

Abstract
HIV transcription is induced by the HIV-1 Tat protein, in concert with cellular co-factors including CDK9, CDK2, NF-κB, and others. The cells of most of the body’s organs are exposed to ~3–6% oxygen, but most in vitro studies of HIV replication are conducted at 21% oxygen. We hypothesized that activities of host cell factors involved in HIV-1 replication may differ at 3% versus 21% O2, and that such differences may affect HIV-1 replication. Here we show that Tat-induced HIV-1 transcription was reduced at 3% O2 compared to 21% O2. HIV-1 replication was also reduced in acutely or chronically infected cells cultured at 3% O2 compared to 21% O2. This reduction was not due the decreased cell growth or increased cellular toxicity and also not due to the induction of hypoxic response. At 3% O2, the activity of CDK9/cyclin T1 was inhibited and Sp1 activity was reduced, whereas the activity of other host cell factors such as CDK2 or NF-κB was not affected. CDK9-specific inhibitor ARC was much less efficient at 3% compared to 21% O2 and also expression of CDK9/cyclin T1-dependent IκB inhibitor α was repressed. Our results suggest that lower HIV-1 transcription at 3% O2 compared to 21% O2 may be mediated by lower activity of CDK9/cyclin T1 and Sp1 at 3% O2 and that additional host cell factors such as CDK2 and NF-κB might be major regulators of HIV-1 transcription at low O2 concentrations.
The role of physiological factors in the regulation of HIV-1 transcription is not well known. Our knowledge about activation of HIV-1 transcription mostly came from cell culture experiments conducted at atmospheric oxygen level (21% O2). In contrast, most organs in the body receive much less oxygen. In the better oxygenated organs (lungs, liver, kidneys, and heart), O2 varies between 4% and 14%; in brain, from 0.5% to 7%; in the eye, from 1 to 5%; and in the bone marrow, from 0% to 4% (reviewed in Ivanovic, 2009). Activities of cellular and viral proteins may differ under the lower than atmospheric oxygen tension. This is exemplified by the iron-responsive proteins, IRP-1 and IRP-2, where IRP-1 is the major sensor of intracellular labile iron in tissue culture experiments at 21% O2 but IRP-2 is the main iron sensor in vivo and in cells cultured at 3% O2 (Meyron-Holtz et al., 2004). Hypoxia can induce some viruses and inhibit the others. Hypoxia (1% O2) induces lytic replication of Epstein-Barr virus (Jiang et al., 2006) and Kaposi sarcoma-associated herpesvirus (Davis et al., 2001) but suppresses replication of oncolytic parvovirus Minute virus of mice (Servais et al., 2006), adenovirus (Shen et al., 2006), or Moloney murine leukemia virus (Puppo et al., 2007).
The HIV-1 promoter contains several binding sites for host transcription factors, including three Sp1 and two NF-κB binding sites (Pereira et al., 2000), but viral Tat protein is absolutely required for expression of the HIV-1 provirus (reviewed in Nekhai and Jeang, 2006). Tat binds to transactivation-responsive (TAR) RNA, a hairpin-looped structure found at the 5′-end of all HIV-1 transcripts, and recruits host cell factors to induce HIV-1 transcription (Nekhai and Jeang, 2006). These factors include CDK9/cyclin T1, which phosphorylates the carboxy-terminal domain (CTD) of RNA polymerase II (RNAP II) and increases the generation of full-length HIV-1 transcripts (reviewed in Price, 2000). Tat also recruits histone acetyl transferases (HATs) to the integrated provirus (Kiernan et al., 1999; Ott et al., 1999; Deng et al., 2000). NF-κB might act in concert with Tat and CDK9/cycin T1 (West et al., 2001) and recruit CDK9/cyclin T1 to HIV-1 LTR in a co-operative manner (Deng et al., 2000; Barboric et al., 2001) likely in part because of the interaction of Tat with the p65 subunit of NF-κB through NFBP protein (Sweet et al., 2005). In the absence of Tat, HIV-1 basal transcription is largely regulated by the Sp1 transcription factor (Jochmann et al., 2009). Also, in the absence of TAR RNA, Cyclin T1 can be targeted to the LTR by Sp1 (Yedavalli et al., 2003). Tat can induce Sp1 phosphorylation by DNA-PK, and such phosphorylation has been shown to increase Sp1’s contribution to HIV-1 transcription (Chun et al., 1998). In fact, a very early study showed that deletion of NF-κB sites did not significantly affect HIV-1 infectivity (Leonard et al., 1989) suggesting that NF-κB might be dispensable for viral replication. We previously showed that CDK2 is also an important regulator of HIV-1 transcription and viral replication (Deng et al., 2002; Nekhai et al., 2002; Ammosova et al., 2005) and that CDK2 may function by phosphorylating Tat (Ammosova et al., 2006).
Primary T cells cultured at 3–6% O2 maintain an intracellular redox environment similar to the in vivo situation, as opposed to T cells cultured at atmospheric 21% O2 in which the intracellular redox state is significantly altered (Atkuri et al., 2007). Recently, peripheral blood mononuclear cell (PBMC) cultured at 5% O2 were found to be efficiently activated by extracellular HIV-1 Tat protein, which mimicked the activation by IL-2 and PMA, and the activated cells were found to support HIV 1 replication (Sahaf et al., 2008). We recently showed that iron chelators inhibit HIV-1 transcription by inhibiting CDK2 activity and disrupting the association of CDK9 and cyclin T (Debebe et al., 2007). In cultured cells, iron chelators deplete iron from cellular enzymes including prolyl hydroxylase, which decreases prolyl hydroxylase activity and increases the protein level of hypoxia-inducible factor-1α (HIF-1α) thus mimicking the effect of hypoxia (reviewed in Sargent et al., 2005). Based on these results we hypothesized that HIV-1 transcription and replication would be reduced in the cells culture at 3% oxygen comparing to atmospheric oxygen concentration. In this article, we compared HIV-1 transcription, HIV-1 replication and cellular activities of host CDK9, CDK2, NF-κB, and Sp1 at 3% O2 and 21% O2. Our results indicate that CDK9 activity, CDK9/Cyclin T association and HIV-1 transcription and replication were lower at 3% O2 than at 21% O2, whereas the activities of CDK2 and NF-κB remained unchanged. Analysis of the HIV-1 promoters with the deletions of NF-κB sites or inactivation of Sp1 sites showed that the inactivation of Sp1 alleviated the inhibitory effect of 3% O2 suggesting that Sp1 activity is compromised at low oxygen. Interestingly, Tat-activated HIV-1 transcription at 21% O2 was much more sensitive to inhibition by ARC, a CDK9 inhibitor, that at 3% O2 and the expression of IκB inhibitor α, a CDK9/cyclin T1-dependent host cell gene, was reduced suggesting that CDK9/cyclin T1 activity is lower at 3% O2.
Materials and Methods
Materials
293T, HeLa, CEM, and THP-1 cells were purchased from ATCC (Manassas, VA). CEM-HIV-1 (LTR) green fluorescent protein (GFP) cells were obtained from the NIH AIDS Research and Reference Reagent Program (Courtesy of Dr. Jacques Corbeil). Chronically HIV-1 infected promonocytic U1 cells, a derivative of non-infected U937 cells and ACH-2 cells, a derivative of CEM T cells were obtained from the NIH AIDS Research and Reference Reagent Program (Courtesy of Dr. Thomas Folks). Recombinant CDK9/cyclin T1 was purified as previously described (Peng et al., 1998). Recombinant CDK2/cyclin E was expressed and purified as described (Deng et al., 2002). Histone H1 was purchased from Upstate Cell Signalling Solutions (Charlottesville, VA). Anti-Flag monoclonal antibodies, protein (G) and protein (A) agarose were purchased from Sigma (Atlanta, GA). All radioactive reagents were purchased from GE Health Care Life Sciences (Piscataway, NJ). Antibodies against CDK9, CDK2, and Cyclin T1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). HIF-1α antibodies were purchased from BD Transduction Laboratories (San Jose, CA). Horseradish peroxidase (HRP)-conjugated F(ab)2 fragment was purchased from Amersham Biosciences (Piscataway, NJ). HIV-1 Tat was expressed in Escherichia coli and purified on Aquapore RP-300 column (Applied Biosystems, Foster City, CA) by reverse-phase chromatography as we described. All other inorganic reagents were purchased from Fisher Scientific (Fair Lawn, NJ) or Sigma (St Louis, MO). 4-Amino-6-hydrazino-7-beta-D-Ribofuranosyl-7H-Pyrrolo (2,3-d)-pyrimidine-5-Carboxamide (ARC) (Radhakrishnan and Gartel, 2006) was a gift from Dr. Andrei L. Gartel (Departments of Medicine, Microbiology and Immunology, University of Illinois at Chicago).
