

Abstract
AIM: To study the mechanisms by which Campylobacter jejuni (C. jejuni) causes inflammation and diarrhea. In particular, direct interactions with intestinal epithelial cells and effects on barrier function are poorly under-stood.
METHODS: To model the initial pathogenic effects of C. jejuni on intestinal epithelium, polarized human colonic HCA-7 monolayers were grown on permeabilized filters and infected apically with clinical isolates of C. jejuni. Integrity of the monolayer was monitored by changes in monolayer resistance, release of lactate dehydrogenase, mannitol fluxes and electron microscopy. Invasion of HCA-7 cells was assessed by a modified gentamicin protection assay, translocation by counting colony forming units in the basal chamber, stimulation of mediator release by immunoassays and secretory responses in monolayers stimulated by bradykinin in an Ussing chamber.
RESULTS: All strains translocated across monolayers but only a minority invaded HCA-7 cells. Strains that invaded HCA-7 cells destroyed monolayer resistance over 6 h, accompanied by increased release of lactate dehydrogenase, a four-fold increase in permeability to [3H] mannitol, and ultrastructural disruption of tight junctions, with rounding and lifting of cells off the filter membrane. Synthesis of interleukin (IL)-8 and prostaglandin E2 was increased with strains that invaded the monolayer but not with those that did not.
CONCLUSION: These data demonstrate two distinct effects of C. jejuni on colonic epithelial cells and provide an informative model for further investigation of initial host cell responses to C. jejuni.
Keywords: Campylobacter jejuni, Cell invasion, Cell culture, Chloride secretion, Colonocyte, HCA-7 cells, Membrane permeability, Monolayer, Mucosal barrier, Ussing chamber
INTRODUCTION
Campylobacters are small (1.5-6.0 μm long and 0.2-0.5 μm wide) Gram-negative spiral rods. Campylobacter jejuni (C. jejuni), a foodborne organism contracted from untreated water, milk and meat, especially chicken, is one of the most important causes of bacterial diarrhea worldwide[1-4]. The clinical spectrum ranges from non-inflammatory watery diarrhea to an acute entero-colitis with neutrophilic invasion of the mucosa and bloody diarrhea mimicking ulcerative colitis.
Much work has been conducted on laboratory strains such as NCT11168, which has been completely genotyped. This has allowed a number of virulence factors to be identified, including a number of flagellar proteins, which not only enable chemotaxis towards mucus and amino acids and epithelial cell invasion[5-7], but also facilitate secretion of non-flagellar virulence proteins[6], O-linked glycosylation, which is required for optimal flagella function[7], proteins secreted via flagella that result in epithelial cell invasion and apoptosis[8-10], a cytolethal distending toxin (CLDT)[11] with DNAse activity[12], associated with apoptosis[13] and secretion of interleukin (IL)-8 and other chemokines[14,15] and a lipo-oligosaccharide that resembles human neuronal gangliosides, which may pre-dispose to autoimmune phenomena such as Guillain-Barre syndrome[16].
Clinical isolates vary in the extent to which they express these virulence factors. C. jejuni display heterogeneity in its ability to invade cells of the intestinal epithelial layer[17-21]. Estimates of the proportion of clinical isolates that are characterized by toxin production range from 12% to 100%[22]. Cell death may occur by a variety of mechanisms, not all involving CLDT[23]. Release of chemokines such as IL-8 seems to occur by CLDT-dependent and independent mechanisms[14], and it is unclear how far inflammatory responses to C. jejuni infection, such as secretion of chemokines, correlate with reported differences in the ability of the bacteria to invade epithelial cells, or how much this is due to responses of cells in the lamina propria, such as macrophages. Similarly, the extent to which secretory responses by epithelial cells can maintain a secretory diarrhea, which is characteristic particularly of childhood infection, is also unclear. One possibility is that C. jejuni induces synthesis of pro-secretory compounds such as prostaglandins directly in epithelial cells[24-27]. An earlier study that failed to show this might have been flawed because it used CaCo2 cells, which do not readily express cyclo-oxygenases (COXs) or synthesize prostaglandins[28].
In order to investigate the direct effects of C. jejuni on colonic epithelial function in vitro, we therefore exposed the well-differentiated colonocyte line HCA-7, clone 29 to a panel of primary clinical isolates. Our data suggest two distinct patterns of interaction between C. jejuni and colonic epithelium, with a minority of strains invading colonic epithelial cells, which causes barrier destruction and induces elaboration of potentially pro-inflammatory mediators.
MATERIALS AND METHODS
Bacterial strains
Nineteen consecutive C. jejuni clinical strains from community patients with acute bacterial enteritis were isolated and characterized by the Laboratory of Public Health, University Hospital, Nottingham, UK. Of these, three fresh clinical isolates (strains 2801055, 2102011 and 1702030) were used for detailed studies along with the laboratory strain 12189 (a kind gift from Dr. J Ketley), originally isolated from a patient with diarrhea and passaged several times in the laboratory[28]. Strains were grown microaerobically for 48 h on chocolate agar plates prior to inoculation into tissue culture flasks containing liquid medium (RPMI 1640 supplemented with 4% Isosensitest broth and 5% FCS) (Sigma, UK). A stock of the original isolates was aliquoted and frozen at -80°C. For study of bacterial-epithelial cell interactions, bacteria were resuspended in 4% Isosensitest broth with 5% FCS. After static microaerobic incubation for 24 h, bacteria were pelleted, washed and resuspended in PBS.
