

Abstract
Neutrophil (PMN) infiltration and associated release of serine proteases contribute to epithelial injury during active phases of mucosal disorders such as inflammatory bowel disease. Previous studies have demonstrated that PMN contact with basolateral surfaces of intestinal epithelial cells in the presence of a chemoattractant results in disruption of barrier function even without transmigration. Similarly, serine protease-mediated activation of epithelial protease-activated receptors (PARs) has been shown to increase permeability. In this study, we assessed whether transmigrating PMNs can regulate barrier function through epithelial PAR activation. Transepithelial resistance (TER) decreased significantly after PMN contact with basolateral surfaces of T84 monolayers or after incubation with PMN elastase and proteinase-3, but not cathepsin G. Inhibition of PMN serine proteases, but not selective inhibition of elastase or cathepsin G, prevented the fall in TER induced by PMN contact and blocked PMN transepithelial migration. Basolateral, but not apical, PAR-1 and -2 activation with selective agonists also decreased TER. PAR-1 and -2 were localized intracellularly and in close proximity to lateral surfaces beneath tight junctions, and expression was increased in colonic mucosa from individuals with Crohn’s disease. Combined, but not individual, transfection with small interfering RNAs targeted against epithelial PAR-1 and -2, prevented the fall in TER induced by PMN contact. Furthermore, basolateral PAR-1 and -2 activation induced phosphorylation of myosin L chain kinase and regulatory myosin L chain. Lastly, epithelial PAR-1 and -2 knockdown decreased the rate of PMN transepithelial migration. These results suggest that protease-mediated epithelial PAR-1 and -2 activation, by migrating PMNs, induces signaling events that increase epithelial permeability thereby facilitates PMN transepithelial migration.
Neutrophil (polymorphonuclear leukocyte; PMN3) accumulation at intestinal mucosal surfaces is a characteristic hallmark of many inflammatory conditions of the intestine. Epithelial injury, disease activity, and patient symptoms have been shown to correlate with the histological finding of extensive PMN migration across the epithelium (1). In addition, studies have indicated that high-density PMN flux across epithelial monolayers mimicking active inflammation, in either the apical-to-basolateral direction or the more physiologically relevant basolateral-to-apical direction, results in disruption of epithelial permeability (2) and produces multifocal wounds (3). Consequently, the loss of epithelial barrier function results in increased luminal Ag penetration and subsequent perpetuation of the inflammatory response. Conversely, low-density PMN migration, which occurs during immune surveillance, is generally believed to be a rapid process that does not damage the integrity of epithelial monolayers (4, 5). Indeed, interactions between the transmigrating PMN and the epithelium result in signaling events which in turn may be amplified and prolonged during high-density PMN transmigration (5, 6). These observations suggest that under physiological conditions, intercellular junctions transiently loosen to allow passage of circulating cells, while at the same time maintaining barrier function. However, the mechanisms that regulate epithelial permeability during low- and high-density PMN transmigration remain poorly defined. Previously, we have demonstrated that high-density PMN transmigration increases paracellular permeability in a contact-dependent manner and activates signaling events in epithelial monolayers in a polarized manner before transmigration in the physiologically relevant basolateral-to-apical direction (6). However, the PMN and epithelial receptors that mediate these contact-dependent signaling events and alter epithelial permeability have remained to be elusive.
Among the candidate PMN surface proteins that may initiate such epithelial signaling events, several structurally similar serine proteases that possess antimicrobial activity (serpocidins) and are contained within the azurophil (primary) granules, have been shown to undergo limited exocytosis and mobilize to the cell surface upon activation (7, 8). Indeed, PMN serpocidins have recently been shown to activate protease-activated receptors (PARs; Refs. 9-12), a unique class of G-protein-coupled signaling receptors that have also been reported to induce epithelial cell apoptosis (10, 13) and regulate epithelial barrier function in vitro and in vivo (13-15). Cleavage of the PAR extracellular N terminus by proteases such as thrombin, trypsin, and tryptase (16) has been shown to allow an exposed tethered ligand to bind and activate the cleaved receptor. Despite these observations, the role of leukocyte proteases and PAR signaling in the regulation of epithelial barrier function at sites of inflammation remains incompletely understood but nevertheless remains to be an intriguing hypothesis that needs to be further elucidated. In this study, we determined that basolateral activation of PAR-1 and -2 increases epithelial permeability and that this event regulates the disruption in epithelial barrier function induced by PMN contact and subsequent PMN transepithelial migration.
Materials and Methods
Reagents
Abs were obtained from Invitrogen/Zymed Laboratories (occludin, claudin-1, claudin-4, and actin), Santa Cruz Biotechnology (phosphorylated myosin L chain kinase (MLCK; Tyr464), and Cell Signaling Technology (dually phosphorylated myosin L chain; Thr18 and Ser19). The goat polyclonal Ab to thrombin receptor or PAR-1 (C-18) was obtained from Santa Cruz Biotechnology, and the rabbit polyclonal Ab to PAR-2 (B5/A5) was kindly provided by Dr. Morley Hollenberg (University of Calgary, Calgary, Canada). The serine proteases human neutrophil elastase and proteinase-3 were obtained from Elastin Products. Human cathepsin G, the broad spectrum metalloproteinase inhibitor galardin (N-[(2R)-2-(hydroxamidocarbonylmethyl)-4-methylpentanoyl]-l-tryptophan methylamide, and GM6001 were obtained from Calbiochem. The specific elastase inhibitor N-(methoxysuccinyl)alanyl alanylprolylvalyl chloromethyl ketone (AAPV), cathepsin G inhibitor α1-antichymotrypsin, and the serine protease inhibitor 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF) were obtained from Sigma-Aldrich. The specific PAR-1 (TFLLR-NH2), PAR-2 (SLIGRL-NH2), and PAR-4 (AYPGKF-NH2) agonists and their inactive controls RLLFT-NH2, LRGILS-NH2, and YAPGKF-NH2 were obtained from Dr. Denis McMaster (Peptide Synthesis Facility, University of Calgary). Peptides were >95% pure by HPLC analysis and mass spectroscopy criteria.