Cell culture
To achieve 3% O2 and 1% O2 concentrations, cells were placed in a modular incubator chamber purchased from Billups~Rothenberg (Del Mar, CA). The chamber was then purged with 80 L (20 L/min for 4 min) of a gas mixture containing either, 92% N2, 3% O2, and 5% CO2 to achieve 3% O2 or 96% N2, 1% O2, and 5% CO2 to achieve 1% O2. After purging, the chamber was placed at 37°C and incubated for the indicated amount of time. CEM-GFP cells were cultured and maintained in RPMI Medium 1640 containing 10% fetal bovine serum (FBS), with 1% antibiotic solution (penicillin and streptomycin), and 500 μg/ml G418 (Invitrogen, Rockville, MD). 293T and HeLa cells were cultured in Dulbecco’s Modified Eagles Medium (DMEM) containing 10% FBS (Gibco-BRL, Rockville, MD) and 1% glutamine (Gibco-BRL). The viability of CEM-GFP cells was determined by trypan blue exclusion assay. Cell cultures were treated for 24 h under 21% O2, 3% O2, or 1% O2. To induce HIV-1 in U1 and ACH-2 cells, the cells were treated with TNF-α (10 μg/ml) for 48 h, and then washed with phosphate-buffered saline (PBS). After virus stimulation, the cells were cultured under 21% O2 or 3% O2 for 48, 72, and 96 h. Supernatants were collected and analyzed for p24 by ELISA in μQuant Facility (University of Maryland).
Peripheral blood mononuclear cells (PBMCs) isolation
PBMCs were isolated from 10 ml of whole blood collected in EDTA containing vacutainer tubes. The Howard University IRB committee approved the IRB protocol and the donor signed the consent from. The whole blood was diluted (1:1) with PBS and applied on Ficoll-Hypaque and centrifuged at 400g for 30 min. PBMCs were removed from the interphase and further washed with PBS. The PMBCs were cultured with 2.5 μg/ml phytohemagglutinin (PHA, Invitrogen) and 50 U/ml IL-2 at 3% O2 for 24 h to activate the cells as previously described. The activated PBMCs were infected with VSVG-pseudotyped HIV-1 Luc virus for 24 h and then incubated for 72 h at 3% O2 and 21% O2. Cells were washed and lysed in luclite buffer (Perkin-Elmer, Waltham, MA) and luminescence was measured on Labsystems Luminoscan RT (Perkin-Elmer).
Induction of HIV-1 transcription with Ad-Tat
The E1-deleted recombinant Adeno virus carrying Tat was generated as previously described (Ammosova et al., 2003). CEM-GFP cells were infected in a 96-well plate containing 400,000 cells per well. After 18 h of incubation, the cells were visually analyzed using Olympus CKX41 fluorescent microscope with blue filter. Then 10 μl aliquots were removed, supplemented with trypan blue and counted to determine the cellular viability. The remaining cells were precipitated by centrifugation, lysed for 20 min at room temperature in 50 μl of lysis buffer, containing 20 mM HEPES at pH 7.9, 0.1% NP-40, and 5 mM EDTA transferred into 150 μl of PBS. The fluorescence was measured with 480 nm excitation and 510 nm emission on Luminescence Spectrometer LS50B (Perkin-Elmer) equipped with the robotic 96-well scanner.
Induction of HIV-1 transcription with recombinant Tat protein
HIV-1 Tat was expressed in E. coli using the pGEM2 Tat bacterial expression vector (obtained from the NIH AIDS Research and Reference Reagent Program, courtesy of Dr. Richard Gaynor) and purified on Aquapore RP-300 column (Applied Biosystems) by reversed-phase chromatography. Purified Tat was dissolved in RPMI media and added to CEM-GFP cells plated in a 96-well plate at 400,000 cells per well in the presence of 10 μM chloroquine (Frankel and Pabo, 1988). At 24 h incubation fluorescence was measured as described above.
Plasmids
The HIV-1 reporter contained HIV-1 LTR (−138 to +82) followed by a nuclear localization signal (NLS) and the LacZ reporter gene (courtesy of Dr. Michael Emmerman, Fred Hutchinson Cancer Institute, Seattle, WA) (Kimpton and Emerman, 1992). The HIV-1 reporter plasmid without TAR contained a deletion of +19 to +87 nucleotides of LTR introduced by restriction digestion with BglII (Ammosova et al., 2003). The pGEM2 Tat bacterial expression vector (courtesy of Dr. Richard Gaynor), pHEF-VSVG expression vector (courtesy of Dr. Lung-Ji Chang), and pNL4-3.Luc.R−E− (Courtesy of Dr. Nathaniel Landau) were obtained from the NIH AIDS Research and Reference Reagent Program. Corp., Carlsbad, CA). WT HIV-1 LTR (−105 to +77) followed by the luciferase reporter gene (κB SP WT), HIV-1 (−105 to +77) with Sp1 inactivated sites followed by the luciferase reporter gene (Sp I +II +III) and HIV-1 (−81 to +77) with NF-κB-deleted sites followed by the luciferase reporter gene were kindly provide by Dr. Manuel López-Cabrera (Unidad de Biología Molecular, Hospital Universitario de la Princesa, Madrid, Spain; Gomez-Gonzalo et al., 2001).
Transient transfections
293T or HeLa cells were cultured in DMEM containing 10% FBS. Co-transfections with Tat-expressing vector and HIV-1 LTR-LacZ or HIV-1 LTRΔTAR were performed at 75% confluence using Lipofectamine and Plus reagents (Invitrogen) and the indicated reporter plasmids. After transfection the cells were cultured for an additional 48 h and β-galactosidase activity was analyzed using quantitative o-nitrophenyl-β-D-galactopyranoside (ONPG)-based assay (Ammosova et al., 2003). Transfections were normalized using EGFP.
β-Galactosidase assays
Cells were washed with PBS and lysed for 20 min at room temperature in 50 μl of lysis buffer, containing 20 mM HEPES at pH 7.9, 0.1% NP-40, and 5 mM EDTA. Subsequently, 100 μl of ONPG solution (72 mM Na2PO4 at pH 7.5, 1 mg/ml ONPG, 12 mM MgCl2, 180 mM 2-mercaptoethanol) was added and incubated at room temperature until a yellow color was developed. The reaction was stopped by addition of 100 μl of 1 M Na2CO3. The 96-well plate was analyzed in a micro plate reader at 414 nm (Lab Systems Multiscan MS, Perkin-Elmer, Waltham, MA).
Analysis of Tat expression
HeLa cells were infected with Ad-Tat at ~10 Pfu per cell to achieve high level of Tat expression in infected HeLa cells. Additionally, 239T cells were transfected with Flag-Tat expression vector using lipofectamine. At 48 h post-infection or transfection, the cells were washed with PBS and lysed in whole cell lysis buffer (50 mM Tris–HCl, pH 7.5, 0.5 M NaCl, 1% NP-40, 0.1% SDS) supplemented with protease cocktail (Sigma). After 10 min on ice, cellular material was scraped and then centrifuged at 14,000 rpm, 4°C for 30 min. The supernatant was recovered and protein concentration was measured by Bradford assay (Bio-Rad, Hercules, CA). Equal amounts of protein lysate was resolved on 15% Tris–tricine SDS–PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Allen, TX). The membrane was analyzed with anti-Flag monoclonal antibodies and with anti-α-tubulin monoclonal antibodies.
Preparation of pseudotyped HIV-1 virus expressing luciferase
293T cells were grown on 100 mm plates and transfected using Ca-Phosphate method (Ammosova et al., 2003) with VSVG-expressing vector (gpHEF-VSVG) and pNL4-3.Luc.R−E− molecular clone that contained two nonsense frame shifts in Env and Vpr genes and Luciferase gene cloned in place of nef (Connor et al., 1995; He and Landau, 1995). At 72 h post-transfection the media was collected, briefly centrifuged at 1,000g for 10 min and then the virus was collected by centrifugation at 4°C for 6 h at 14,000g. The precipitated virus was resuspended in PBS containing 10% glycerol, aliquoted, and stored at −70°C.
Luciferase assay
CEM T cells, THP-1 cells or activated PBMC were infected with VSVG-pseudotyped pNL4-3.Luc.R−E− virus and then cultured at 0.5 × 106 cells/ml in 6-well plates as indicated. Then the cells were collected, washed with PBS and resuspended in 100 μl of PBS. Then 100 μl of reconstituted luclite buffer (luclite kit, Perkin-Elmer) was added to each well. After 10 min incubation the lysates were transferred into the white plates (Perkin-Elmer) and luminescence was measured on Labsystems Luminoscan RT (Perkin-Elmer).
