

Abstract
Humans with the Lesch-Nyhan syndrome have an X-chromosomal mutant gene that causes severe neurological and developmental abnormalities. The patients are deficient in hypoxanthine-guanine phosphoribosyltransferase, which converts hypoxanthine to inosinic acid, a major precursor of adenine and guanine nucleotides. Paradoxically, the enzyme defect causes hypernormal de novo synthesis of inosinic acid, which manifests itself as excesses of hypoxanthine, xanthine, and uric acid. The first step in the de novo pathway is thought to be rate-limiting, due to feedback repression by adenine and guanine nucleotides. The derepressed rate of purine production in mutants and their failure to thrive could result from reduction in the amounts of nucleotides derived from inosinic acid to levels that are inadequate for normal feedback control and for nucleic acid synthesis needed in growth. Studies with cultured cells, reported here, support the interpretation that mutants are, in effect, nucleotide-deficient. Skin fibroblasts from patients fail to proliferate in media that do not contain supplementary adenine or folic acid, a participant in two stages of purine biosynthesis. The folic acid requirement of mutant cells is at least 50-fold greater than that of normal cells, which can synthesize all the nucleotides needed for growth without exogenous adenine. Both folic acid and adenine supplements are thought to provide mutant cells with the means of making more inosinic acid available for conversion to adenine and guanine nucleotides. It is not clear why the availability of inosinate or its conversion to other nucleotides is impaired. Therapy with adenine or folic acid begun at the time of birth may avert development of the disease in mutant males.