Cell culture
Cultures of T84 (2) and SK-CO15 (17) intestinal epithelial monolayers were grown as previously described. For PMN transepithelial migration experiments, monolayers were grown on 0.33-cm2 collagen-coated polycarbonate membranes with 0.4- or 5.0-μm pore sizes (Costar). Monolayer confluence was determined by monitoring transepithelial electrical resistance (TER) to passive ion flow, and cultures were used when TER values exceeded 1000 Ω/cm2 at 7–10 days after seeding.
PMN isolation
Normal human PMNs were isolated as described previously (18) with approval from the Emory University Institutional Review Board on human subjects. Briefly, citrated peripheral blood from healthy human volunteers was subjected to RBC-dextran 500 (Amersham-Pharmacia Biotech) sedimentation followed by density sedimentation using Ficoll-Paque (Amersham) according to the manufacturer’s instructions. The purified PMNs were resuspended in calcium- and magnesium-free HBSS containing 0.4 g/L KCl, 0.06 g/L KH2PO4, 0.35 g/L NaHCO3, 8.0 g/L NaCl, 0.048 g/L Na2HPO4, 1.0 g/L glucose, and 10 mM HEPES at 4°C. For all experiments, PMNs were suspended in HBSS containing 0.185 g/L CaCl2·2H2O and 0.098 g/L MgSO4.
PMN-stimulated transepithelial permeability assay
Inverted T84 or SK-CO15 monolayers grown on 0.33-cm2 filters with 0.4-μm pores were washed and equilibrated in HBSS for 1 h at 37°C. The upper reservoir was emptied and replenished with HBSS or 10−6 M fMLP (180 μl for 0.33 cm2). The lower reservoir was filled with HBSS or 10−6 M fMLP (500 μl for 0.33 cm2), and the initial TER was recorded. Next, PMNs (106 for 0.33 cm2) or HBSS were added to the upper reservoir, and the monolayers were incubated at 37°C followed by TER measurements at the indicated times.
Immunohistochemistry
Colorectal mucosal tissue samples were derived from anonymous surgical resection specimens and approved by the Emory University Institutional Review Board on human subjects. T84 monolayers or colorectal mucosal tissue samples were fixed with 100% methanol for 20 min at −20°C and then blocked in 2% FBS for 1 h at room temperature. Samples were incubated with primary Abs for 1 h at room temperature and then probed with secondary Abs conjugated with Alexa 488 or Alexa 568 (1/1000; Molecular Probes) for 1 h at room temperature. Stained monolayers were mounted in ProLong Gold Antifade medium (Invitrogen) and examined using a Zeiss 510 confocal microscope (Zeiss). Images were processed and analyzed using LSM5 browser software (Zeiss).
RNA interference
Custom chemically synthesized duplex small interfering RNA (siRNA) against PAR-1 (F2R) GGUUGAAACAUAUCUCUUATT and PAR-2 (F2RL1) GGAUGUGGAACCUGUUUAAUU were obtained from Dharmacon. In addition, control and validated siRNA against cyclophilin B was obtained from Dharmacon. All of the siRNA transfections were performed using HiPerFect (Qiagen) in Opti-MEM I medium (Invitrogen) according to the manufacturer’s protocol with a final siRNA concentration of 50 nM. Experiments were performed 72 h after transfection when TER exceeded 1000 Ω/cm2.
RNA extraction and RT-PCR
Total RNA was isolated from cultured SK-CO15 epithelial cells using TRIzol reagent (Invitrogen) following the manufacturer’s protocol. Purity was checked by using the OD260:OD280 ratio. Total RNA was reverse transcribed using the Superscript III One-Step RT-PCR System with platinum TaqDNA polymerase (Invitrogen). In a total volume of 50 μl, the reaction mixture contained 25 μl of 2× reaction mix, 100 ng of RNA, 1 μl of 10 μM sense and antisense primers, and 1 μl of Superscript III reverse transcriptase-platinum Taq mix. The reaction mixtures were subjected to cDNA synthesis for 30 min at 58°C and denaturation for 2 min at 94°C. PCR mixtures were then directly amplified by 30 cycles (15 s at 94°C, 30 s at 54°C, 30 s at 68°C). PCR amplification was followed by a final extension period of 5 min at 68°C. PCR products were analyzed by 2% agarose gel electrophoresis with ethidium bromide. Oligonucleotide primers for human PAR-1 (accession number NM001992; sense 5′-CTTCTCAGGAACCCCAATGA-3′, antisense 5′-GCCAGACAAGTGAAGGAAGC-3′), human PAR-2 (accession number NM005242, sense 5′-TGCTAGCAGCCTCTCTCTCC-3′, antisense 5′-TTGAACATGTCTCCCACCAA-3′), and human GAPDH (accession number NM002046, sense 5′-ACCACAGTCCATGCCATCAC-3′, antisense 5′-TCCACCACCCTGTTGCTGTA-3′) were obtained from Integrated DNA Technologies.
Western blot analysis
Monolayers were harvested with a rubber cell scraper into RIPA cell lysis buffer (pH 7.4) containing 20 mM Tris, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS, protease inhibitor mixture (1/100; Sigma-Aldrich), phosphatase inhibitor mixtures 1 and 2 (1/100; Sigma-Aldrich), 100 mM phenanthroline, 80 mM PMSF, 11 μM tosylphenylalanylchloromethyl ketone, 100 mM iodoacetamide, and 132 mM N-ethylmaleimide. Lysates were then cleared by centrifugation (10 min at 14,000 × g and 4°C) diluted with Laemmli sample buffer, and boiled for 5 min. Equal protein amounts (10 μg) were run on a polyacrylamide gel, transferred to nitrocellulose, and then analyzed by Western blot as per standard methods.