Immunoblots and kinase assay
Cells were washed with PBS, and whole-cell lysates were prepared from the cells cultured at 3% O2 and 21% O2 using whole cell lysis buffer (50 mM Tris–HCl, pH 7.5, 0.5 M NaCl, 1% NP-40, 0.1% SDS) supplemented with protease cocktail (Sigma). Protein concentration of lysates was measured by Bradford assay (Bio-Rad) and 30–50 μg of total protein was subjected to electrophoresis on 10% SDS–PAGE. The gels were transferred onto PVDF membrane (Millipore), and analyzed for CDK2, CDK9, and Cyclin T1. The blots were developed and quantified using ChemiDoc XRS Station (Bio-Rad). Immunoprecipitations were carried out with 400 μg of the cell lysate and 800 ng of antibodies combined with 50 μl of 50% slurry of protein A agarose for 2 h at 4°C in a TNN Buffer containing 50 mM Tris–HCl, pH 7.5, 0.15 M NaCl, and 1% NP-40. The agarose beads were precipitated, washed with TNN buffer and divided into two parts, and used for the kinase assay and Western blotting. Kinase assay was performed at 30°C for 30 min in a kinase assay buffer (50 mM HEPES-KOH, pH 7.9, 10 mM MgCl2, 6 mM EGTA, 2.5 mM DTT) containing 1 μg of histone H1 as a substrate, 200 μM cold ATP, and 5 μCi of (γ-32P) ATP. CTD phosphorylation assay was performed at 30°C for 30 min in 20 μl of kinase assay buffer (50 mM HEPES-KOH, pH 7.9, 10 mM MgCl2, 6 mM EGTA, 2.5 mM DTT) containing 4 μg of CTD substrate, 250 μM cold ATP, and 5 μCi of (γ-32P)ATP. Recombinant CDK2/cyclin E or CDK9/cyclin T1 were used as controls. Reaction was stopped with SDS-loading buffer and resolved on 10% PAGE. Dried gel was exposed to Phosphor Imager screen. In parallel, the immunoprecipitations were resolved on 10% Tris–glycine SDS–PAGE, transferred to PVDF membranes (Millipore) and immunoblotted with appropriate antibodies.
Cell proliferation assay
CEM cells or THP1 cells were grown in 96-well plates at 21% O2 or 3% O2 then supplemented with 0.2 μM Calcein-AM (Molecular probes, Invitrogen, Rockville, MD) for 10 min at 37°C and 21% O2. Fluorescence was measured with 495 nm excitation and 515 nm emission on Luminescence Spectrometer LS50B (Perkin-Elmer) equipped with the robotic 96-well scanner.
Analysis of NF-κB activity
293T cells were transfected at 30% confluency in 96-well plates with CMV-EGFP reporter in combination with NF-κB driven e-selectin-Luciferase reporter (gift from Dr. Chou-Zen Giam, Uniformed Services University of the Health Sciences, Bethesda, MD), or pNL4-3.Luc.R−E− (HIV-1 promoter) for 3 h at 37°C by lipofectamine and then simultaneously placed at 3% O2 and 21% O2 for 48 h. The media was then removed from the cells and equal amounts of 1 × PBS and Luciferase buffer were added to measure luciferase activity. Following luciferase activity, GFP was measured at excitation 495 and emission 515.
Analysis of HIV-1 promoters with the deletion of NF-κB and inactivation of Sp1 sites
293T cells were transfected at 30% confluency in 96-well with vectors in which luciferase reporter was driven by WT HIV-1 LTR (−105 to +77), HIV-1 LTR (−105 to +77) with Sp1 inactivated sites or HIV-1 LTR (−81 to +77) with NF-κB-deleted sites (courtesy of Dr. Manuel López-Cabrera, Unidad de Biología Molecular, Madrid, Spain). The cells were transfected for 3 h at 37°C by lipofectamine and Plus and then simultaneously placed at 3% O2 and 21% O2 for 18 h. The media was then removed from the cells and equal amounts of 1 × PBS and Luciferase buffer were added to measure luciferase activity.
RT-PCR analysis of expression of CDK9/cyclin T1-dependent genes
Total RNA was isolated with TRIzol reagent from 293T cells culture for 48 h at 3% O2 and 21% O2 using the Invitrogen protocol (Invitrogen™, Life technologies, Carlsbad, CA). Cells were lysed in 1 ml TRIzol/10 cm2 culture dish. Purified total RNA was quantified by absorbance at 260 nm and quality checked on a 1% agarose gel. Reverse Transcription (RT) was performed using the Superscript™ RT-PCR kit (Invitrogen, Life Technologies). Random hexamers were used for RT reaction. Primers for PCR of individual target sequences were designed for NF-κB inhibitor alpha (forward: GCCTGGACTCCATGAAAGAC, reversed: GTCTGCTGCAGGTTGTTCTG); Gro-beta (forward: CTGCTGCTCCTGCTCCTG, reverse: AGACAAGCTTTCTGCCCATT), IL8 (forward: GTGCAGTTTTGCCAAGGAGT, reverse: ACTTCTCCACAACCCTCTGC); HLA-DRA (forward: TCCCGAGCTCTACTGACTCC, reverse: CAGACCGTCTCCTTCTTTGC), and β-actin (forward: GCGGGAAATCGTGCGTGCGTGACATT, reverse:GATGGAGTTGAAGGTAGTTTCGTG). Primers were generated to amplify a product no larger than 500 nucleotides, incorporating two different exons, and spanning no <0.5 kb of intron nucleotides. Amplification of target cDNA was performed in an amplification format of melting at 95°C for 15 sec, annealing at 55°C for 30 sec, and extension at 72°C for 45 sec for 27 cycles.
Results
HIV-1 transcription is reduced at 3% versus 21% O2 concentration
To study the effect of O2 concentration on HIV-1 transcription, we used CEM T cells containing an integrated GFP under the control of HIV-1 LTR (CEM-GFP). These cells have a low level of GFP expression in the absence of HIV-1 Tat protein (Nekhai et al., 2007). We examined and compared HIV-1 transcription at two different O2 concentrations: 21% O2 and 3% O2. Transcription of GFP from HIV-1 LTR was induced by infection with recombinant adenovirus expressing Tat (Ad-Tat) and GFP fluorescence measured as we previously described (Debebe et al., 2007; Nekhai et al., 2007). While Ad-Tat-induced HIV-1 LTR-driven transcription of GFP in CEM-GFP cells cultured at 21% O2, there was less induction of GFP at 3% O2 (Fig. 1A, twofold difference, P <0.05, N = 4). Because the level of Tat protein produced by Ad-Tat in CEM-GFP cells was insufficient for quantification by Western blot, direct comparison of Tat levels at 3% and 21% oxygen was not possible. To rule out the possibility that lower oxygen concentration reduced the level of adenovirus infection and/or level of Tat expression from adenoviral vector, we treated CEM-GFP cells with purified recombinant Tat protein added to the media in the presence of chloroquine (Frankel and Pabo, 1988) (Fig. 1B). Chloroquine, a lysosome inhibitor, was added to prevent Tat protein degradation after its endocytosis by the cells. HIV-1 LTR-dependent GFP expression induced by recombinant Tat was significantly lower (about twofold, P <0.05, N = 3) in CEM-GFP cells cultured at 3% O2 compared to 21% O2 (Fig. 1B).
Fig. 1.

Less efficient HIV-1 transcription at 3% O2 compared to 21% O2. A: HIV-1 transcription in CEM GFP cells infected with Ad-Tat. CEM GFP cells were grown in 96-well plates, and different amount of the cells were infected with Adeno-Tat and cultured at either atmospheric 21% O2 or 3% O2 conditions. After 18 h of incubation, the cells were lysed and GFP fluorescence was measured on a luminescence spectrometer. The results are representative of four experiments. B: HIV-1 transcription in CEM GFP cells treated with purified Tat protein. CEM-GFP cells were grown in 96-well plates and treated with purified recombinant Tat protein dissolved in RPMI media supplemented with 100 μM chlorquine to induce HIV-1 transcription. After 18 h treatment under 3% O2 or 21% O2, the cells were lysed and GFP fluorescence was measured on a luminescence spectrometer. The results are representative of three experiments.
To investigate if HIV-1 transcription at 3% oxygen depended on the presence of the TAR region, 293T cells and HeLa cells were transiently transfected with HIV-1 LTR-LacZ reporter plasmid and Tat-expression vector (Ammosova et al., 2003). Tat-induced transcription was less efficient in cells cultured at 3% O2 versus 21% O2 (2.7-fold reduction for 293T cells and 1.7-fold for HeLa cells, P <0.05, N = 3) (Fig. 2A,B, lane 2). However, basal HIV-1 transcription and transcription from HIV-1 LTR with the TAR-coding sequence deleted (HIV-1 LTRΔTAR) were not significantly changed (Fig. 2A,B, lanes 3 and 4). These results indicate that the lower HIV-1 transcription at 3% oxygen concentrations was likely to be connected to changes in the function of Tat.
Fig. 2.