The relevant gene is X-linked and shows clonal, single-allele-expression: phenotypically normal and phenotypically mutant clones have been derived from females heterozygous for the mutant gene. The phenotypically mutant heterozygous clones have the same requirement for adenine or folic acid as cells from hemizygous mutant males, an indication that the normal allele is repressed in these clones. The adenine-folic acid requirement of mutant cells provides a method of direct, clonal selection for rare, phenotypically normal cells in mutant populations, which is applicable to the single-active-X problem and other in vitro genetic studies.
Full text
PDF

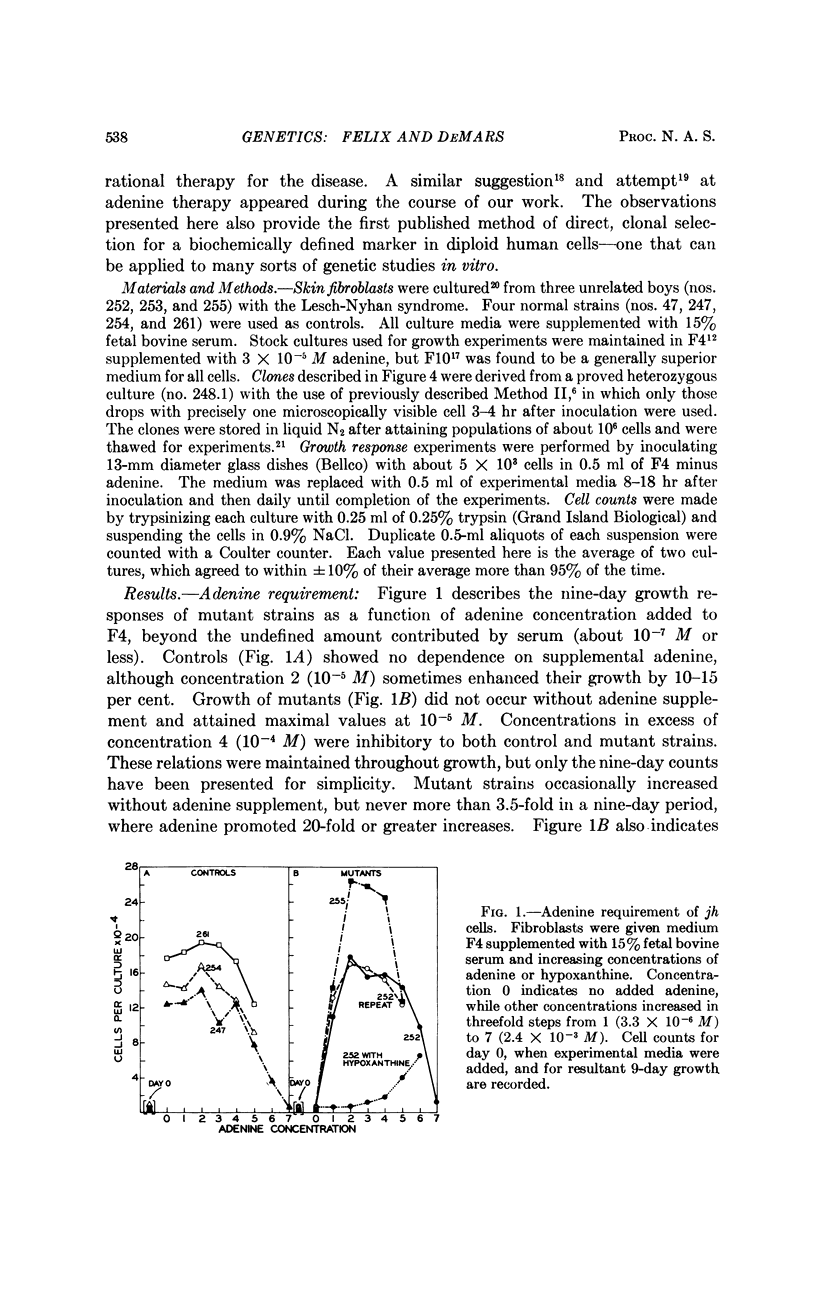
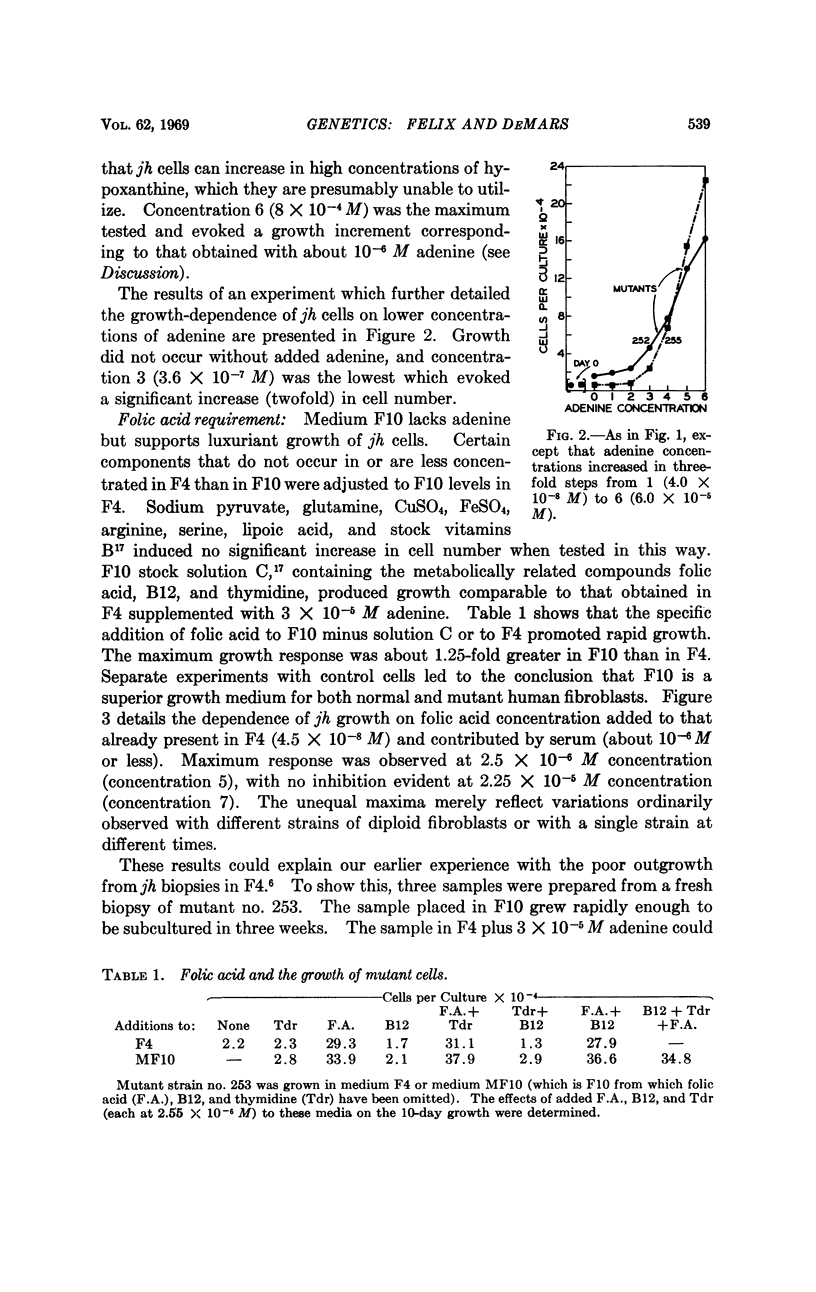
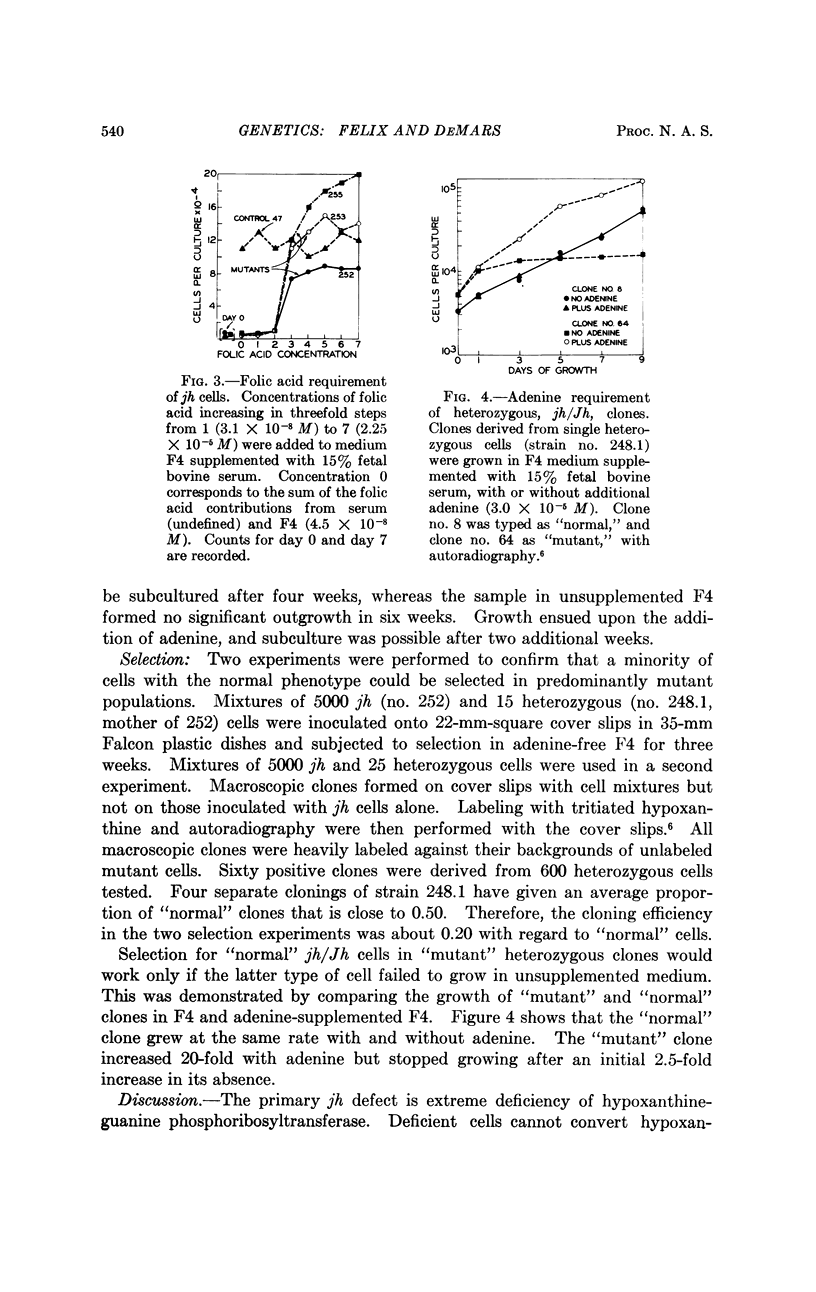



Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- CASKEY C. T., ASHTON D. M., WYNGAARDEN J. B. THE ENZYMOLOGY OF FEEDBACK INHIBITION OF GLUTAMINE PHOSPHORIBOSYLPYROPHOSPHATE AMIDOTRANSFERASE BY PURINE RIBONUCLEOTIDES. J Biol Chem. 1964 Aug;239:2570–2579. [PubMed] [Google Scholar]
- DeMars R. A temperature-sensitive glucose-6-phosphate dehydrogenase in mutant cultured human cells. Proc Natl Acad Sci U S A. 1968 Oct;61(2):562–569. doi: 10.1073/pnas.61.2.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMars R. The single-active-X: functional differentiation at the chromosome level. Natl Cancer Inst Monogr. 1967 Sep;26:327–351. [PubMed] [Google Scholar]