PMN transepithelial migration
PMN transmigration experiments were performed with inverted T84 or SK-CO15 monolayers as described previously (18). Briefly, the upper reservoir was filled with 180 μl of HBSS and 20 μl of PMNs (50 × 106/ml). The assay was initiated upon addition of 500 μl of 10−6 M fMLP to the lower reservoir followed by incubation of the plates at 37°C. Every 30 min, the monolayers were transferred to new reservoirs containing 10−6 M fMLP and incubated at 37°C. At the end of the indicated time, migrated PMNs in the lower reservoirs and unmigrated PMNs in the upper reservoirs were quantified by assaying for myeloperoxidase (2). PMN migration into the lower reservoirs is expressed as a percentage of total applied PMNs per transwell.
Statistical analysis
Results were expressed as means ± SEM, and unless indicated, data were pooled from three independent experiments done in duplicate. Data was compared by one-way ANOVA, followed by Tukey’s test for multiple comparison analysis where applicable. Statistical significance was established at p < 0.05.
Results
PMN serine proteases increase paracellular permeability
Given that PMN proteases have been shown to play multiple roles in microbial killing and cell signaling, we sought to determine whether they may play a functional role in enhancing epithelial permeability. Confluent, high-resistance T84 monolayers were incubated with 1 U/ml purified PMN proteases and evaluated for their effects on epithelial barrier function. Specifically, incubation with PMN elastase and proteinase-3, but not cathepsin G, decreased TER by ~40% baseline after 30 s (Fig. 1).
FIGURE 1.
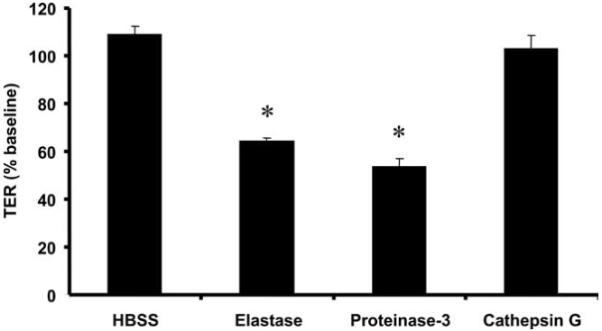
PMN serine proteases elastase and proteinase-3, but not cathepsin G, decrease TER. Purified PMN elastase, proteinase-3, and cathepsin G (1 U/ml) were added to the basolateral aspect of inverted T84 monolayers cultured on 0.4-μm pore size filters. TER was measured after 30 s of incubation and is expressed as the percentage of the baseline TER at the start of the assay. Values are means ± SEM, n = 3 per group; *, p < 0.05 compared with vehicle control (HBSS). Data were generated from three independent experiments done in duplicate.
To further establish the role of PMN-derived proteases in mediating paracellular permeability, PMNs were pretreated with a variety of protease inhibitors, washed with HBSS vehicle, and then incubated with inverted T84 epithelial monolayers cultured on 0.4-μm pore size supports in the presence of an fMLP gradient. Pretreatment with the broad-spectrum water-soluble serine protease inhibitor AEBSF prevented the decrease in TER (Fig. 2A). Conversely, pretreatment with individual inhibitors to metalloproteinases (GM6001), PMN elastase (AAPV), or cathepsin G (α1-antichymotrypsin) failed to prevent the decrease in TER induced by PMN contact (Fig. 2A). Incubation of epithelial monolayers with the inhibitors alone did not affect TER (data not shown). We also conducted studies to determine whether the increase in epithelial permeability induced by PMN contact was due to gross cleavage of barrier-forming proteins. We observed that the disruption in epithelial barrier function induced by PMN contact was not accompanied by gross structural alterations to occludin; ZO-1; ZO-2; JAM-A; claudin-1, -2, or -5; α- and β-catenin; E-cadherin; and α-actinin as assessed by immunofluorescence (6), nor were there any changes to occludin, claudin-1 or claudin-4, and E-cadherin by Western blotting analysis (data not shown).
FIGURE 2.
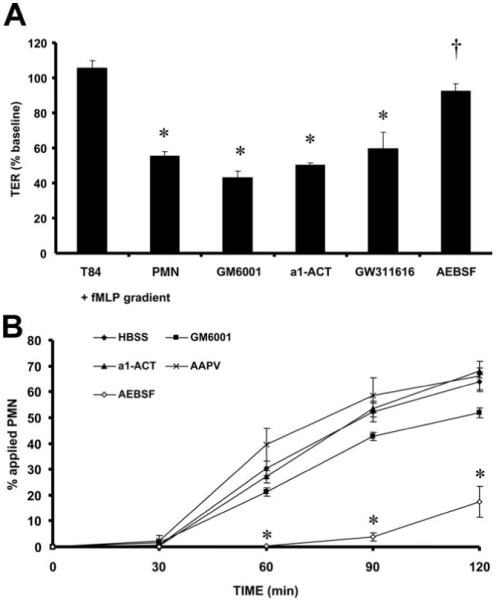
Inhibition of PMN serine proteases prevents the fall in TER induced by PMN contact with epithelial monolayers. PMNs (106 cells) were pretreated for 1 h with inhibitors to metalloproteinases (25 μM GM6001), cathepsin G (50 μM α1-ACT), elastase (50 μM AAPV), or serine proteases (200 μM AEBSF). After pretreatment, PMNs were added to the basolateral aspect of inverted T84 monolayers cultured on 0.4-μm pore size filters to assay transepithelial permeability after 3 h (A) or 5.0-μm pore size filters to assay PMN transepithelial migration (B). TER is expressed as the percentage of baseline TER at the start of the assay. Transepithelial migration is expressed as the percentage of applied PMN. Values are means ± SEM, n = 3 per group; *, p < 0.05 compared with control [A, Inverted T84 monolayers incubated with a transepithelial fMLP gradient (T84 + fMLP gradient); B, HBSS; PMNs pretreated with HBSS alone)]; †, p < 0.05 compared with PMNs added to the basolateral aspect of epithelial monolayers in the presence of a transepithelial fMLP gradient (PMN + fMLP gradient). Data were generated from three independent experiments done in duplicate.