Less efficient HIV-1 transcription in 293T and HeLa cells at 3% compared to 21%O2. 293Tcells(A) or HeLa cells(B)were grownin96-well plates and transiently transfected with HIV-1 LTR-LacZ vector without (lane 1) or with Tat-expressing vector (lane 2). The cells were also transfected with TAR deleted construct of HIV-1LTR-LacZ(HIV-1 LTRΔTAR) without (lane 3) or with Tat-expressing vector (lane 4). The cells were cultured at 21% O2 or3%O2 for18 h. Then the cells were lysed and analyzed for β-galactosidase activity. In parallel, MTT assay was performed and used for normalization. The results are representative of three experiments. C: 3% versus 21% O2 does not affect Tat expression. HeLa cells were infected with recombinant adenovirus expressing Flag-tagged Tat (Ad-Tat) (lanes 3 and 4). 293T cells were transfected with Flag-Tat expression vector (lanes 7 and 8). Mock-infection was carried out with Ad-GFP (lanes 1 and 2);and mock-transfection was carried out with an empty vector (lanes5 and 6). Cells wereincubatedat21%O2 (lanes1,3,5, and 7)or 3% O2 (lanes2,4,6, and 8). At 48 h post-infection or transfection, the cells were lysed and equal amount of lysate was resolved by 15% Tris–tricine SDS–PAGE, and immunoblotted with anti-Flag or anti-α-tubulin monoclonal antibodies. The results are representative of two experiments.
To determine whether oxygen tension influenced the expression of Tat, we compared the expression of Tat protein in HeLa cells infected with Adeno-Tat and in 293T cells transiently transfected with Flag-Tat expression vector at 3% O2 and 21% O2 (Fig. 2C). In both cell lines there was no difference in Tat protein expression in the cells cultured at 21% O2 versus 3% O2 (Fig. 2C, compare lanes 3 and 7, to the lanes 4 and 8). Taken together, these results suggested that host cell factors involved in Tat-mediated HIV-1 transcription may be regulated differently at 3% O2 compared to 21% O2, if cellular viability is not compromised.
Growth of cultured T cells and monocytes is not lower at 3% O2 concentration
To exclude the possibility that HIV-1 transcription is less at 3% O2 because cultured cells have lower degrees of growth or survival at this tension, we analyzed viability and proliferation of CEM T cells cultured for 24 h at 3% and 21% O2. There was no significant difference in the viability of CEM-GFP cells cultured at 3% O2 or 21% O2 as determined by trypan blue exclusion assay (Fig. 3A). Analysis of cell proliferation using calcein-AM uptake assay showed increased proliferation of CEM T cells (Fig. 3B) and a slight increase in proliferation of promonocytic THP-1 cells at 3% O2 (Fig. 3C). Thus, lower degree of HIV-1 transcription at 3% O2 was not likely to be due to the poorer cellular proliferation or increased cellular death.
Fig. 3.

Effect of O2 concentration on cell viability and proliferation. A: Determination of CEM cells viability by trypan blue exclusion assay. CEM-GFP cells were grown in a 96-well plate, infected with Ad-Tat, and cultured at 21% O2 or physiological 3% O2 for 18 h. Cells were then supplemented with Trypan Blue and counted to determine cell viability by Trypan blue exclusion assay. The results are representative of three experiments. B and C: Determination of cellular proliferation by calcein-AM uptake assay. CEM cells (part B) or THP-1 cells (part C) were seeded in 96-well plates with threefold increment, cultured for the indicated amount of days at 21% O2 or 3% O2. The cells were supplemented with calcein-AM and calcein fluorescence was measured as described in Materials and Methods Section. The results are representative of three experiments.
3% O2 concentration leads to reduced HIV-1 replication compared to 21% O2
We next examined the effect of oxygen concentration on HIV-1 replication in acute and chronic HIV-1 infection. Because oxygen concentration can affect variable responses in different types of cells, we compared the effects of 3% and 21% O2 on one round of HIV-1 replication in three different types of HIV-1 target cells: CEM T lymphocytes, promonocytic THP-1 cells, and primary human PBMCs. The cells were infected with pseudotyped HIV-1 virus containing luciferase in place of nef gene (HIV-1 Luc) (Connor et al., 1995; He and Landau, 1995). CEM T cells and THP-1 cells were infected overnight with the virus and then cultured for 72 h at 3% O2 or 21% O2. Primary PBMC were first activated with 2.5 μg/ml PHA and 50 U/ml IL-2 at 3% O2 for 24 h (Sahaf et al., 2008) and then infected overnight with the virus and cultured for 72 h at 3% O2 or 21% O2. Luciferase activity was approximately twofold lower in all HIV-1 Luc infected cells that were cultured at 3% O2 as compared to the cells cultured at 21% O2 (Fig. 4A, compare lanes 2 and 4, P <0.05, N = 3).
Fig. 4.
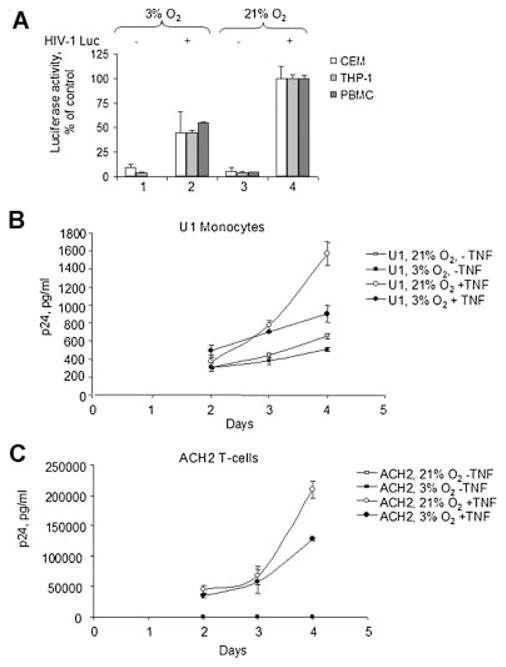
HIV-1 replication in 3% and 21% O2. A: CEM cells, THP-1 cells or human peripheral blood mononuclear cells (PBMCs) were infected with pseudotyped HIV-1 Luc virus as described in Materials and Methods Section and then cultured at 21% O2 (lane 4) or 3% O2 (lane 2) for72 h. Then the cells were lysed and luciferase activity was measured as described in Materials and Methods Section. Lanes 1 and 3 are uninfected controls. The results are quadruplicates and representative of three experiments. B and C: Chronically HIV-1 infected promonocytic U1 cells (B), a derivative of non-infected U937 cells and infected ACH-2 cells (C), a derivative of CEM T cells, were induced with TNF-α for 48 h. After the induction, the cells were cultured at 21% O2 or physiological 3% O2 for48, 72, and 96 h. Following treatment, culture media was collected and p24 determine by ELISA assays as described in Materials and Methods Section. The results are quadruplicates and representative of two experiments.
We next examined the effect of O2 concentration on HIV-1 replication in promonocytic and T lymphocyte cell lines that were chronically infected with HIV-1. Promonocytic U1 cell line was a derivative of non-infected U937 cell line and ACH-2 cell line was derived from CEM T cell line. In both cell lines, HIV-1 replication was induced with TNF-α for 48 h and after that the cells were continuously cultured either at 3% O2 or 21% O2. Both U1 and ACH-2 cells produced low levels of p24 in the absence of TNF-α treatment (Fig. 4B,C). Neither U937 nor CEM T non-infected control cell lines produced any detectable amount of p24 (not shown). Induction of HIV-1 replication with TNF-α resulted in a significant increase of p24 production in both U1 and ACH-2 cells cultured at 21% O2, most markedly at day 4 post-induction (Fig. 4B,C). In contrast, p24 levels were significantly lower at day 4 in U1 and ACH-2 cells induced with TNF-α and cultured at 3% O2 (Fig. 4B,C, 1.7- to 1.8-folds less, P <0.05, N = 3). Taken together, these results indicate that HIV-1 replication is lower cells cultured at 3% O2 compared to 21% O2.
HIF-1α is induced at 1% but not 3% O2
Hypoxia or ischemia, a local decrease in oxygen tension that occurs in many pathological processes, affects many cellular genes through the stabilization of the HIF-1α transcriptional factor (Kenneth and Rocha, 2008). To study if HIF-1α was involved in the downregulation of Tat-induced HIV-1 transcription, we analyzed the effect of various oxygen concentrations on HIF-1α expression in 293T cells. As a positive control, we used 0.3 nM CoCl2 (Hwang et al., 2006) that substitutes for iron in prolylhydroxylase and induces the expression of HIF-1 α in 293T cells (Fig. 5A, lane 1). HIF-1α expression was markedly increased in the cells cultured for 4 h at 1% O2, but not at 3% O2 or 21% O2 (Fig. 5A, lanes 2–4). Surprisingly, HIF-1α expression was not detected in the cells cultured for 24 h at 1% O2 (Fig. 5A, lane 6), and was not observed in the cells cultured for 24 h at 3% O2 or 21% O2 (Fig. 5A, lanes 5 and 7). Similar results were observed in CEM T cells (not shown). These results indicate that HIF-1 α is expressed when the cells are cultured at 1% O2 but not at 3% O2, and that its expression is transient in nature and might be an early response for the cell when exposed to hypoxic conditions. Thus, HIF-1α is unlikely to be the factor that leads to reduction of HIV-1 transcription and replication at 3% O2 concentration compared to 21% O2.