The bacteria were diluted in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma, UK) to different concentrations (105-108 bacteria/mL). Determination of the number of bacteria was standardized by optical density at a wavelength of 570 nm. Growth curves for the different strains and titration and culture of serial dilutions was used to establish the number of bacteria in culture. This was done by incubating the different strains grown on a chocolate agar plates in Isosensitest broth (5% FCS) overnight. After centrifugation (7000 r/min, 5 min) and washing in PBS, bacteria were resuspended in 1 mL PBS, and serial diluted. Each dilution was measured in a spectrophotometer at a wavelength of 540 nm. Each sample was then serially diluted in a 96-well plate and dilutions incubated on chocolate agar plates. After overnight growth, bacterial colonies were counted and plotted against absorbance. The results were used to establish bacterial numbers before incubating epithelial cells.
Cell culture
Tumor-derived colonic epithelial cells, HCA-7, colony 29 (a kind gift from Susan Kirkland) were cultured as described previously[29]. Briefly, cells were grown in DMEM with 10% FCS, glutamine (0.29 mg/mL), ampicillin (8 μg/mL), penicillin (40 μg/mL), streptomycin (368 μg/mL) and non-essential amino acids in an atmosphere of 5% CO2 at 37°C. For studies of bacterial cell interactions, normal medium was replaced with antibiotic-free medium for at least 24 h. Tests for mycoplasma contamination were not performed. For electrophysiological studies, cells (105 cells/membrane) were seeded on Snapwell or Transwell filters (polycarbonate membrane, pore size 0.45 μm, surface area 1 cm2; Costar UK Ltd) and formed confluent monolayers within 8-10 d, as assessed by an epithelial volt-ohmmeter (EVOM; World Precision Instruments, Stevenage, UK)[29].
Effect of C. jejuni on barrier function
Bacteria were grown and added to the apical side of monolayers grown on Transwell or Snapwell filters at a concentration of 104-108 bacteria/monolayer. Transepithelial resistance was measured with an EVOM at various timepoints up to 24 h after inoculation with different concentrations of bacteria. These data were supported by detailed studies of filter-grown monolayers voltage-clamped in Ussing chambers (World Precision Instruments) and by assessment of [3H] mannitol flux[26].
Ussing chamber studies
Confluent HCA-7 cell monolayers were inoculated on the apical side with varying amounts of strains of C. jejuni. After different time periods, filters were placed in an Ussing chamber and voltage-clamped by continuous application of a short circuit current (SCC)[30]. Resistance (Ω cm2) was measured under basal conditions and the change in short circuit current (∆SCC μA/cm2) after basolateral stimulation by bradykinin (10-6 mol/L), and finally after similar stimulation by carbachol (10-4 mol/L).
Invasion assay
Invasion of epithelial cells was investigated using a gentamicin invasion assay[18]. Bacteria were grown in 4% Isosensitest broth and 5% FCS. After static microaerobic incubation for 24 h, bacteria were pelleted, washed and resuspended in PBS. Bacterial number was assessed spectrophotometrically and 106 organisms added to the cell monolayer. Infected monolayers were incubated for up to 6 h at 37°C, washed and covered with tissue culture medium containing gentamicin (100 μg/mL). After 90 min, the integrity of the monolayer was checked microscopically, then washed in PBS and flooded with 1 mL 10 mL/L Triton X-100 for 5 min, to release intracellular bacteria. Dilutions and viable counts were made of the bacteria within the lysed monolayer. Positive control was a Yersinia enterocolitica invasive strain 8081c, and the negative control was Escherichia coli non-invasive strain HB101. When it became clear that monolayer destruction occurred with some bacteria, we modified the assay to correct for the number of remaining cells per monolayer. We calculated the number of cells remaining per monolayer by using a hemocytometer, before lysing to obtain counts of intracellular protected bacteria.
Lactate dehydrogenase (LDH) release
HCA-7 cells were incubated with C. jejuni for 4 h. Media from the apical reservoir of bacterium-exposed and control monolayers were then collected and analyzed for spontaneous LDH using a colometric assay (Sigma)[31]. In addition, total intracellular LDH concentration from HCA-7 cells was determined by addition of 1 mL 0.1% Triton X-100 to wells of bacterium-exposed and control monolayers. After vigorous pipetting to ensure lysis of all cells, the homogenate was then collected from each well and was also assayed for LDH.
[3H] mannitol flux
Mannitol flux studies were performed in an Ussing chamber[32]. Mannitol (final concentration 5 mmol/L) was added to both sides of the monolayer in Krebs solution. After equilibration of the epithelial monolayer, [3H] mannitol (1 μCi/mL) was added to the apical side. Radioactivity was counted on samples from the basolateral compartment at 15-min intervals for 60 min. Basolateral chamber volume was maintained by replacing the sample aliquot with an equal volume of fresh Krebs buffer. Apical to basolateral flux, expressed as mol·h/cm2, was calculated by relating the accumulation of tritium in the basolateral chamber compared to the apical.
Electron microscopy (EM)
Filters with HCA-7 cells inoculated with 107 bacteria for 6 h where fixed by immersion in 2.5% glutaraldehyde (in 0.1 mol/L cacodylate buffer, pH 7.4). Subsequent processing was performed as described previously[25].
Bacterial translocation
Translocation was measured by inoculating the apical side of monolayers with 107 bacteria/0.5 mL. Medium from the basolateral compartment was collected after 2, 4, 6 and 8 h inoculation, diluted 10-fold and cultured on agar plates[33,34]. After each collection, membranes were transferred to fresh wells to establish the number of bacteria translocating over each 2-h period. The number of bacteria was established by counting colony-forming units at each time interval to establish the total number of bacteria translocated. Colony forming units where counted after microaerophilic incubation for 24 h to assess the time course of bacterial translocation.
IL-8 measurement
Level of IL-8 in the basolateral supernatant after 6 h incubation with different strains of C. jejuni and of control monolayers was measured using a quantitative ELISA[14] (Amersham, UK). Samples were pipetted into wells coated with specific antibody for IL-8, followed by incubation with a biotinilated antibody reagent. After extensive washing, a streptavidin-horseradish peroxidase conjugate was added and developed with 3,3',5,5'-tetramethylbenzidine substrate. After terminating the reaction, the optical density was read at 450 nm. Detection level was 25-1000 pg/mL.