Because our data suggested that PMN serine proteases can increase epithelial permeability independent of transmigration, we hypothesized that these mediators may play an important role in regulating PMN transepithelial migration after protease release. PMNs were pretreated with the protease inhibitors mentioned above and were assayed for their ability to migrate across inverted T84 monolayers cultured on 5.0 μm pore-size supports in the presence of an fMLP gradient. Pretreatment of PMNs with the serine protease inhibitor AEBSF, which potently prevented the fall in TER, also dramatically inhibited PMN transepithelial migration as compared with pretreatment with the general metalloproteinase inhibitor GM6001, the cathepsin G inhibitor α1-antichymotrypsin, and the PMN elastase inhibitor AAPV (Fig. 2B). These findings suggest that multiple PMN serine proteases may regulate PMN-epithelial signaling events that control paracellular permeability which precedes PMN transepithelial migration through the paracellular space between epithelial cells.
PAR-1 and -2 regulate the fall in TER induced by PMN contact with the basolateral surfaces of epithelial cells
Given the above data suggesting that PMN proteases increase epithelial permeability independent of proteolytic cleavage of apical junction proteins, we investigated their role(s) in cell signaling. Recent studies have determined that proteases may act as signaling molecules through their ability to activate a class of transmembrane, cell surface G protein-coupled receptors termed PARs. PARs are activated by a tethered ligand which is liberated through the proteolytic cleavage of their extracellular N terminus and have been reported to regulate permeability in other cellular systems (13-15). Because activation of PAR-1, -2, and -4 induces cellular signaling responses coupled with our previous observation that basolateral, but not apical, application of PMNs decreases TER (6), we tested whether specific peptide agonists to these receptors have the capability to decrease TER in a polarized manner. Experiments were performed by adding specific PAR-1 TFLLR-NH2, PAR-2 SLIGRL-NH2, and PAR-4 AYPGKF-NH2 agonists and their respective reverse control peptides RLLFT-NH2, LRGILS-NH2, and YAPGKF-NH2 to the apical or basolateral compartment of T84 monolayers cultured on 0.4-μm pore size filters. Apical addition of the specific PAR-1 (TFLLR-NH2), PAR-2 (SLIGRL-NH2), and PAR-4 (AYPGKF-NH2) agonists and their respective reverse control peptides failed to decrease TER in confluent T84 monolayers (Fig. 3A). However, basolateral addition of the specific PAR-1 TFLLR-NH2 or PAR-2 SLIGRL-NH2 agonists, but not the PAR-4 agonist AYPGKF-NH2, potently decreased baseline TER by ~40% within 30 s (Fig. 3A). Similar results were obtained with the SK-CO15 cell line (data not shown). These results suggest that the polarized increase in epithelial permeability induced by PMN contact is dependent on the activation of PAR-1 and -2 at the basolateral surfaces of the epithelium.
FIGURE 3.
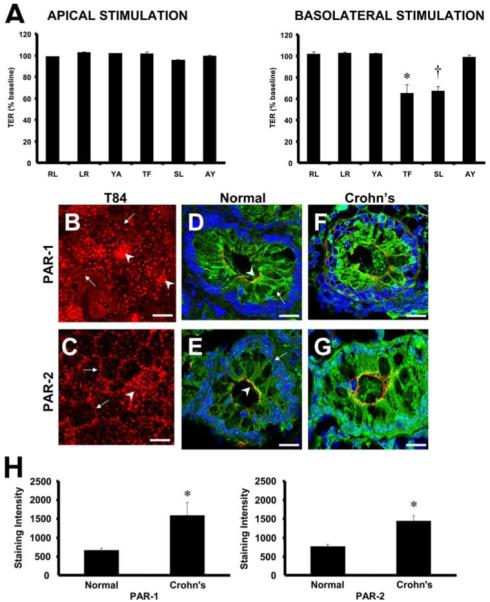
Basolateral activation of PAR-1 and -2 regulates the fall in TER induced by PMN contact. A, T84 monolayers were cultured in either the upright (apical stimulation) or inverted (basolateral) orientation on 0.4-μm pore size filters, and TER was measured 30 s after addition of 50 μM RLLFT-NH2 (RL; PAR-1 control peptide), TFLLR-NH2 (TF; PAR-1 agonist), LRGILS-NH2 (LR; PAR-2 control peptide), SLIGRL-NH2 (SL; PAR-2 agonist), YAPGKF-NH2 (YA; PAR-4 control peptide), or AYPGKF-NH2 (AY; PAR-4 agonist) to the upper chamber. TER is expressed as the percentage of the baseline TER at the start of the assay. Values are means ± SEM, n = 3 per group; *, p < 0.05 compared with control (RLLFT-NH2; RL); †, p < 0.05 compared with control (LR). Data were generated from three independent experiments done in duplicate. To assess the cellular localization of PAR-1 and -2, T84 monolayers cultured on 0.4-μm pore size filters were fixed and processed for immunofluorescence microscopy. En face confocal image of epithelial monolayer demonstrates that PAR-1 (B) and PAR-2 (C) are localized within intracellular pools (arrowhead) and in close proximity to the lateral surface (arrow). Furthermore, colonic mucosal tissues from normal noninflammatory bowel disease and Crohn’s disease patients were fixed and processed for confocal immunofluorescence microscopy. Confocal images obtained from colonic mucosa of normal noninflammatory bowel disease patients indicates that PAR-1 (green, arrow; D) and PAR-2 (green, arrow; E) are localized beneath the tight junctional protein occludin (red, arrowhead). Colonic mucosa obtained from Crohn’s disease patients exhibited increased intensities of PAR-1 (F) and PAR-2 (G) which were quantified by image analysis software (H). Values are means ± SEM from tissue sections obtained from four normal and four Crohn’s disease patients. *, p < 0.05 compared with normal colon samples. Bar, 20 μm.