- EAGLE H. Amino acid metabolism in mammalian cell cultures. Science. 1959 Aug 21;130(3373):432–437. doi: 10.1126/science.130.3373.432. [DOI] [PubMed] [Google Scholar]
- HAM R. G. An improved nutrient solution for diploid Chinese hamster and human cell lines. Exp Cell Res. 1963 Feb;29:515–526. doi: 10.1016/s0014-4827(63)80014-2. [DOI] [PubMed] [Google Scholar]
- HENDERSON J. F., KHOO K. Y. ON THE MECHANISM OF FEEDBACK INHIBITION OF PURINE BIOSYNTHESIS DE NOVO IN EHRLICH ASCITES TUMOR CELLS IN VITRO. J Biol Chem. 1965 Jul;240:3104–3109. [PubMed] [Google Scholar]
- Henderson J. F. Rational approaches to the treatment of the Lesch-Nyhan syndrome. Fed Proc. 1968 Jul-Aug;27(4):1105–1106. [PubMed] [Google Scholar]
- Kelley W. N., Rosenbloom F. M., Henderson J. F., Seegmiller J. E. A specific enzyme defect in gout associated with overproduction of uric acid. Proc Natl Acad Sci U S A. 1967 Jun;57(6):1735–1739. doi: 10.1073/pnas.57.6.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LYON M. F. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. 1961 Apr 22;190:372–373. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- Migeon B. R., Der Kaloustian V. M., Nyhan W. L., Yough W. J., Childs B. X-linked hypoxanthine-guanine phosphoribosyl transferase deficiency: heterozygote has two clonal populations. Science. 1968 Apr 26;160(3826):425–427. doi: 10.1126/science.160.3826.425. [DOI] [PubMed] [Google Scholar]
- Nyhan W. L. Clinical features of the Lesch-Nyhan syndrome. Introduction--clinical and genetic features. Fed Proc. 1968 Jul-Aug;27(4):1027–1033. [PubMed] [Google Scholar]