PGE2 measurement
Aliquots of supernatant were prepared in the same way as for IL-8 release. PGE2 levels were measured by ELISA (Amersham), based on competition between unlabeled PGE2 and a fixed quantity of peroxidase-labeled PGE2 for a limited amount of PGE2-specific antibody[35].
Statistical analysis
Analysis of variance was used in experiments where time or dose was varied, to investigate the influence of cellular invasive and non-invasive bacteria and the interaction with time. For simple comparisons, unpaired t tests were used. P < 0.05 was considered statistically significant. Statistical analysis was performed using PRISM (Graphpad, San Diego, CA, USA) or Statistical Package for the Social Sciences (SPSS, Chicago, IL, USA).
RESULTS
Transepithelial resistance
Fourteen of the 19 clinical strains tested, as well as the laboratory isolate 12189 had no effect on transepithelial resistance up to 24 h. In contrast, the other five fresh clinical isolates abrogated monolayer resistance entirely by 6 h. These differences did not correlate with rates of translocation. Detailed studies, with strain 2801055 which abrogated transepithelial resistance, showed that decrease in transepithelial resistance varied with bacterial load and was time dependent, starting after 2 h of inoculation with 108 bacteria/0.5 mL (Figure 1). With strains 12189 or 2102011, no changes were seen at any time with any of the bacterial loads inoculated (Figure 1). Measurements in Ussing chambers mirrored those obtained with the EVOM with the different strain types. There was no significant difference in electrical resistance in monolayers infected with the strains 12189 and 2102011 at 4 and 8 h. However, strain 2801055 showed a time-dependent decrease in transepithelial resistance, with resistance falling to 71.3% and 17.7% of control at 4 and 8 h respectively (P < 0.05 vs control at 8 h).
Figure 1.
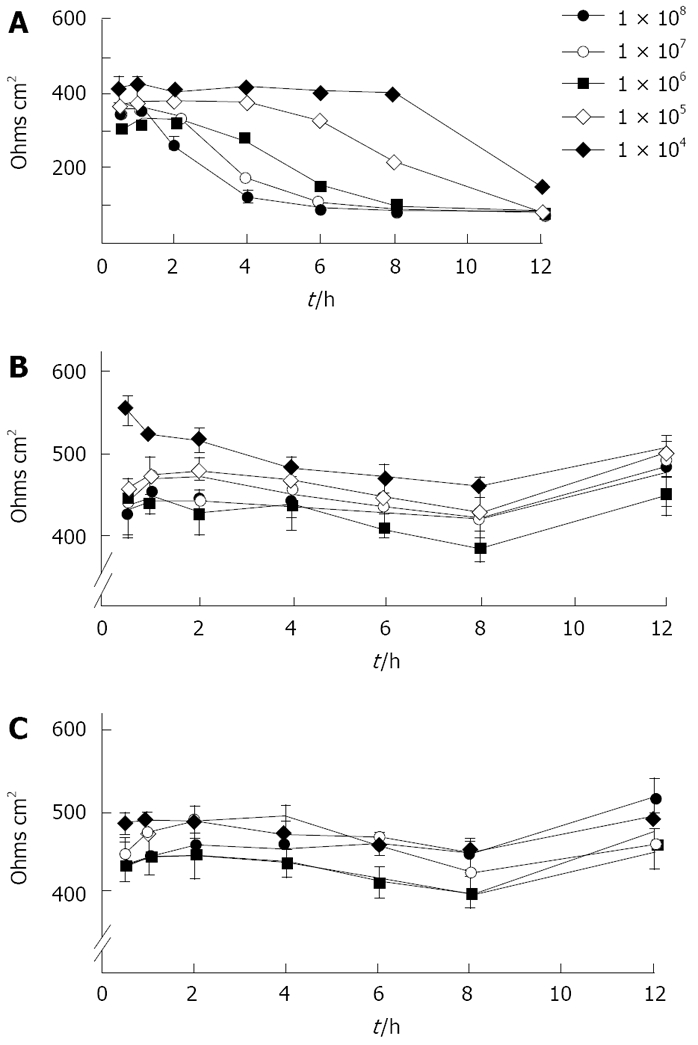
Time course and dose-response of changes in transepithelial resistance after inoculation of T84 cells with three different strains of C jejuni. A: Strain 2801055 reduced resistance to baseline over 6 h in a time- and dose-dependent manner [data are mean ± SE, n = 3 for each bacterial concentration; P < 0.05 compared with controls (uninfected monolayers)]. Baseline resistance across the filter membrane was 100 Ω.cm2; B: Strain 12189 had no effect on resistance across the monolayer; C: Strain 2102011 had no effect on resistance across the monolayer.
Invasion of epithelial cells by C. jejuni
The standard gentamicin protection assay showed that 1.15% ± 0.05% of the positive control strain Y. enterocolitica invaded HCA-7 cells vs 0.0005% with the negative control strain E. coli HB101. Overall, C. jejuni showed levels of HCA-7 invasion that were intermediate between these values. Analysis of variance showed that the total number of C. jejuni per monolayer was higher for those that abrogated monolayer resistance compared to those that did not (P = 0.033), and that this increased with time (P = 0.02). When the number of bacteria was related to the number of HCA7 cells remaining in the monolayer at the time of assessment, there was a substantial and highly significant difference between strains that destroyed and did not destroy the monolayer (Figure 2).
Figure 2.

Invasion of monolayers of HCA-7 cells determined by a gentamicin protection assay, corrected for the number of cells remaining per monolayer. C. jejuni strain 2801055 invaded the cells of the monolayer in a time-dependent manner, while strains 12189 and 2102011 showed no cellular invasion. Data are mean ± SE, n = 4. Analysis of variance showed that the total number of C. jejuni per monolayer was higher for those strains that abrogated transepithelial resistance compared to those that did not (P = 0.033), and that this increased with time (P = 0.02).