Fig. 5.

Effect of O2 concentration on HIF-1α, CDK2, and NF-κB. A: EffectofO2 concentration on the expression of HIF-1α.293Tcellswere grown at 21% O2, 3% O2, or 1% O2 for the indicated amount of time. Where shown, the cells were treated with 0.3 nM CoCl2. The equal amount of protein lysate was resolved on 10% SDS–PAGE and immunoblotted with the HIF-1α antibodies. The results are representative of four experiments. B: Effect of O2 concentration on cellular activity of CDK2. 293T cells were grown in 100 mm plates at 21% O2, 3% O2, or 1% O2 for the indicated amount of time. Where indicated, the cells were treated with 100 μM ICL670 or 0.3 nM CoCl2. The cells were lysed and CDK2 was immunoprecipitated as described in Materials and Methods Section. The precipitated material was divided in half and one portion was incubated with histone H1 in the presence of γ-(32P) ATP. Kinase reactions were resolved on 10% SDS–PAGE and exposed to the Phosphor Imager screen. The second half of the precipitated material was resolved on 10% SDS–PAGE and immunoblotted with anti-CDK2 antibodies. Lane 1, preimmune IgGs were used for the immunoprecipitation. The results are representative of three experiments. C: Effect of O2 concentration on NF-κB-driven transcription. 293T cells were transiently transfected with CMV-EGFP reporter in combination with e-selectin-Luciferase (an NF-κB driven promoter), or pNL4-3.Luc.R−E− (HIV-1 promoter). The cells were cultured at21% O2 or3%O2 for48 h. Then the cells were lysed and luciferase activity was measured. In parallel, EGFP fluorescence was measured and used for normalization. The results are representative of three experiments.
O2 concentration does not have a significant effect on CDK2 activity
Our results supported the hypothesis that the observed effect of oxygen concentration was due to altered regulation of host factors involved in HIV-1 transcription. We previously demonstrated that CDK2 plays an important role in Tat-activated HIV-1 transcription. Also, we and others showed that iron chelators which mimic the effect of hypoxia potently inhibit cellular activity of CDK2 (Debebe et al., 2007; Pahl et al., 2007). So we hypothesized that decreasing O2 concentration from 21% to 3% might have an effect similar to that of iron chelators and lead to reduced CDK2 activity in cells. To analyze CDK2 activity we use the Histone H1 phosphorylation assay (described in Materials and Methods Section). As we previously described, treatment with the iron chelator ICL670 inhibited CDK2 activity (Fig. 5B, lane 5). Interestingly, treatment with CoCl2 also inhibited CDK2 activity (Fig. 5B, lane 9) suggesting that hypoxia might indeed inhibit CDK2. In agreement with this observation, CDK2 activity was markedly lower at 1% versus 21% O2 both at 4 and 24 h of treatment (Fig. 5B, lanes 4 and 8). In contrast, there was almost no difference in CDK2 activity at 3% versus 21% O2 at 4 h treatment (Fig. 5B, compare lanes 2 and 3) and no difference at 24 h treatment (Fig. 5B, compare lanes 6 and 7).
NF-κB activity is higher at 3% compared to 21% O2
NF-κB activity has been shown to be affected by hypoxia (reviewed in Taylor, 2008). To determine whether oxygen concentration has an effect on NF-κB activity, we analyzed the activity of NF-κB-dependent e-selectin promoter (Fu et al., 2003) at 3% O2 and 21% O2. The activity of e-selectin promoter was slightly higher at 3% O2 as compared to 21% O2, while the activity of HIV-1 LTR promoter was lower (Fig. 5C, lanes 1 and 2). This result suggests that NF-κB activity is the same at 3% and 21% O2 and that changes in NF-κB activity are not likely to be the cause for lower HIV-1 transcription at 3% O2.
Sp1-dependent Tat-induced transcription is inhibited at 3% O2
Sp1 activity is has important for HIV-1 basal transcription (Jochmann et al., 2009) and Tat-induced HIV-1 transcription (Chun et al., 1998). To determine whether oxygen concentration has an effect on Sp1-driven and NF-κB-driven HIV-1 transcription, we analyzed the activity of HIV-1 promoters with the deletion of NF-κB sites or inactivation of Sp1 sites (Gomez-Gonzalo et al., 2001) at 3% O2 and 21% O2. In the absence of Tat, basal the activity of WT HIV-1 LTR promoter at 3% O2 was lower less than twofold (Fig. 6A, lane 1), whereas the activity of HIV-1 LTR without NF-κB sites was about twofold lower (Fig. 6A, lane 2), and the activity of HIV-1 LTR with inactivated Sp1 sites—about threefold lower (Fig. 6A, lane 3). Tat-induced transcription was lower at 3% O2 by about fourfold for WT HIV-1 LTR and HIV-1 LTR without NF-κB sites (Fig. 6B, lanes 1 and 2). Inactivation of Ap1 sites reduced Tat-transactivation but also made it less sensitive to 3% O2 with only twofold reduction in Tat-transactivation (Fig. 6B, lane 3). These results indicate that Sp1 sites are the most critical for the basal and Tat-induced HIV-1 transcription and that the reduction of Tat-induced transcription is likely due to the deregulation of Sp1 activity, although both Sp1 and NF-κB are required for the optimal HIV-1 promoter activity.
Fig. 6.

Effect of O2 concentration on basal and Tat-induced transcription of HIV-1 LTR with deletion of NF-κB sequences and inactivation of Sp1 sites. 293T cells were transiently transfected with indicated HIV-1 LTR-Luciferase reporters without Tat (A) or with Tat (B). Lane 1, WT HIV-1 LTR (−105 to +77). Lane 2, HIV-1 LTR (−81 to +77) with NF-κB-deleted sites. Lane 3, HIV-1 LTR (−105 to +77) with Sp1 inactivated sites. The cells were cultured at 21% O2 or 3% O2 for 18 h. Then the cells were lysed and luciferase activity was measured.
CDK9/cyclin T1 activity is lower in cells cultured at 3% compared to 21% O2
Next, we analyzed the effect of O2 concentration on the activity of CDK9/cyclin T1. HIV-1 transcription in CEM-GFP cells, HeLa-MAGI cells, and 293T cells at 21% O2 is strongly dependent on CDK9 as we recently showed using a specific CDK9 inhibitor, ARC, and a dominant-negative CDK9 (Nekhai et al., 2007). Expression of both CDK9 and cyclin T1 were not different in cells cultured at 3% or 21% O2 (Fig. 7A,B). To analyze the activity of CDK9/cyclin T1, Cyclin T1 was immunoprecipitated and the activity of co-precipitated CDK9 was assayed with GST-CTD as substrate. CTD kinase activity of CDK9/cyclin T1 was markedly lower in 293T cells cultured at 3% versus 21% O2 (Fig. 7C, lane 2). In contrast, there was no significant difference in total CDK9 kinase activity when CDK9 was immunoprecipitated with anti-CDK9 antibodies from lysates of 293T cells cultured at 21% O2 and 3% O2 and the kinase activity was analyzed with GST-CTD as substrate (Fig. 7D, lanes 2 and 3). Our recent study showed a reduced association of CDK9 and cyclin T1 in the cells treated with iron chelators. To analyze whether the association of CDK9 and cyclin T1 was different between 3% and 21% O2, we immunoprecipitated CDK9 and determined the amount of co-precipitated cyclin T1 by immunoblotting. While the amount of CDK9 precipitated with anti-CDK9 antibodies was the same at 21% O2 and 3% O2 (Fig. 7E, lanes 2 and 3), the amount of cyclin T1 co-precipitating with CDK9 at 3% O2 was significantly lower (Fig. 7F, lane 3). These results indicate that CDK9/cyclin T1 may not associate properly in the cells cultured at 3% O2 and that this may cause lower CDK9/cyclin T1 activity but not lower overall activity of CDK9. To analyze directly the contribution of CDK9 to the regulation of HIV-1 transcription at 3% O2, we used a CDK9 inhibitor, ARC, that we previously showed to inhibit HIV-1 transcription at 21% O2 (Nekhai et al., 2007). 293T cells were transiently transfected with HIV-1 LTR-LacZ reporter plasmid and Tat-expression vector, treated with different concentrations of ARC and cultured at 3% O2 versus 21% O2. While at 21 O2, ARC efficiently inhibited Tat-induced HIV-1 transcription at sub-micromolar concentrations (IC50 = 0.17 μM, Fig. 7G), the inhibition of HIV-1 transcription at 3% O2 required much higher ARC concentrations (IC50 = 3 μM, Fig. 7G). This result suggests that HIV- 1 transcription might be much less dependent on CDK9 at 3% O2. To determine if transcription of CDK9/cyclin T1-dependent host cell genes might be affected, we analyzed the expression of IκBα, Gro-β, and IL-8 all shown to be induced by CDK9/cyclin T1 (Nowak et al., 2008), and HLA-DRA that was shown to be repressed by CDK9/cyclin T1 (Kanazawa and Peterlin, 2001). Expression of IκBα was reduced at 3% O2 whereas the expression of Gro-β and HLA-DRA was not changed (Fig. 7H), and the expression of IL-8 was not detectable (not shown). This results is consistent with the possibility that CDK9/cyclin T1 expression is reduced at 3% O2 as the expression of at least one CDK9/cyclin T1-dependent gene was reduced.