To further support the role of basolateral activation of PAR-1 and -2 in regulating epithelial permeability, we conducted immunofluorescence microscopy studies to evaluate the expression and localization of these receptors on epithelial monolayers. T84 monolayers cultured on 0.4-μm pore-size filters were fixed in absolute methanol and immunostained for PAR-1 (Santa Cruz; Ref. 19) or PAR-2 (from Dr. Morley Hollenberg; Ref. 20). As shown in Fig. 3, B and C, en face confocal images revealed abundant levels of both PAR-1 and -2 within intracellular pools as previously shown by other studies which indicated that these receptors undergo trafficking between cytosolic compartments and the cell surface (21, 22). In addition to assessing the localization of PAR-1 and -2 in vitro, human colonic tissue specimens were immunostained to assess the localization of PAR-1 and -2 under normal physiological or inflammatory conditions (Crohn’s disease). In a similar pattern seen in vitro, PAR-1 and -2 were abundantly expressed within the cytoplasm beneath the tight junctions in normal tissues (Fig. 3, D and E). However, tissues obtained from Crohn’s disease patients exhibited enhanced staining intensities of both PAR-1 and -2 as compared with normal tissues (Fig. 3, F-H). In addition, no leukocyte infiltrate was observed in the samples. These observations suggest that localization of PAR-1 and -2 to the basolateral surfaces of epithelial cells may partially explain the polarized decrease in TER induced by PMN contact. Furthermore, the abundant intracellular staining of PAR-1 and -2 may indicate that these receptors can be replenished to the cell surface from intracellular compartments to maintain cell signaling, particularly under inflammatory conditions.
Because the above data indicate that basolateral activation of epithelial PAR-1 and -2 decreases TER, we used an siRNA-based approach to examine whether the loss of endogenous expression of these receptors can prevent the loss of epithelial barrier function induced by PMN contact. Instead of T84 cells, which exhibit low transfection efficiency, subconfluent SK-CO15 epithelial cell monolayers were transfected with siRNA directed against PAR-1 and/or -2. As shown in Fig. 4A, PAR-1 or -2 mRNA was significantly reduced by treatment with PAR-1 or -2 siRNA. To determine whether the loss of epithelial PAR-1 or -2 affected the ability of basolaterally applied PMNs to disrupt barrier function, TER measurements were performed in cell monolayers treated with siRNA. As shown in Fig. 4B, a combination of PAR-1 and PAR-2 siRNA treatment was required to prevent the decrease in TER induced by PMN contact. To demonstrate that these effects were not simply the result of a general effect of protein knockdown, the cells were transfected with siRNA against an unrelated protein, cyclophilin B. As shown in Fig. 4B, siRNA against cyclophilin B did not prevent the decrease in TER induced by PMN contact. To reflect the total siRNA concentration used for combined PAR-1 and PAR-2 knockdown, pretreatment with 100 nM cyclophilin B also failed to reverse the fall in TER induced by PMN contact (data not shown). Furthermore, transfection with the siRNA alone did not affect TER. These results suggest that PMN contact with basolateral surfaces with epithelial cells disrupt barrier function through activation of either PAR-1 or -2.
FIGURE 4.
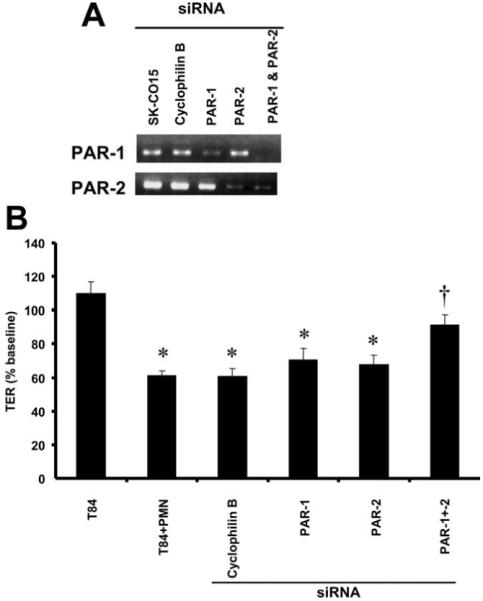
Knockdown of epithelial PAR-1 and -2 prevents the fall in TER induced by PMN contact. Inverted SK-CO15 monolayers cultured on 0.4-μm pore size filters were transfected with siRNA targeting cyclophilin B (control siRNA), PAR-1, PAR-2, or PAR-1 and -2. Semiquantitative RT-PCR analysis revealed decreased PAR-1 and -2 mRNA after transfection with PAR-1 and -2 siRNA, respectively. B, After epithelial siRNA pretreatment, PMNs were added to the basolateral aspect (upper compartment) of the monolayers cultured on 0.4-μm pore size filters while fMLP was added to the lower compartment. TER was measured after 3 h and is expressed as the percentage of baseline TER prior to addition of PMNs or vehicle. Values are means ± SEM, n = 3 per group; *, p < 0.05 compared with untransfected epithelial monolayers incubated with an fMLP gradient; †, p < 0.05 compared with PMNs added to the basolateral aspect of epithelial monolayers transfected with cyclophilin B siRNA or to untransfected epithelial monolayers. Data were generated from three independent experiments done in duplicate.
Activation of PAR-1 and -2 stimulates MLCK and myosin L chain phosphorylation
Because our results suggested that the fall in TER induced by PMN contact with the basal aspect of epithelial cells is mediated by activation of PAR-1 and -2, we performed experiments to probe the downstream mechanism for such effects. We previously demonstrated that PMN contact with the basolateral surfaces of epithelial cells enhances myosin L chain phosphorylation (6). Interestingly, phosphorylation of MLCK has been shown to result in contraction of the epithelial perijunctional actin-myosin ring which regulates the passage of solutes through the paracellular space (23). Specifically, phosphorylation of the 215 kDa isoform of MLCK accounts for >95% of myosin L chain phosphorylation activity (24), and phosphorylation of myosin L chain at Thr18 and Ser19 has been shown to induce such contractile events (23). In a similar manner, PMN-stimulated monolayers exhibited enhanced phosphorylation of the 215 kDa MLCK isoform and subsequent phosphorylation of myosin L chain (Fig. 5A).