LDH release
LDH release from monolayers inoculated with the cell-invasive strain 2801055 was significantly increased after 24 h compared to inoculation with the non-invasive strains 12189 and 210211 (n = 4 per experiment, P < 0.05). LDH release with these strains did not differ from that seen with uninfected monolayers (Figure 3A).
Figure 3.
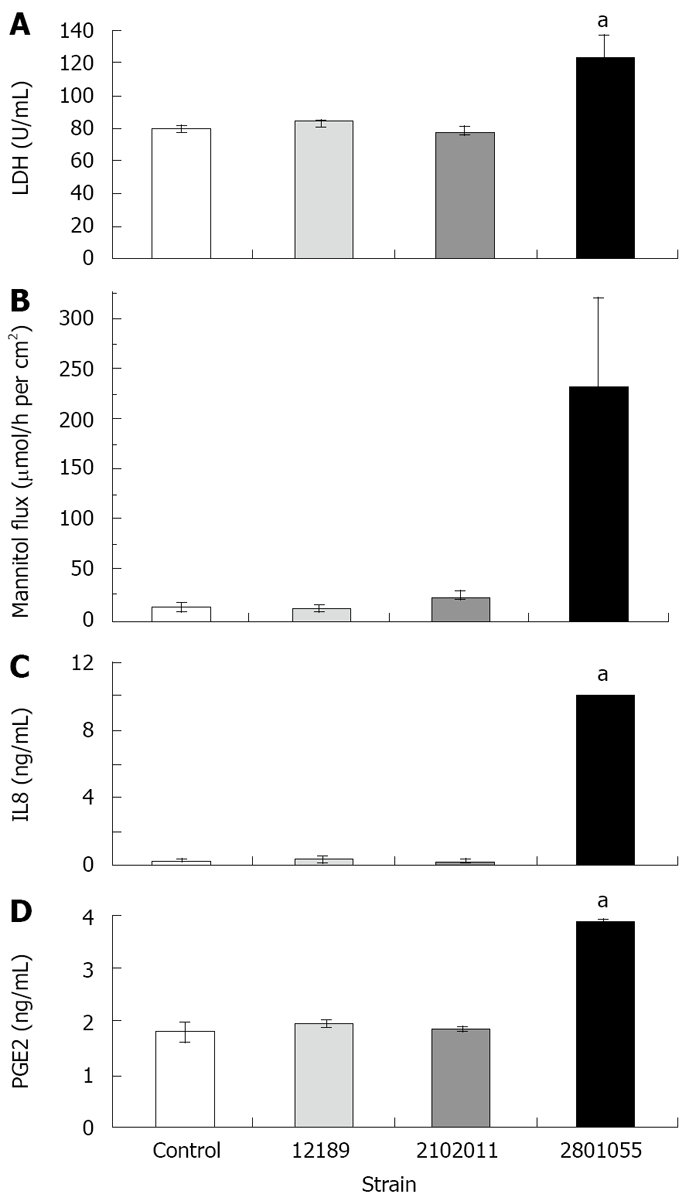
HCA-7 monolayer release of LDH, [3H] mannitol flux and release of IL8 and PGE2 after inoculation with different strains of C. jejuni. All data are mean ± SE at 4 h, (n = 4). A: Strain 2801055 showed a significantly higher LDH release after 4 h (aP < 0.05) incubation compared to the other two strains, which were similar to control values; B: Strain 2801055 induced a significantly higher flux rate across the monolayer after 4 h (aP < 0.05) incubation compared to the other two strains, which were similar to control values; C: Strain 2801055 showed a significantly higher IL8 release after 4 h (aP < 0.05) incubation compared to the other two strains, which were similar to control values; D: Strain 2801055 showed a significantly higher PGE2 release after 4 h (aP < 0.05) incubation compared to the other two strains, which were similar to control values.
Transepithelial mannitol flux
Cumulative flux data show that strains 12189 and 210211 did not change the flux of [3H] mannitol, which indicated an intact paracellular resistance, whilst strain 2801055 increased the flux significantly (Figure 3B).
EM studies
In keeping with electrophysiological results, strains 12189 and 2102011 did not affect cellular morphology (Figure 4A and B). Monolayers infected with these strains showed close cell-to-cell contact, but bacteria were occasionally located in the pores of the filter membrane (Figure 4C). In contrast, strain 2801055 showed marked cellular and tight junction destruction at 6 h (Figure 4D). There was cell rounding and condensation of the plasma membrane (Figure 4E). In addition, cells were lifted off the filter membrane and multiple bacteria were seen between epithelial cells and the filter (Figure 4E and F).
Figure 4.