Fig. 7.

Effect of O2 concentration on cellular activity of CDK9. A and B: The effect of O2 concentration on the expression of CDK9 and cyclin T1. 293T cells were grown in 100 mm plates at 21% O2 (lane 1) or 3% O2 (lane 2) for 18 h. The equal amount of protein lysate was resolved on 10% SDS–PAGE and immunoblotted with the indicated antibodies. C: O2 concentration and CDK9/cyclin T1 activity. Cyclin T1 was immunoprecipitated from cell lysates prepared as in part A. The immunoprecipitated material was incubated with recombinant GST-CTD in the presence of γ-(32P) ATP. Kinase reactions were resolved on 10% SDS–PAGE and exposed to the Phosphor Imager screen. Lane 1, cells treated at 21% O2. Lane 2, cells treated at 3% O2. Lane3, preimmune IgGs were used for the immunoprecipitation. Lane 4, recombinant CDK9/cyclin T1 was used to phosphorylate GST-CTD. D: O2 and total CDK9 activity. CDK9 was immunoprecipitated from 293T cell lysates prepared as in part A. The immunoprecipitated material was incubated with recombinant GST-CTD in the presence of γ-(32P) ATP. Kinase reactions were resolved on 10% SDS–PAGE and exposed to the Phosphor Imager screen. Lane 1, preimmune IgGs were used for the immunoprecipitation. Lane 2, cells treated at 21% O2. Lane 3, cells treated at 3% O2. E and F: O2 concentration and dissociation of CDK9 and cyclin T1. CDK9 was immunoprecipitated from 293T cell lysates prepared as in part A. The immunoprecipitated material was resolved on10% SDS–PAGE and immunoblotted with antibodies against CDK9(part E) or cyclin T1 (part F). Lane 1, preimmune IgGs were used for the immunoprecipitation. Lane 2, cells treated at 21% O2. Lane 3, cells treated at 3% O2. All experiments are representative of at least three different experiments. G: O2 concentration and inhibition of HIV-1 transcription by ARC.293Tcells were grown in 96-well plates and transiently transfected with HIV-1LTR-LacZ vector with Tat-expressing vector (lane 2). The cells were treated with indicated concentrations of ARC and cultured at 21% O2 or 3% O2 for 18 h. Then the cells were lysed and analyzed for β-galactosidase activity. The results are triplicates and representative of two experiments. H: O2 concentration and expression of CDK9/cyclin T1-dependent genes. Semi-quantitative RT-PCR analysis of CDK9/cyclin T1-dependent genes. Total RNA was isolated from 293T cells cultured at 21% O2 and 3%, reverse transcribed and amplified with primers for IκBα, Gro-β and HLA-DRA and β-actin as described in Materials and Methods Section. PCR products were resolved on 2% agarose gel and visualized with ethidium bromide staining. No RT, control in which RNA was not reverse transcribed.
Taken together, our results demonstrate that reduced HIV-1 transcription and replication at 3% O2 compared to 21% O2 may be related to less efficient association of CDK9 with Cyclin T1 and resultant lower CDK9 activity.
Discussion
In this study we showed that HIV-1 transcription and viral replication is less efficient in cells cultured at 3% O2 concentration compared to 21% O2. CD4+ T cells largely reside within lymphatic tissues and in the gastrointestinal tract where CD4+ depletion occurs (Brenchley et al., 2004). HIV-1 latency in non-dividing resting PBMC has long been recognized as a major obstacle for the success of highly active anti-retroviral therapy (HAART) (Han et al., 2007). In the individuals treated with HAART, viremia is reduced to non-detectable levels, at the same time HIV-1 that resides in peripheral tissues is able to escape HAART, which is evidenced by the release of trace levels of HIV-1 virions (Shen and Siliciano, 2008). The mechanisms of HIV-1 latency in quiescent T cells include recruitment of histone deacetylase (HDAC) to the HIV-1 promoter (Coull et al., 2000; Lehrman et al., 2005); cellular miRNA targeting of the 3′ end of HIV-1 (Huang et al., 2007; Klase et al., 2007) or cellular miRNA hsa-miR29a targeting Nef (Ahluwalia et al., 2008); lack of sustained Tat production (Williams et al., 2007) and likely other yet unrecognized pathways. Recently, HIV-1 latency in actively dividing T cell lines was attributed to a stochastic expression of viral genes showing that latency is not restricted to resting T cells, but might be an intrinsic property of HIV-1 (Jeeninga et al., 2008). Although our results do not indicate that physiological 3% O2 promotes latency, our data suggest that HIV-1 transcription is less efficient at lower oxygen and it might be regulated by a different set of host cell factors and might be less dependent on CDK9/cyclin T1. Although the regulation of the activity of CDK9/cyclin T1 has been thoroughly investigated (reviewed in Zhou and Yik, 2006), the reduced association of CDK9 with cyclin T1 that we now report has not been previously observed. We are currently investigating the possible cause of the relative dissociation of CDK9 and cyclin T1 at 3% versus 21% O2. Treatment of latently infected resting CD4+ T cells with hexamethylene bisacetamide was reported to induce HIV-1 production and Sp1-dependent CDK9 recruitment to HIV-1 promoter (Choudhary et al., 2008) suggesting that low availability of CDK9 might indeed reduce HIV-1 replication. In a recent study, protein phosphatase 2B (PP2B) and protein phosphatase 1α (PP1α) were shown to disrupt co-operatively the interaction between CDK9/cyclin T1 and 7SK RNA/HEXIM1 in part via dephosphorylation of CDK9 by PP1α (Chen et al., 2008). Hypoxia was previously shown to decrease PP1 expression (Taylor et al., 2000) and PP1 activity (Comerford et al., 2006) suggesting that the deregulation of PP1 might have a potential link to the deregulation of CDK9 activity. We found that inactivation of Sp1-binding sites alleviated hypoxia-mediated inhibition of Tat-induced HIV-1 transcription. Sp1 activity is regulated by PP1 and thus changes in PP1 activity in hypoxia might deregulate Sp1 activity and potentially affect HIV-1 transcription. Our analysis of expression of CDK9/cyclin T1-dependent genes showed a decrease in the expression of IκBα but not Gro-β or HLA-DRA genes suggesting a lack of general inhibitory effect or activation effect on cellular gene expression at 3% O2. IκBα associates with NF-κB and its phosphorylation by IκB kinase targets IκBα for degradation (Karin and Ben-Neriah, 2000). Reduction in IκBα expression might explain an increase in NF-κB activity at 3% O2, and further support our conclusion that HIV-1 transcription reduction is not linked to the reduction of NF-κB activity.
Our previous studies point to CDK2 as a critical regulator of HIV-1 transcription (reviewed in Nekhai and Jeang, 2006). We recently showed that iron chelators inhibit HIV-1 transcription by reducing the activity of CDK2 and the association of CDK9 and cyclin T1 (Debebe et al., 2007). Our current results suggest that CDK2 activity is not reduced at 3% oxygen compared to 21% oxygen. We hypothesize that inhibition of CDK2 may be more effective than inhibition of CDK9 in preventing HIV replication at 3% oxygen levels and plan to investigate this hypothesis employing small molecule inhibitors and iron chelators. It was shown that CDK2 knock-out mice are viable although CDK2 was required for germ cell development as CDK2(−/−) mice were sterile (Berthet et al., 2003). In the absence of CDK2, CDK1 compensates for its loss, however, the impairment of DNA repair activity makes CDK2(−/−) mice more sensitive to lethal irradiation (Satyanarayana et al., 2008). Interestingly, while CDK2 activity is needed for proliferation of cultured lymphocytes, proliferation of human marrow cells is driven by CDK1/cyclin A or CDK1/cyclin B (Xie et al., 2008). These recent studies show that CDK2 may be dispensable for the proliferation and survival of some cells in vivo and if so, inhibitors of CDK2 might be considered as a novel class of anti-HIV-1 therapeutics.
All in all, our study points to the possibility that HIV-1 replication can be altered as a result of intrinsic changes of CDK9/cyclin T1 activity and Sp1 activity in cells according to the level of oxygen in the body.
Acknowledgments
Contract grant sponsor: National Institutes of Health;
Contract grant numbers: 2 R25 HL003679-08, 2MO1 RR10284, 1SC1GM082325-01, RO1 DK49419, RO1 HL55605.
Contract grant sponsor: National Heart, Lung, and Blood Institute.
Contract grant sponsor: The Office of Research on Minority Health.