FIGURE 5.
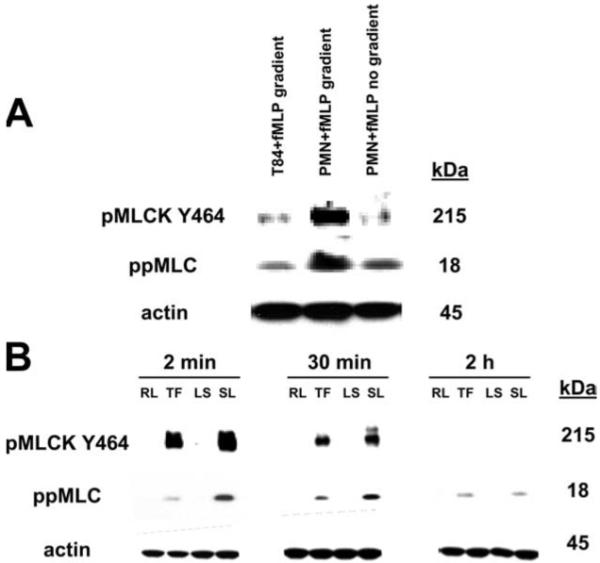
Phosphorylation of MLCK and myosin L chain is enhanced upon PMN contact with the basolateral aspect of epithelial cells or activation of PAR-1 and -2. A, PMNs were added to the basolateral aspect of inverted T84 epithelial monolayers cultured on 0.4 μm pore-size supports and induced to migrate in the basolateral to apical direction in the presence of a transepithelial fMLP gradient (PMN + fMLP gradient). Samples were incubated at 37°C until TER had decreased by 50% of the baseline TER. TER did not decrease when T84 monolayers were incubated with an fMLP transepithelial gradient (T84 + fMLP gradient) or basolaterally applied PMNs directly stimulated with fMLP (PMN + fMLP; no gradient). Whole-cell lysates of inverted T84 monolayers, were run on SDS-polyacrylamide gels and Western blotted for phosphorylated MLCK (Tyr494), myosin L chain (Thr18 and Ser19), and actin. Epithelial monolayers stimulated with PMN contact exhibited enhanced MLCK and myosin L chain phosphorylation. B, SDS-PAGE and Western blot analysis of phosphorylated MLCK (Tyr494), myosin L chain (Thr18 and Ser19), and actin. Whole-cell lysates were obtained from inverted T84 epithelial monolayers cultured on 0.4-μm pore size supports that were stimulated basolaterally with 50 μM of RLLFT-NH2 (RL; PAR-1 control peptide), TFLLR-NH2 (TF; PAR-1 agonist), LRGILS-NH2 (LR; PAR-2 control peptide), SLIGRL-NH2 (SL; PAR-2 agonist), YAPGKF-NH2 (YA; PAR-4 control peptide), or AYPGKF-NH2 (AY; PAR-4 agonist) for 2 min, 30 min, and 2 h at 37°C.
To determine whether activation of PAR-1 or PAR-2 can similarly induce changes in the myosin L chain phosphorylation cascade, monolayers were stimulated with specific agonists to PAR-1 (TFLLR-NH2), PAR-2 (SLIGRL-NH2), or their respective control peptides (RLLFT-NH2, LRGILS-NH2) for various times (2 min, 30 min, and 2 h) and then prepared for biochemical analysis. As shown in Fig. 5B, an increase in phosphorylation of the 215 kDa isoform of MLCK was observed in monolayers stimulated with PAR-1 and -2 agonists. Furthermore, the increase in MLCK phosphorylation induced by PAR-1 and -2 activation was accompanied by an increase in myosin L chain phosphorylation (Fig. 5B). These findings suggest that the PMN-induced fall in TER is mediated by the activation of either PAR-1 or -2 followed by subsequent phosphorylation of MLCK and myosin L chain.
PAR-1 and -2 regulate PMN transepithelial migration
Given that the above data implicate PAR-1 and -2 in the regulation of PMN-induced changes in epithelial permeability, we next sought to determine whether these receptors regulate PMN transepithelial migration. We reasoned that early signaling events between migrating PMNs and the basolateral aspect of the intestinal epithelium might serve to loosen the epithelial barrier and facilitate subsequent transmigration through the paracellular space. If this hypothesis is true, then inhibition of PAR-1- and -2-mediated signaling events should inhibit translocation of PMNs across epithelial monolayers. To test this hypothesis, inverted SK-CO15 monolayers cultured on 5.0-μm pore size supports were pretreated with siRNA directed against PAR-1, PAR-2, or a combination of both before use in transmigration assays. In a similar manner shown by the decrease in TER induced by PMN contact, pretreatment of epithelial monolayers with a combination of both PAR-1 and PAR-2 siRNA significantly decreased the rate of PMN transepithelial migration (Fig. 6). These findings strongly implicate PAR-1 and -2 as mediators of PMN-epithelial signaling and as regulators of PMN transepithelial migration.
FIGURE 6.
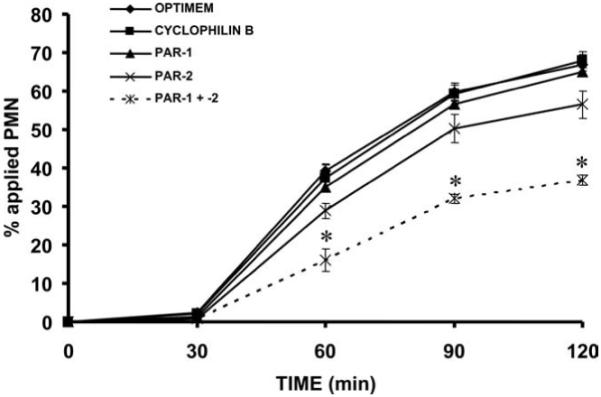
Knockdown of epithelial PAR-1 and -2 inhibits PMN transepithelial migration. Inverted T84 monolayers cultured on 5.0-μm pore size filters were transfected with siRNA targeting cyclophilin B (control siRNA), PAR-1, PAR-2, or PAR-1 and -2. After epithelial siRNA pretreatment, PMNs were added to the basolateral aspect of the monolayers to assay for PMN transepithelial migration. Values are means ± SEM, n = 3 per group; *, p < 0.05 compared with epithelial monolayers transfected with cyclophilin B siRNA or to untransfected epithelial monolayers. Data were generated from three independent experiments done in duplicate.