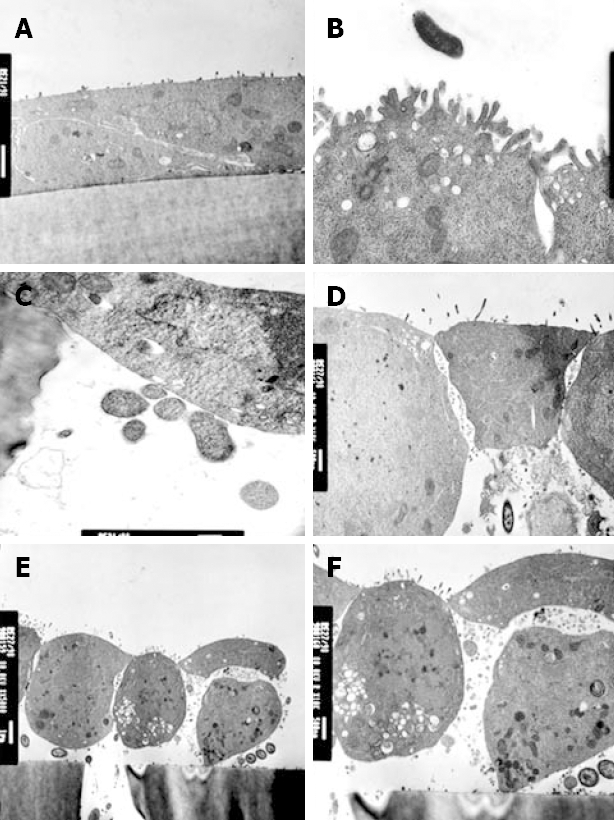
Comparison of the effects of strains 12189 and 2801055 on transmission electron micrograph appearance of monolayers of HCA-7 cells. A: Following inoculation of strain 12189, 107 bacteria/0.5 mL, on the apical side for 6 h, there was no disruption of monolayer integrity; B: There were normal tight junctions and normal apical microvilli; C: Close cell-to-cell contact is seen with occasional bacteria located in the pores of the filter membrane; D: Following inoculation of strain 12189, 107 bacteria/0.5 mL, on the apical side for 6 h, monolayer integrity was compromised with disruption of tight junctions; E: Monolayers showed condensation of the plasma membrane with lifting and rounding of cells off the supporting membrane. C. jejuni are also seen beneath a cell which is lifting off the membrane; F: Changes are seen at higher power.
Bacterial translocation
All strains tested translocated across the monolayer to become detectable after 2 h, with no obvious inter-strain differences.
IL-8 production
IL-8 release from HCA-7 monolayers increased in response to inoculation with the invasive strain 2801055, whereas IL-8 release with the non-invasive strains 12189 and 210211 was not significantly different from that seen in uninfected monolayers (Figure 3C).
PGE2 release
There was a two-fold rise in PGE2 released after inoculation with the invasive strain 2801055 (n = 4, P < 0.05). By contrast, PGE2 levels for strains 12189 and 2102011 were similar to spontaneous PGE2 production in control monolayers (Figure 3D).
Bradykinin- and carbachol-induced secretion
Bradykinin 10-6 mol/L administered basolaterally induced a ∆SCC of 19.83 ± 3.25 μAmp/cm2. ∆SCC responses to bradykinin were significantly enhanced by 30% compared with control (uninfected) monolayers at 4 h (n = 4, P < 0.05 for all 3 strains Figure 5A), but were lost at later time points, when there was a significant reduction with strain 2801055. The early increase was not seen in response to carbachol (Figure 5B), and at 8 h was significantly decreased with strains 2801055 and 2102011 (n = 4, P < 0.05).
Figure 5.
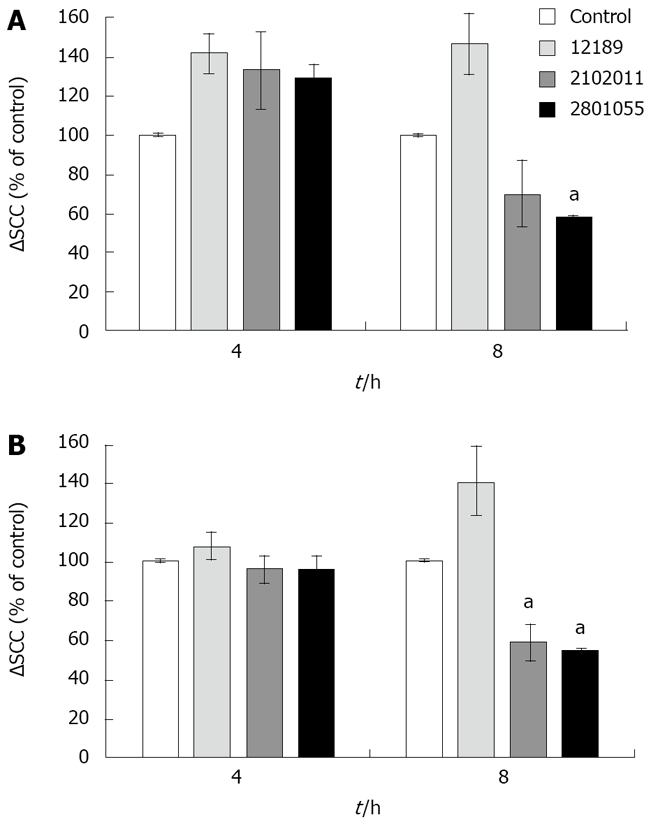
ΔSCC after stimulation with bradykinin (10-6 mol/L) and carbachol (10-4 mol/L) in monolayers inoculated with different strains of C. jejuni. Data are mean ± SE (n = 4) and are shown as percentage of control. A: Reduced chloride secretory response (represented by SCC) to bradykinin after 8 h inoculation with strain 2801055. aP < 0.05; B: Reduced chloride secretory response to carbachol after 8 h inoculation with strains 2801055 and 2102011. aP < 0.05.
DISCUSSION
In this study, we showed two distinct patterns of interaction between clinical isolates of C. jejuni and a colonic epithelial cell line. Strains that invaded epithelial cells were shown to destroy them, as demonstrated by a fall in transepithelial resistance and release of LDH. These processes were accompanied by release of IL-8 and PGE2. Strains that did not invade epithelial cells did not affect barrier properties or increase mediator production.
Translocation to the lamina propria[2,33] and a consequent interaction between bacterial antigens and antigen presenting cells, immunocytes and macrophages in the lamina propria[36], is likely also to result in production of cytokines and chemokines. However our data show that epithelial cells can themselves be a source of mediators that could influence inflammatory and secretory processes in the case of strains that invade epithelial cells.