The authors thank Dr. Anna Suter (Novartis, Pharma AG, Ltd., Basel) for the gift of ICL670. We thank Dr. Andrei L. Gartel (Departments of Medicine, Microbiology and Immunology, University of Illinois at Chicago) for the gift of ARC (4-Amino-6-hydrazino-7-beta-D-Ribofuranosyl-7H-Pyrrolo (2,3-d)-pyrimidine-5-Carboxamide). We also thank Dr. Michael Emmerman (Fred Hutchinson Cancer Institute, Seattle, WA) for the HIV-1 LTR LacZ expression vector. We thank the NIH AIDS Research and Reference Reagent Program for CEM–HIV-1 LTR-GFP cells (courtesy of Dr. Jacques Corbeil), pGEM2 Tat bacterial expression vector (courtesy of Dr. Richard Gaynor), pHEF-VSVG expression vector (courtesy of Dr. Lung-Ji Chang), and pNL4-3.Luc.R−E− (courtesy of Dr. Nathaniel Landau). We thank Dr. Chou-Zen Giam (Uniformed Services University of the Health Sciences, Bethesda, MD) for the gift of e-selectin-Luiferase reporter. We thank Zufan Debebe for help in preparation of VSVG-pseudotyped HIV-1 Luc viruses. This project was supported by NIH Research Grant 2 R25 HL003679-08 funded by the National Heart, Lung, and Blood Institute and The Office of Research on Minority Health (to V.R.G.); by Howard University General Clinical Research Center grant from the NIH No.2MO1 RR10284 (to V.R.G.), by NIH Grant 1SC1GM082325-01 (to S.N.), and by Grants RO1 DK49419 and RO1 HL55605 (to P.E.R.). The authors would like to thank members of Dr. Victor Gordeuk’s laboratory at the Center for Sickle Cell Disease, Howard University for valuable discussions.
Literature cited
- Ahluwalia JK, Khan SZ, Soni K, Rawat P, Gupta A, Hariharan M, Scaria V, Lalwani M, Pillai B, Mitra D, Brahmachari SK. Human cellular microRNA hsa-miR-29a interferes with viral nef protein expression and HIV-1 replication. Retrovirology. 2008;5:117. doi: 10.1186/1742-4690-5-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammosova T, Jerebtsova M, Beullens M, Voloshin Y, Ray PE, Kumar A, Bollen M, Nekhai S. Nuclear protein phosphatase-1 regulates HIV-1 transcription. J Biol Chem. 2003;278:32189–32194. doi: 10.1074/jbc.M300521200. [DOI] [PubMed] [Google Scholar]
- Ammosova T, Berro R, Kashanchi F, Nekhai S. RNA interference directed to CDK2 inhibits HIV-1 transcription. Virology. 2005;341:171–178. doi: 10.1016/j.virol.2005.06.041. [DOI] [PubMed] [Google Scholar]
- Ammosova T, Berro R, Jerebtsova M, Jackson A, Charles S, Klase Z, Southerland W, Gordeuk VR, Kashanchi F, Nekhai S. Phosphorylation of HIV-1 Tat by CDK2 in HIV-1 transcription. Retrovirology. 2006;3:78. doi: 10.1186/1742-4690-3-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkuri KR, Herzenberg LA, Niemi AK, Cowan T. Importance of culturing primary lymphocytes at physiological oxygen levels. Proc Natl Acad Sci USA. 2007;104:4547–4552. doi: 10.1073/pnas.0611732104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell. 2001;8:327–337. doi: 10.1016/s1097-2765(01)00314-8. [DOI] [PubMed] [Google Scholar]
- Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 knockout mice are viable. Curr Biol. 2003;13:1775–1785. doi: 10.1016/j.cub.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, Nguyen PL, Khoruts A, Larson M, Haase AT, Douek DC. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200:749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Liu M, Li H, Xue Y, Ramey WN, He N, Ai N, Luo H, Zhu Y, Zhou N, Zhou Q. PP2B and PP1{alpha} cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev. 2008;22:1356–1368. doi: 10.1101/gad.1636008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary SK, Archin NM, Margolis DM. Hexamethylbisacetamide and disruption of human immunodeficiency virus type 1 latency in CD4(+) T cells. J Infect Dis. 2008;197:1162–1170. doi: 10.1086/529525. [DOI] [PubMed] [Google Scholar]
- Chun RF, Semmes OJ, Neuveut C, Jeang KT. Modulation of Sp1 phosphorylation by human immunodeficiency virus type 1 Tat. J Virol. 1998;72:2615–2629. doi: 10.1128/jvi.72.4.2615-2629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comerford KM, Leonard MO, Cummins EP, Fitzgerald KT, Beullens M, Bollen M, Taylor CT. Regulation of protein phosphatase 1gamma activity in hypoxia through increased interaction with NIPP1: Implications for cellular metabolism. J Cell Physiol. 2006;209:211–218. doi: 10.1002/jcp.20726. [DOI] [PubMed] [Google Scholar]
- Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- Coull JJ, Romerio F, Sun JM, Volker JL, Galvin KM, Davie JR, Shi Y, Hansen U, Margolis DM. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol. 2000;74:6790–6799. doi: 10.1128/jvi.74.15.6790-6799.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DA, Rinderknecht AS, Zoeteweij JP, Aoki Y, Read-Connole EL, Tosato G, Blauvelt A, Yarchoan R. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood. 2001;97:3244–3250. doi: 10.1182/blood.v97.10.3244. [DOI] [PubMed] [Google Scholar]
- Debebe Z, Ammosova T, Jerebtsova M, Kurantsin-Mills J, Niu X, Charles S, Richardson DR, Ray PE, Gordeuk VR, Nekhai S. Iron chelators ICL670 and 311 inhibit HIV-1 transcription. Virology. 2007;367:324–333. doi: 10.1016/j.virol.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, de la Fuente C, Fu P, Wang L, Donnelly R, Wade JD, Lambert P, Li H, Lee CG, Kashanchi F. Acetylation of HIV-1 Tat by CBP/P300 increases transcription of integrated HIV-1 genome and enhances binding to core histones. Virology. 2000;277:278–295. doi: 10.1006/viro.2000.0593. [DOI] [PubMed] [Google Scholar]
- Deng L, Ammosova T, Pumfery A, Kashanchi F, Nekhai S. HIV-1 Tat interaction with RNA polymerase II C-terminal domain (CTD) and a dynamic association with CDK2 induce CTD phosphorylation and transcription from HIV-1 promoter. J Biol Chem. 2002;277:33922–33929. doi: 10.1074/jbc.M111349200. [DOI] [PubMed] [Google Scholar]
- Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- Fu DX, Kuo YL, Liu BY, Jeang KT, Giam CZ. Human T-lymphotropic virus type I tax activates I-kappa B kinase by inhibiting I-kappa B kinase-associated serine/threonine protein phosphatase 2A. J Biol Chem. 2003;278:1487–1493. doi: 10.1074/jbc.M210631200. [DOI] [PubMed] [Google Scholar]
- Gomez-Gonzalo M, Carretero M, Rullas J, Lara-Pezzi E, Aramburu J, Berkhout B, Alcami J, Lopez-Cabrera M. The hepatitis B virus X protein induces HIV-1 replication and transcription in synergy with T-cell activation signals: Functional roles of NF-kappaB/NF-AT and SP1-binding sites in the HIV-1 long terminal repeat promoter. J Biol Chem. 2001;276:35435–35443. doi: 10.1074/jbc.M103020200. [DOI] [PubMed] [Google Scholar]
- Han Y, Wind-Rotolo M, Yang HC, Siliciano JD, Siliciano RF. Experimental approaches to the study of HIV-1 latency. Nat Rev Microbiol. 2007;5:95–106. doi: 10.1038/nrmicro1580. [DOI] [PubMed] [Google Scholar]
- He J, Landau NR. Use of a novel human immunodeficiency virus type 1 reporter virus expressing human placental alkaline phosphatase to detect an alternative viral receptor. J Virol. 1995;69:4587–4592. doi: 10.1128/jvi.69.7.4587-4592.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, Huang W, Squires K, Verlinghieri G, Zhang H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med. 2007;13:1241–1247. doi: 10.1038/nm1639. [DOI] [PubMed] [Google Scholar]
- Hwang II, Watson IR, Der SD, Ohh M. Loss of VHL confers hypoxia-inducible factor (HIF)-dependent resistance to vesicular stomatitis virus: Role of HIF in antiviral response. J Virol. 2006;80:10712–10723. doi: 10.1128/JVI.01014-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovic Z. Hypoxia or in situ normoxia: The stem cell paradigm. J Cell Physiol. 2009;219:271–275. doi: 10.1002/jcp.21690. [DOI] [PubMed] [Google Scholar]
- Jeeninga RE, Westerhout EM, van Gerven ML, Berkhout B. HIV-1 latency in actively dividing human T cell lines. Retrovirology. 2008;5:37. doi: 10.1186/1742-4690-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JH, Wang N, Li A, Liao WT, Pan ZG, Mai SJ, Li DJ, Zeng MS, Wen JM, Zeng YX. Hypoxia can contribute to the induction of the Epstein-Barr virus (EBV) lytic cycle. J Clin Virol. 2006;37:98–103. doi: 10.1016/j.jcv.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Jochmann R, Thurau M, Jung S, Hofmann C, Naschberger E, Kremmer E, Harrer T, Miller M, Schaft N, Sturzl M. O-linked N-acetylglucosaminylation of Sp1 inhibits the human immunodeficiency virus type 1 promoter. J Virol. 2009;83:3704–3718. doi: 10.1128/JVI.01384-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa S, Peterlin BM. Combinations of dominant-negative class II transactivator, p300 or CDK9 proteins block the expression of MHC II genes. Int Immunol. 2001;13:951–958. doi: 10.1093/intimm/13.7.951. [DOI] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Kenneth NS, Rocha S. Regulation of gene expression by hypoxia. Biochem J. 2008;414:19–29. doi: 10.1042/BJ20081055. [DOI] [PubMed] [Google Scholar]