- RUSSELL L. B. Mammalian X-chromosome action: inactivation limited in spread and region of origin. Science. 1963 May 31;140(3570):976–978. doi: 10.1126/science.140.3570.976. [DOI] [PubMed] [Google Scholar]
- Rosenbloom F. M., Henderson J. F., Caldwell I. C., Kelley W. N., Seegmiller J. E. Biochemical bases of accelerated purine biosynthesis de novo in human fibroblasts lacking hypoxanthine-guanine phosphoribosyltransferase. J Biol Chem. 1968 Mar 25;243(6):1166–1173. [PubMed] [Google Scholar]
- Russell J. D., DeMars R. UDP-glucose: alpha-D-galactose-1-phosphate uridylytransferase activity in cultured human fibroblasts. Biochem Genet. 1967 Jun;1(1):11–24. doi: 10.1007/BF00487732. [DOI] [PubMed] [Google Scholar]
- SZYBALSKI W., SZYBALSKA E. H. Drug sensitivity as a genetic marker for human cell lines. Med Bull (Ann Arbor) 1962 Sep-Oct;28:277–293. [PubMed] [Google Scholar]
- Salzmann J., DeMars R., Benke P. Single-allele expression at an X-linked hyperuricemia locus in heterozygous human cells. Proc Natl Acad Sci U S A. 1968 Jun;60(2):545–552. doi: 10.1073/pnas.60.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seegmiller J. E., Rosenbloom F. M., Kelley W. N. Enzyme defect associated with a sex-linked human neurological disorder and excessive purine synthesis. Science. 1967 Mar 31;155(3770):1682–1684. doi: 10.1126/science.155.3770.1682. [DOI] [PubMed] [Google Scholar]
- van der Zee S. P., Schretlen E. D., Monnens L. A. Megaloblastic anaemia in the Lesch-Nyhan syndrome. Lancet. 1968 Jun 29;1(7557):1427–1427. doi: 10.1016/s0140-6736(68)92002-3. [DOI] [PubMed] [Google Scholar]