Although a paracellular route has been invoked to explain the ability of C. jejuni to invade the mucosa and achieve systemic infection, intracellular bacteria have been reported and a paracellular route of invasion inferred[37]. Using a modified gentamicin protection assay that allows for cell death, we were able to confirm cellular invasion by C. jejuni, and showed that this was associated with cytotoxicity and elaboration of pro-inflammatory and pro-secretory molecules. As yet it is uncertain whether intracellular invasion directly causes the associated release of IL-8 and PGE2, or whether this is a secondary consequence of cell death. A direct specific effect is possible since we have reported that IL-8 synthesis and PGE2 release from epithelial monolayers also occur in response to treatment with a boiled cell-free extract of C. jejuni[38], with induction of COX-2[39] and activation of nuclear factor-κB (NF-κB) and other relevant signaling pathways[40].
Enhanced release of IL-8 and other chemokines would play an important role in chemoattraction of neutrophils that characterizes some clinical infections with C. jejuni. Whether the increased prostaglandin synthesis that we observed with cellular invasion is sufficient under some circumstances to induce secretory diarrhea is more difficult to evaluate. We showed an early increase in bradykinin-induced secretion, as indicated by changes in SCC, with all strains tested, including non-invasive strains that did not stimulate prostaglandin synthesis. This increase in bradykinin-induced chloride secretion may therefore occur by prostaglandin-independent mechanisms. Direct epithelial action, via cAMP, cGMP, calcium mobilization, or induction of galanin or inducible nitric oxide are alternative mechanisms that are activated directly by bacterial enterotoxins or via signaling mechanisms that include NF-κB, which we and others have shown are upregulated by components of C. jejuni[24-27,40-43]. Destruction of the monolayer by strains that did invade epithelial cells and stimulate prostaglandin synthesis makes it difficult to evaluate whether enhanced prostaglandin synthesis by epithelial cells contributed to secretory diarrhea in these cases.
Campylobacter, like Salmonella, Yersinia, Shigella and Listeria, is an organism capable of translocation, as demonstrated by the current and previous monolayer studies and clinical features that include septicemia, Guillain-Barre syndrome and meningitis[16,33,34,41]. Previous studies have left unclear whether the main route is transcellular or paracellular. Both have been described. Our data suggest the translocation across the monolayer is common since all of the 19 clinical strains isolated from patients and one laboratory strain showed this property, regardless of whether they invaded epithelial cells and/or destroyed the monolayer. This suggests a dominant paracellular route of translocation, which is supported by our EM observations, which showed bacteria in the paracellular space. This appeared to occur efficiently, as judged by the rate of accumulation of bacteria on the basolateral side, and without gross disruption of tight or adherence junctions, as judged by the unchanged transepithelial resistance and mannitol permeability, seen with strains that translocated without epithelial cell invasion.
In the case of bacteria that invaded and destroyed the epithelial monolayer, translocation could be a crude consequence of its destruction, although bacteria that destroyed the monolayer did not appear to translocate faster than those that did not. Translocation results may differ according to the cell type used for epithelial monolayers. In previous studies that used differentiated CaCo2 cells, cellular invasion was not accompanied by the complete abrogation of monolayer resistance seen in our study and not all strains translocated[34]. We chose to use HCA7 cells in preference to CaCo2 cells because they have a differentiated large rather than small bowel phenotype and because they are capable of expressing COX-2 under induction conditions. Our data suggest that this is an informative model and cell line to study disease pathogenesis and signaling mechanisms[40].
COMMENTS
Background
Campylobacter jejuni (C. jejuni) is the commonest cause of bacterial diarrhea worldwide but its mode of pathogenesis is not clear.
Research frontiers
Since this work was done, the Campylobacter genome has been sequenced. Work following from the current study has investigated gene expression and has shown that chemokines play a central role.
Innovations and breakthroughs
The paper underlines the importance of allowing for cell destruction when doing gentamicin assays. Unlike many previous studies, this one used clinical isolates, which showed that there were two distinct patterns for the effect of C. jejuni on colonic epithelial cells. The cells themselves have the capacity to generate chemoattractant molecules, without necessary involvement of immune and other cells in the lamina propria.
Applications
Showing that Campylobacter spp. are cell invasive and stimulate production of chemoattractant mediators points to possible targets for treatment.
Terminology
Ussing chamber: Monolayers are grown on a permeable filter. When cells form tight junctions they cause resistance to an electrical current passed through the monolayer.
Peer review
The majority of previous studies regarding C. jejuni have been performed using a laboratory strain NCT11168. This manuscript is considered to contain attractive information for enhancing the understanding the mechanism of C. jejuni infection. It’s an interesting paper.