Discussion
In this study, we report a mechanism by which basolaterally applied PMNs increase epithelial permeability and how this early process facilitates PMN transepithelial migration in the physiologically relevant basolateral-to-apical direction. Our data suggest that PMN contact with basolateral surfaces of epithelial cells reduces TER in a manner that is independent of proteolytic degradation of apical junction proteins and that the PMN proteases elastase and proteinase-3, but not cathepsin G, increase epithelial permeability. The functional decrease in TER induced by PMN contact and subsequent PMN transepithelial migration were dependent on PMN serine proteases. Investigation into protease-mediated signaling events has revealed that activation of basolaterally expressed PAR-1 and -2 disrupt epithelial barrier function and immunolocalization studies revealed that PAR-1 and -2 are present in intracellular compartments in close proximity to the lateral epithelial surfaces, and are up-regulated in Crohn’s disease. Combined inhibition of PAR-1 and -2 using siRNA prevented the fall in TER induced by PMN contact. We further report that the decrease in TER induced by PMN contact and PAR-1 and -2 activation results in an increase in MLCK and myosin L chain phosphorylation. Lastly, combined inhibition of epithelial PAR-1 and -2 using siRNA results in inhibition of PMN transepithelial migration. This is the first report demonstrating that PMN-induced alterations in epithelial barrier function and subsequent PMN transepithelial migration are dependent on PAR-1 and -2.
Previously, we have demonstrated that multiple apical junction proteins did not exhibit gross morphological changes or differences in localization patterns in monolayers stimulated with PMN contact (6). To further characterize the effects of PMN contact on epithelial monolayers, the present study reports that the reduction in TER induced by PMNs is associated neither with proteolytic degradation nor with changes in total apical junction protein levels. Past studies have shown that endothelial cadherin is degraded during PMN adhesion to endothelial monolayers (25-27). Furthermore, it has been suggested that PMN-mediated cleavage of endothelial cadherin may be due to PMN membrane-bound proteases, which increase in expression after limited degranulation from primary granules upon stimulation (8, 27). A major difference between transendothelial and transepithelial migration is the physical distance required by PMNs to traverse the paracellular space. Endothelial apical junction proteins are readily accessed by transmigrating and adherent PMNs, whereas epithelial apical junction proteins can be accessed only after significant migration has been made through the paracellular space, a condition that is also not permissible with filter-inhibited PMN transmigration. Another report has demonstrated that soluble E-cadherin fragments have been detected in supernatants obtained during PMN transepithelial migration (28), thus further suggesting that direct contact between PMNs and epithelial cells in the paracellular space may facilitate the cleavage of apical junction proteins. Furthermore, addition of purified elastase to the apical compartment of epithelial monolayers resulted in the cleavage of E-cadherin, but only after opening of tight junctions (28). Despite the lack of morphological and biochemical evidence of apical junction protein changes, we cannot exclude the possibility of other molecular alterations in these proteins. However, these studies collectively suggest that physical disruption of epithelial intercellular junctions by transmigrating PMNs within the paracellular space and direct release of PMN proteases into this microenvironment is required to induce cleavage of E-cadherin. Indeed, our model of filter-inhibited PMN transmigration limits the physical interactions between PMNs or PMN membrane-bound proteases and epithelial intercellular junctions. Therefore, it is likely that the functional decrease in TER induced by PMN contact is not due to gross morphological changes to apical junctional proteins, but rather an induction of epithelial signaling events.
Given that total protein levels and localization of several apical junction proteins did not change in monolayers subjected to PMN contact, we shifted our investigation into the role of PMN proteases in inducing a functional response in epithelial monolayers. Initial experiments revealed that the decrease in TER is achieved more rapidly (within 30 s) with the use of soluble PMN elastase and proteinase-3 as compared with PMN contact which may involve the delayed delivery of membrane-bound proteases through 0.4-μm pores. In addition, only broad inhibition of serine proteases prevents the PMN-stimulated decrease in TER and inhibits PMN transepithelial migration. It was previously reported that inhibition of elastase with DMP-777 attenuated transepithelial migration and the fall in TER during this event (28). Although we were unable to obtain DMP-777, our investigations using AAPV or GW311616A (data not shown) at multiple concentrations failed to support the role of PMN elastase in mediating PMN-stimulated epithelial permeability and transepithelial migration. In a similar manner, a previous study reported that inhibition of PMN elastase with AAPV failed to prevent the increase in permeability induced by prolonged PMN contact with the epithelium (5). Furthermore, PMNs isolated from PMN elastase-deficient mice did not exhibit any defects in transendothelial migration (26). Our results are consistent with sufficient redundancy exhibited by PMN proteases and that broad inhibition is required to prevent epithelial permeability induced by PMN contact and subsequent PMN transepithelial migration. Indeed, nonspecific inhibition of serine proteases with diisopropylphosphate significantly inhibited PMN transepithelial migration and the accompanying fall in TER (28). Regardless of these findings, it still remains to determine the PMN and epithelial cell surface receptors that are responsible for the polarized decrease in epithelial barrier function induced by PMN contact.