- Kiernan RE, Vanhulle C, Schiltz L, Adam E, Xiao H, Maudoux F, Calomme C, Burny A, Nakatani Y, Jeang KT, Benkirane M, Van Lint C. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999;18:6106–6118. doi: 10.1093/emboj/18.21.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimpton J, Emerman M. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. J Virol. 1992;66:2232–2239. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klase Z, Kale P, Winograd R, Gupta MV, Heydarian M, Berro R, McCaffrey T, Kashanchi F. HIV-1 TAR element is processed by dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol Biol. 2007;8:63. doi: 10.1186/1471-2199-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrman G, Hogue IB, Palmer S, Jennings C, Spina CA, Wiegand A, Landay AL, Coombs RW, Richman DD, Mellors JW, Coffin JM, Bosch RJ, Margolis DM. Depletion of latent HIV-1 infection in vivo: A proof-of-concept study. Lancet. 2005;366:549–555. doi: 10.1016/S0140-6736(05)67098-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard J, Parrott C, Buckler-White AJ, Turner W, Ross EK, Martin MA, Rabson AB. The NF-kappa B binding sites in the human immunodeficiency virus type 1 long terminal repeat are not required for virus infectivity. J Virol. 1989;63:4919–4924. doi: 10.1128/jvi.63.11.4919-4924.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyron-Holtz EG, Ghosh MC, Rouault TA. Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science. 2004;306:2087–2090. doi: 10.1126/science.1103786. [DOI] [PubMed] [Google Scholar]
- Nekhai S, Jeang KT. Transcriptional and post-transcriptional regulation of HIV-1 gene expression: Role of cellular factors for Tat and Rev. Future Micorbiol. 2006;1:417–426. doi: 10.2217/17460913.1.4.417. [DOI] [PubMed] [Google Scholar]
- Nekhai S, Zhou M, Fernandez A, Lane WS, Lamb NJ, Brady J, Kumar A. HIV-1 Tat-associated RNA polymerase C-terminal domain kinase, CDK2, phosphorylates CDK7 and stimulates Tat-mediated transcription. Biochem J. 2002;364:649–657. doi: 10.1042/BJ20011191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekhai S, Bhat UG, Ammosova T, Radhakrishnan SK, Jerebtsova M, Niu X, Foster A, Layden TJ, Gartel AL. A novel anticancer agent ARC antagonizes HIV-1 and HCV. Oncogene. 2007;26:3899–3903. doi: 10.1038/sj.onc.1210158. [DOI] [PubMed] [Google Scholar]
- Nowak DE, Tian B, Jamaluddin M, Boldogh I, Vergara LA, Choudhary S, Brasier AR. RelA Ser276 phosphorylation is required for activation of a subset of NF-kappaB-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol Cell Biol. 2008;28:3623–3638. doi: 10.1128/MCB.01152-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M, Schnolzer M, Garnica J, Fischle W, Emiliani S, Rackwitz HR, Verdin E. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr Biol. 1999;9:1489–1492. doi: 10.1016/s0960-9822(00)80120-7. [DOI] [PubMed] [Google Scholar]
- Pahl PM, Reese SM, Horwitz LD. A lipid-soluble iron chelator alters cell cycle regulatory protein binding in breast cancer cells compared to normal breast cells. J Exp Ther Oncol. 2007;6:193–200. [PubMed] [Google Scholar]
- Peng J, Zhu Y, Milton JT, Price DH. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998;12:755–762. doi: 10.1101/gad.12.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira LA, Bentley K, Peeters A, Churchill MJ, Deacon NJ. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res. 2000;28:663–668. doi: 10.1093/nar/28.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DH. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol. 2000;20:2629–2634. doi: 10.1128/mcb.20.8.2629-2634.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puppo M, Bosco MC, Federico M, Pastorino S, Varesio L. Hypoxia inhibits Moloney murine leukemia virus expression in activated macrophages. J Leukoc Biol. 2007;81:528–538. doi: 10.1189/jlb.0506361. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan SK, Gartel AL. A novel transcriptional inhibitor induces apoptosis in tumor cells and exhibits antiangiogenic activity. Cancer Res. 2006;66:3264–3270. doi: 10.1158/0008-5472.CAN-05-3940. [DOI] [PubMed] [Google Scholar]
- Sahaf B, Atkuri K, Heydari K, Malipatlolla M, Rappaport J, Regulier E, Herzenberg LA. Culturing of human peripheral blood cells reveals unsuspected lymphocyte responses relevant to HIV disease. Proc Natl Acad Sci USA. 2008;105:5111–5116. doi: 10.1073/pnas.0712363105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent PJ, Farnaud S, Evans RW. Structure/function overview of proteins involved in iron storage and transport. Curr Med Chem. 2005;12:2683–2693. doi: 10.2174/092986705774462969. [DOI] [PubMed] [Google Scholar]
- Satyanarayana A, Hilton MB, Kaldis P. p21 Inhibits Cdk1 in the absence of Cdk2 to maintain the G1/S phase DNA damage checkpoint. Mol Biol Cell. 2008;19:65–77. doi: 10.1091/mbc.E07-06-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servais C, Caillet-Fauquet P, Draps ML, Velu T, de Launoit Y, Brandenburger A. Hypoxic-response elements in the oncolytic parvovirus Minute virus of mice do not allow for increased vector production at low oxygen concentration. J Gen Virol. 2006;87:1197–1201. doi: 10.1099/vir.0.81754-0. [DOI] [PubMed] [Google Scholar]
- Shen L, Siliciano RF. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J Allergy Clin Immunol. 2008;122:22–28. doi: 10.1016/j.jaci.2008.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen BH, Bauzon M, Hermiston TW. The effect of hypoxia on the uptake, replication and lytic potential of group B adenovirus type 3 (Ad3) and type 11p (Ad11p) Gene Ther. 2006;13:986–990. doi: 10.1038/sj.gt.3302736. [DOI] [PubMed] [Google Scholar]
- Sweet T, Sawaya BE, Khalili K, Amini S. Interplay between NFBP and NF-kappaB modulates tat activation of the LTR. J Cell Physiol. 2005;204:375–380. doi: 10.1002/jcp.20419. [DOI] [PubMed] [Google Scholar]
- Taylor CT. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J Physiol. 2008;586:4055–4059. doi: 10.1113/jphysiol.2008.157669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CT, Furuta GT, Synnestvedt K, Colgan SP. Phosphorylation-dependent targeting of cAMP response element binding protein to the ubiquitin/proteasome pathway in hypoxia. Proc Natl Acad Sci USA. 2000;97:12091–12096. doi: 10.1073/pnas.220211797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Lowe AD, Karn J. Activation of human immunodeficiency virus transcription in T cells revisited: NF-kappaB p65 stimulates transcriptional elongation. J Virol. 2001;75:8524–8537. doi: 10.1128/JVI.75.18.8524-8537.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SA, Kwon H, Chen LF, Greene WC. Sustained induction of NF-kappa B is required for efficient expression of latent human immunodeficiency virus type 1. J Virol. 2007;81:6043–6056. doi: 10.1128/JVI.02074-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie DX, Yao J, Zhang P, Li XL, Feng YD, Wu JH, Tao DD, Hu JB, Gong JP. Are progenitor cells pre-programmed for sequential cell cycles not requiring cyclins D and E and activation of Cdk2? Cell Prolif. 2008;41:265–278. doi: 10.1111/j.1365-2184.2008.00518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yedavalli VS, Benkirane M, Jeang KT. Tat and trans-activation-responsive (TAR) RNA-independent induction of HIV-1 long terminal repeat by human and murine cyclin T1 requires Sp1. J Biol Chem. 2003;278:6404–6410. doi: 10.1074/jbc.M209162200. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Yik JH. The Yin and Yang of P-TEFb regulation: Implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol Mol Biol Rev. 2006;70:646–659. doi: 10.1128/MMBR.00011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]