Footnotes
Supported by The Medical Research Council (UK), No. G9716348
Peer reviewer: Atsushi Mizoguchi, Assistant Professor, Department of Experimental Pathology, Massachusetts General Hospital, Simches 8234, 185 Cambridge Street, Boston MA 02114, United States
S- Editor Li DL L- Editor Kerr C E- Editor Ma WH
References
- 1.Snelling WJ, Matsuda M, Moore JE, Dooley JS. Campylobacter jejuni. Lett Appl Microbiol. 2005;41:297–302. doi: 10.1111/j.1472-765X.2005.01788.x. [DOI] [PubMed] [Google Scholar]
- 2.Young KT, Davis LM, Dirita VJ. Campylobacter jejuni: molecular biology and pathogenesis. Nat Rev Microbiol. 2007;5:665–679. doi: 10.1038/nrmicro1718. [DOI] [PubMed] [Google Scholar]
- 3.Dorrell N, Wren BW. The second century of Campylobacter research: recent advances, new opportunities and old problems. Curr Opin Infect Dis. 2007;20:514–518. doi: 10.1097/QCO.0b013e3282a56b15. [DOI] [PubMed] [Google Scholar]
- 4.Jorgensen F, Bailey R, Williams S, Henderson P, Wareing DR, Bolton FJ, Frost JA, Ward L, Humphrey TJ. Prevalence and numbers of Salmonella and Campylobacter spp. on raw, whole chickens in relation to sampling methods. Int J Food Microbiol. 2002;76:151–164. doi: 10.1016/s0168-1605(02)00027-2. [DOI] [PubMed] [Google Scholar]
- 5.Yao R, Burr DH, Guerry P. CheY-mediated modulation of Campylobacter jejuni virulence. Mol Microbiol. 1997;23:1021–1031. doi: 10.1046/j.1365-2958.1997.2861650.x. [DOI] [PubMed] [Google Scholar]
- 6.Guerry P. Campylobacter flagella: not just for motility. Trends Microbiol. 2007;15:456–461. doi: 10.1016/j.tim.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Guerry P, Ewing CP, Schirm M, Lorenzo M, Kelly J, Pattarini D, Majam G, Thibault P, Logan S. Changes in flagellin glycosylation affect Campylobacter autoagglutination and virulence. Mol Microbiol. 2006;60:299–311. doi: 10.1111/j.1365-2958.2006.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konkel ME, Kim BJ, Rivera-Amill V, Garvis SG. Bacterial secreted proteins are required for the internaliztion of Campylobacter jejuni into cultured mammalian cells. Mol Microbiol. 1999;32:691–701. doi: 10.1046/j.1365-2958.1999.01376.x. [DOI] [PubMed] [Google Scholar]
- 9.Ziprin RL, Young CR, Byrd JA, Stanker LH, Hume ME, Gray SA, Kim BJ, Konkel ME. Role of Campylobacter jejuni potential virulence genes in cecal colonization. Avian Dis. 2001;45:549–557. [PubMed] [Google Scholar]
- 10.Poly F, Ewing C, Goon S, Hickey TE, Rockabrand D, Majam G, Lee L, Phan J, Savarino NJ, Guerry P. Heterogeneity of a Campylobacter jejuni protein that is secreted through the flagellar filament. Infect Immun. 2007;75:3859–3867. doi: 10.1128/IAI.00159-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson WM, Lior H. A new heat-labile cytolethal distending toxin (CLDT) produced by Campylobacter spp. Microb Pathog. 1988;4:115–126. doi: 10.1016/0882-4010(88)90053-8. [DOI] [PubMed] [Google Scholar]
- 12.Lara-Tejero M, Galan JE. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science. 2000;290:354–357. doi: 10.1126/science.290.5490.354. [DOI] [PubMed] [Google Scholar]
- 13.Hickey TE, Majam G, Guerry P. Intracellular survival of Campylobacter jejuni in human monocytic cells and induction of apoptotic death by cytholethal distending toxin. Infect Immun. 2005;73:5194–5197. doi: 10.1128/IAI.73.8.5194-5197.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hickey TE, McVeigh AL, Scott DA, Michielutti RE, Bixby A, Carroll SA, Bourgeois AL, Guerry P. Campylobacter jejuni cytolethal distending toxin mediates release of interleukin-8 from intestinal epithelial cells. Infect Immun. 2000;68:6535–6541. doi: 10.1128/iai.68.12.6535-6541.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu L, Hickey TE. Campylobacter jejuni induces secretion of proinflammatory chemokines from human intestinal epithelial cells. Infect Immun. 2005;73:4437–4440. doi: 10.1128/IAI.73.7.4437-4440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu RK, Usuki S, Ariga T. Ganglioside molecular mimicry and its pathological roles in Guillain-Barre syndrome and related diseases. Infect Immun. 2006;74:6517–6527. doi: 10.1128/IAI.00967-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perera VN, Nachamkin I, Ung H, Patterson JH, McConville MJ, Coloe PJ, Fry BN. Molecular mimicry in Campylobacter jejuni: role of the lipo-oligosaccharide core oligosaccharide in inducing anti-ganglioside antibodies. FEMS Immunol Med Microbiol. 2007;50:27–36. doi: 10.1111/j.1574-695X.2007.00225.x. [DOI] [PubMed] [Google Scholar]
- 18.Oelschlaeger TA, Guerry P, Kopecko DJ. Unusual microtubule-dependent endocytosis mechanisms triggered by Campylobacter jejuni and Citrobacter freundii. Proc Natl Acad Sci USA. 1993;90:6884–6888. doi: 10.1073/pnas.90.14.6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harvey P, Battle T, Leach S. Different invasion phenotypes of Campylobacter isolates in Caco-2 cell monolayers. J Med Microbiol. 1999;48:461–469. doi: 10.1099/00222615-48-5-461. [DOI] [PubMed] [Google Scholar]
- 20.Kopecko DJ, Hu L, Zaal KJ. Campylobacter jejuni--microtubule-dependent invasion. Trends Microbiol. 2001;9:389–396. doi: 10.1016/s0966-842x(01)02107-2. [DOI] [PubMed] [Google Scholar]
- 21.Van Deun K, Haesebrouck F, Heyndrickx M, Favoreel H, Dewulf J, Ceelen L, Dumez L, Messens W, Leleu S, Van Immerseel F, et al. Virulence properties of Campylobacter jejuni isolates of poultry and human origin. J Med Microbiol. 2007;56:1284–1289. doi: 10.1099/jmm.0.47342-0. [DOI] [PubMed] [Google Scholar]
