–32. doi: 10.1038/sj.gt.3302495. [DOI] [PubMed] [Google Scholar]
- 172.Zhou J, Yockman JW, Kim SW, Kern SE. Intracellular kinetics of non-viral gene delivery using polyethylenimine carriers. Pharm Res. 2007;24:1079–87. doi: 10.1007/s11095-006-9229-5. [DOI] [PubMed] [Google Scholar]
- 173.Akita H, Ito R, Khalil IA, Futaki S, Harashima H. Quantitative three-dimensional analysis of the intracellular trafficking of plasmid DNA transfected by a nonviral gene delivery system using confocal laser scanning microscopy. Mol Ther. 2004;9:443–51. doi: 10.1016/j.ymthe.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 174.Hama S, Akita H, Ito R, Mizuguchi H, Hayakawa T, Harashima H. Quantitative comparison of intracellular trafficking and nuclear transcription between adenoviral and lipoplex systems. Mol Ther. 2006;13:786–94. doi: 10.1016/j.ymthe.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 175.Jiang M, Arzumanov AA, Gait MJ, Milner J. A bi-functional siRNA construct induces RNA interference and also primes PCR amplification for its own quantification. Nucleic Acids Res. 2005;33:e151. doi: 10.1093/nar/gni144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Overhoff M, Wünsche W, Sczakiel G. Quantitative detection of siRNA and single-stranded oligonucleotides: relationship between uptake and biological activity of siRNA. Nucleic Acids Res. 2004;32:e170. doi: 10.1093/nar/gnh168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Detzer A, Overhoff M, Mescalchin A, Rompf M, Sczakiel G. Phosphorothioate-stimulated cellular uptake of siRNA: a cell culture model for mechanistic studies. Curr Pharm Des. doi: 10.2174/138161208786898770. In press. [DOI] [PubMed] [Google Scholar]
- 178.Maxfield FR. Weak bases and ionophores rapidly and reversibly raise the pH of endocytic vesicles in cultured mouse fibroblasts. J Cell Biol. 1982;95:676–81. doi: 10.1083/jcb.95.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Shiraishi T, Pankratova S, Nielsen PE. Calcium ions effectively enhance the effect of antisense peptide nucleic acids conjugated to cationic tat and oligoarginine peptides. Chem Biol. 2005;12:923–9. doi: 10.1016/j.chembiol.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 180.Shiraishi T, Nielsen PE. Enhanced delivery of cell-penetrating peptide-peptide nucleic acid conjugates by endosomal disruption. Nat Protoc. 2006;1:633–6. doi: 10.1038/nprot.2006.92. [DOI] [PubMed] [Google Scholar]
- 181.Takeuchi T, Kosuge M, Tadokoro A, Sugiura Y, Nishi M, Kawata M, et al. Direct and rapid cytosolic delivery using cell-penetrating peptides mediated by pyrenebutyrate. ACS Chem Biol. 2006;1:299–303. doi: 10.1021/cb600127m. [DOI] [PubMed] [Google Scholar]
- 182.Sakai N, Takeuchi T, Futaki S, Matile S. Direct observation of anion-mediated translocation of fluorescent oligoarginine carriers into and across bulk liquid and anionic bilayer membranes. Chembiochem. 2005;6:114–22. doi: 10.1002/cbic.200400256. [DOI] [PubMed] [Google Scholar]
- 183.Lo SL, Wang S. An endosomolytic Tat peptide produced by incorporation of histidine and cysteine residues as a nonviral vector for DNA transfection. Biomaterials. 2008;29:2408–14. doi: 10.1016/j.biomaterials.2008.01.031. [DOI] [PubMed] [Google Scholar]
- 184.Youngblood DS, Hatlevig SA, Hassinger JN, Iversen PL, Moulton HM. Stability of cell-penetrating peptide-morpholino oligomer conjugates in human serum and in cells. Bioconjug Chem. 2007;18:50–60. doi: 10.1021/bc060138s. [DOI] [PubMed] [Google Scholar]
- 185.Berg K, Folini M, Prasmickaite L, Selbo PK, Bonsted A, Engesaeter BO, et al. Photochemical internalization: a new tool for drug delivery. Curr Pharm Biotechnol. 2007;8:362–72. doi: 10.2174/138920107783018354. [DOI] [PubMed] [Google Scholar]
- 186.Bøe Longva AS, Hovig E. Photochemically induced gene silencing using small interfering RNA molecules in combination with lipid carriers. Oligonucleotides. 2007;17:166–73. doi: 10.1089/oli.2007.0076. [DOI] [PubMed] [Google Scholar]
- 187.Oliveira S, Fretz MM, Hogset A, Storm G, Schiffelers RM. Photochemical internalization enhances silencing of epidermal growth factor receptor through improved endosomal escape of siRNA. Biochim Biophys Acta. 2007;1768:1211–7. doi: 10.1016/j.bbamem.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 188.Bonsted A, Wagner E, Prasmickaite L, Høgset Berg K. Photochemical enhancement of DNA delivery by EGF receptor targeted polyplexes. Methods Mol Biol. 2008;434:171–81. doi: 10.1007/978-1-60327-248-3_11. [DOI] [PubMed] [Google Scholar]