- 22.Wassenaar TM. Toxin production by Campylobacter spp. Clin Microbiol Rev. 1997;10:466–476. doi: 10.1128/cmr.10.3.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalischuk LD, Inglis GD, Buret AG. Strain-dependent induction of epithelial cell oncosis by Campylobacter jejuni is correlated with invasion ability and is independent of cytolethal distending toxin. Microbiology. 2007;153:2952–2963. doi: 10.1099/mic.0.2006/003962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eckmann L, Stenson WF, Savidge TC, Lowe DC, Barrett KE, Fierer J, Smith JR, Kagnoff MF. Role of intestinal epithelial cells in the host secretory response to infection by invasive bacteria. Bacterial entry induces epithelial prostaglandin h synthase-2 expression and prostaglandin E2 and F2alpha production. J Clin Invest. 1997;100:296–309. doi: 10.1172/JCI119535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laurent F, Kagnoff MF, Savidge TC, Naciri M, Eckmann L. Human intestinal epithelial cells respond to Cryptosporidium parvum infection with increased prostaglandin H synthase 2 expression and prostaglandin E2 and F2alpha production. Infect Immun. 1998;66:1787–1790. doi: 10.1128/iai.66.4.1787-1790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Resta-Lenert S, Barrett KE. Enteroinvasive bacteria alter barrier and transport properties of human intestinal epithelium: role of iNOS and COX-2. Gastroenterology. 2002;122:1070–1087. doi: 10.1053/gast.2002.32372. [DOI] [PubMed] [Google Scholar]
- 27.Berkes J, Viswanathan VK, Savkovic SD, Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut. 2003;52:439–451. doi: 10.1136/gut.52.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Everest PH, Cole AT, Hawkey CJ, Knutton S, Goossens H, Butzler JP, Ketley JM, Williams PH. Roles of leukotriene B4, prostaglandin E2, and cyclic AMP in Campylobacter jejuni-induced intestinal fluid secretion. Infect Immun. 1993;61:4885–4887. doi: 10.1128/iai.61.11.4885-4887.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beltinger J, Hawkey CJ, Stack WA. TGF-alpha reduces bradykinin-stimulated ion transport and prostaglandin release in human colonic epithelial cells. Am J Physiol. 1999;276:C848–C855. doi: 10.1152/ajpcell.1999.276.4.C848. [DOI] [PubMed] [Google Scholar]
- 30.Ussing HH, Zerahn K. Active transport of sodium as the source of electric current in the short-circuited isolated frog skin. Reprinted from Acta. Physiol. Scand. 23: 110-127, 1951. J Am Soc Nephrol. 1999;10:2056–2065. [PubMed] [Google Scholar]
- 31.Adams RB, Guerrant RL, Zu S, Fang G, Roche JK. Cryptosporidium parvum infection of intestinal epithelium: morphologic and functional studies in an in vitro model. J Infect Dis. 1994;169:170–177. doi: 10.1093/infdis/169.1.170. [DOI] [PubMed] [Google Scholar]
- 32.Terres AM, Pajares JM, Hopkins AM, Murphy A, Moran A, Baird AW, Kelleher D. Helicobacter pylori disrupts epithelial barrier function in a process inhibited by protein kinase C activators. Infect Immun. 1998;66:2943–2950. doi: 10.1128/iai.66.6.2943-2950.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konkel ME, Mead DJ, Hayes SF, Cieplak W Jr. Translocation of Campylobacter jejuni across human polarized epithelial cell monolayer cultures. J Infect Dis. 1992;166:308–315. doi: 10.1093/infdis/166.2.308. [DOI] [PubMed] [Google Scholar]
- 34.Bras AM, Ketley JM. Transcellular translocation of Campylobacter jejuni across human polarised epithelial monolayers. FEMS Microbiol Lett. 1999;179:209–215. doi: 10.1111/j.1574-6968.1999.tb08729.x. [DOI] [PubMed] [Google Scholar]
- 35.Carew MA, Thorn P. Carbachol-stimulated chloride secretion in mouse colon: evidence of a role for autocrine prostaglandin E2 release. Exp Physiol. 2000;85:67–72. [PubMed] [Google Scholar]
- 36.Jones MA, Totemeyer S, Maskell DJ, Bryant CE, Barrow PA. Induction of proinflammatory responses in the human monocytic cell line THP-1 by Campylobacter jejuni. Infect Immun. 2003;71:2626–2633. doi: 10.1128/IAI.71.5.2626-2633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Spreeuwel JP, Duursma GC, Meijer CJ, Bax R, Rosekrans PC, Lindeman J. Campylobacter colitis: histological immunohistochemical and ultrastructural findings. Gut. 1985;26:945–951. doi: 10.1136/gut.26.9.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mellits KH, Mullen J, Wand M, Armbruster G, Patel A, Connerton PL, Skelly M, Connerton IF. Activation of the transcription factor NF-kappaB by Campylobacter jejuni. Microbiology. 2002;148:2753–2763. doi: 10.1099/00221287-148-9-2753. [DOI] [PubMed] [Google Scholar]
- 39.Mellits KH, Mullen J, Wand M, Smith J, Connerton I, Hawkey CJ. Activation of cellular genes by Campylobacter jejuni. Gastroenterol. 2003;148:A1097. [Google Scholar]
- 40.Mellits KH, Connerton IF, Loughlin MF, Clarke P, Smith J, Dillon E, Connerton PL, Hawkey CJ. Induction of a chemoattractant transcriptional response by campylobacter jejuni extract in colonocytes. BMC Microbiology. 2009;9:28. doi: 10.1186/1471-2180-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rinella ES, Eversley CD, Carroll IM, Andrus JM, Threadgill DW, Threadgill DS. Human epithelial-specific response to pathogenic Campylobacter jejuni. FEMS Microbiol Lett. 2006;262:236–243. doi: 10.1111/j.1574-6968.2006.00396.x. [DOI] [PubMed] [Google Scholar]
- 42.Scott RO, Thelin WR, Milgram SL. A novel PDZ protein regulates the activity of guanylyl cyclase C, the heat-stable enterotoxin receptor. J Biol Chem. 2002;277:22934–22941. doi: 10.1074/jbc.M202434200. [DOI] [PubMed] [Google Scholar]
- 43.Matkowskyj KA, Danilkovich A, Marrero J, Savkovic SD, Hecht G, Benya RV. Galanin-1 receptor up-regulation mediates the excess colonic fluid production caused by infection with enteric pathogens. Nat Med. 2000;6:1048–1051. doi: 10.1038/79563. [DOI] [PubMed] [Google Scholar]