- 189.Folini M, Bandiera R, Millo E, Gandellini P, Sozzi G, Gasparini P, et al. Photochemically enhanced delivery of a cell-penetrating peptide nucleic acid conjugate targeting human telomerase reverse transcriptase: effects on telomere status and proliferative potential of human prostate cancer cells. Cell Prolif. 2007;40:905–20. doi: 10.1111/j.1365-2184.2007.00470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Silva JN, Filipe P, Morliere P, Maziere JC, Freitas JP, Cirne de Castro JL, et al. Photodynamic therapies: principles and present medical applications. Biomed Mater Eng. 2006;16:S147–S154. [PubMed] [Google Scholar]
- 191.Brown SB, Brown EA, Walker I. The present and future role of photodynamic therapy in cancer treatment. Lancet Oncol. 2004;5:497–508. doi: 10.1016/S1470-2045(04)01529-3. [DOI] [PubMed] [Google Scholar]
- 192.Oliveira S, Høgset A, Storm G, Schiffelers RM. Delivery of Small Interfering RNA to the Target Cell Cytoplasm Photochemical Internalization Facilitates Endosomal Escape and Imprves Silencing Efficiency, In Vitro and In Vivo. Curr Pharm Des. doi: 10.2174/138161208786898789. In press. [DOI] [PubMed] [Google Scholar]
- 193.Epand RM. Fusion peptides and the mechanism of viral fusion. Biochim Biophys Acta. 2003;1614:116–21. doi: 10.1016/s0005-2736(03)00169-x. [DOI] [PubMed] [Google Scholar]
- 194.Haque ME, Koppaka V, Axelsen PH, Lentz BR. Properties and structures of the influenza and HIV fusion peptides on lipid membranes: implications for a role in fusion. Biophys J. 2005;89:3183–94. doi: 10.1529/biophysj.105.063032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Plank C, Zauner W, Wagner E. Application of membrane-active peptides for drug and gene delivery across cellular membranes. Adv Drug Deliv Rev. 1998;34:21–35. doi: 10.1016/s0169-409x(98)00005-2. [DOI] [PubMed] [Google Scholar]
- 196.Oliveira S, van R I, Kranenburg O, Storm G, Schiffelers RM. Fusogenic peptides enhance endosomal escape improving siRNA-induced silencing of oncogenes. Int J Pharm. 2007;331:211–4. doi: 10.1016/j.ijpharm.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 197.Tu Y, Kim JS. A fusogenic segment of glycoprotein H from herpes simplex virus enhances transfection efficiency of cationic liposomes. J Gene Med. 2008;10:646–54. doi: 10.1002/jgm.1184. [DOI] [PubMed] [Google Scholar]
- 198.Futaki S, Masui Y, Nakase I, Sugiura Y, Nakamura T, Kogure K, et al. Unique features of a pH-sensitive fusogenic peptide that improves the transfection efficiency of cationic liposomes. J Gene Med. 2005;7:1450–8. doi: 10.1002/jgm.796. [DOI] [PubMed] [Google Scholar]
- 199.Kwon EJ, Bergen JM, Pun SH. Application of an HIV gp41-derived peptide for enhanced intracellular trafficking of synthetic gene and siRNA delivery vehicles. Bioconjug Chem. 2008;19:920–7. doi: 10.1021/bc700448h. [DOI] [PubMed] [Google Scholar]
- 200.Michiue H, Tomizawa K, Wei FY, Matsushita M, Lu YF, Ichikawa T, et al. The NH2 terminus of influenza virus hemagglutinin-2 subunit peptides enhances the antitumor potency of polyarginine-mediated p53 protein transduction. J Biol Chem. 2005;280:8285–9. doi: 10.1074/jbc.M412430200. [DOI] [PubMed] [Google Scholar]
- 201.Mustard D, Ritchie DW. Docking essential dynamics eigenstructures. Proteins. 2005;60:269–74. doi: 10.1002/prot.20569. [DOI] [PubMed] [Google Scholar]
- 202.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 203.Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–50. [PubMed] [Google Scholar]
- 204.Pooga M, Hällbrink M, Zorko M, Langel Ü. Cell penetration by transportan. FASEB J. 1998;12:67–77. doi: 10.1096/fasebj.12.1.67. [DOI] [PubMed] [Google Scholar]
- 205.Soomets U, Lindgren M, Gallet X, Hällbrink M, Elmquist A, Balaspiri L, et al. Deletion analogues of transportan. Biochim Biophys Acta. 2000;1467:165–76. doi: 10.1016/s0005-2736(00)00216-9. [DOI] [PubMed] [Google Scholar]
- 206.Tönges L, Lingor P, Egle R, Dietz GP, Fahr A, Bahr M. Stearylated octaarginine and artificial virus-like particles for transfection of siRNA into primary rat neurons. RNA. 2006;12:1431–8. doi: 10.1261/rna.2252206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Nakamura Y, Kogure K, Futaki S, Harashima H. Octaarginine-modified multifunctional envelope-type nano device for siRNA. J Control Release. 2007;119:360–7. doi: 10.1016/j.jconrel.2007.03.010. [DOI] [PubMed] [Google Scholar]