We previously reported that PMN contact induces phosphorylation of multiple proteins in epithelial cells (6), thus indicating the activation of intracellular signaling events. Because the present study indicates that multiple PMN proteases regulate epithelial permeability and PMN transepithelial migration, we sought to determine whether PMN proteases can activate epithelial PARs and contribute to the increase in epithelial permeability before PMN transmigration through the paracellular space. Indeed, neutrophil elastase has been shown to disable PAR-2 function (29) while activating PAR-1 in lung epithelial cells (10). Proteinase-3 activates PAR-2 in oral epithelial cells (12), whereas cathepsin G activates PAR-4 in platelets (9) and PAR-2 in gingival fibroblasts (11). Despite these studies, little is known about the role of PAR activation in the regulation of epithelial barrier function. However, given that PMN serpocidins have been shown to activate PARs, we attempted to further characterize the role of PAR activation in regulating epithelial permeability. Our study demonstrates that basolateral activation of PAR-1 or -2 disrupts epithelial barrier function; this polarized response further supports our previous observations whereby basolateral, but not apical, application of PMNs induces a fall in TER in epithelial monolayers (6). Although apical and basolateral activation of PAR-1 has been shown to increase epithelial permeability in small intestinal duodenal monolayers (13), our studies indicate that only basolateral activation of PAR-1 or -2 in colonic crypt-like T84 epithelial cells is responsible for this activity. Indeed, only basolateral activation of PAR-2 in T84 epithelial cells has been reported to increase permeability and induced changes in ZO-1, occludin, and perijunctional F-actin (14). However, the polarized response to PAR-2 agonists may not be the sole result of intercellular differences, but rather the peculiar nature of the receptor. Indeed, whereas apical and basolateral activation of PAR-1 in small intestinal epithelial cells increased permeability (13), only basolateral activation of PAR-2 elicited this response in the same cell line (15). Further studies are needed to determine differences in PAR distribution along the gastrointestinal tract and whether apical and basolateral activations of PARs are coupled to different signaling pathways.
The role of basolateral PAR-1 and -2 signaling in the regulation of epithelial barrier function is further supported by immunolocalization studies. The results described herein demonstrate that PAR-1 and -2 are expressed within the cytosol and in close proximity to the lateral surfaces beneath the tight junctions. PAR-1 and -2 staining within the cytosol indicates that these receptors are stored within intracellular compartments. Indeed, uncleaved PAR-1 has been shown to constitutively cycle between the cell surface and recycling endosomes, thereby providing a protected intracellular pool that may be mobilized to the cell surface after protease exposure and lead to rapid resensitization (21). On a similar note, uncleaved PAR-2 is localized within a pre-existing Golgi pool but does not undergo constitutive recycling (22). In addition, the present study demonstrates for the first time that epithelial cells of colonic tissue specimens obtained from patients with active Crohn’s disease express higher levels of both PAR-1 and PAR-2. Lending support to our findings, a previous report has demonstrated enhanced levels of PAR-1 in the intestinal mucosa in a rat model of intestinal ischemia-reperfusion (30). Similarly, other studies have described elevated PAR-2 levels in lamina propria mast cells in ulcerative colitis biopsies (31), in isolated human coronary arteries after inflammatory mediator exposure ex vivo (32), in synovial and periarticular tissues in a murine model of adjuvant monoarthritis (33), in keratinocytes of patients with atopic dermatitis (34), and in the bladder in a rat model of cystitis (35). Despite these observations, the role of epithelial PARs during mucosal inflammation has not been thoroughly investigated. It is conceivable that the combination of rapid resensitization of PARs from intracellular compartments, increased PAR levels in inflamed tissues, and the abundance of proteases from neutrophilic exudates may together amplify PAR-mediated disruption of epithelial barrier function and perpetuate the active migration of PMNs through the intestinal epithelium. Given that epithelial cells have been shown to express formylated peptide receptors (36), we also determined whether incubation with fMLP enhanced epithelial PAR expression or induced changes in PAR localization. Treatment with fMLP failed to enhance PAR expression or induce changes to PAR localization. Furthermore, we have also determined that addition of fMLP to both the basolateral and apical aspect in the presence of basolateral PMNs failed to reduce TER (data not shown). Nevertheless, further studies are needed to determine the mechanisms behind the increase in PAR-1 and -2 under inflammatory conditions.
The signaling events that couple PAR-1 and -2 activation with the disruption of epithelial barrier function are poorly understood. However, we have previously shown that PMN contact with epithelial cells increases myosin L chain phosphorylation (6), which is a key event in maintaining actin-myosin ring contraction and increasing paracellular permeability. In the current study, we provide evidence that PMN contact as well as basolateral activation of PAR-1 and -2 enhance MLCK and myosin L chain phosphorylation in epithelial cells. Upon activation, PAR-1 and -2 have been reported to couple to several G proteins including Gαq which in turn induces intracellular calcium release (16). The increase in intracellular calcium activates calmodulin which is critical for the activity of calcium-calmodulin kinases such as MLCK (37), which accounts for >95% of myosin L chain-phosphorylating activity in confluent epithelial monolayers (24). MLCK activity, as measured by myosin L chain phosphorylation, can be further enhanced through tyrosine phosphorylation by p60src (38), which may be the mechanism observed in our system. Indeed, a past study has demonstrated that PAR-1-mediated chloride secretion is induced through a p60src-dependent mechanism (39). Lending further support for our results, pharmacological inhibition of MLCK has been shown to prevent the increase in epithelial permeability induced by PAR-1 (13) and PAR-2 activation (15). Furthermore, pharmacological inhibition of MLCK and inhibition of intracellular calcium/calmodulin activity in endothelial cells have been shown to decrease PMN transendothelial migration (40). Although our results strongly suggest that the PAR-1- and -2-mediated disruption in epithelial barrier function occurs in a MLCK-dependent manner, myosin L chain phosphorylation can also occur downstream of the calcium-independent Rho effector pathway. Further studies investigating the role of PAR signaling in the activation of RhoA-related pathways and regulation of barrier function in epithelial cells should provide important mechanistic insights. Nevertheless, our results indicate that the contraction of the actin-myosin ring followed by increased paracellular permeability induced by the phosphorylation of regulatory myosin L chain represents a major signaling pathway during PMN transepithelial migration.
Given that basolateral activation of epithelial PAR-1 and -2 decreases TER, we initiated a series of experiments to determine whether the fall in TER induced by PMN contact and subsequent PMN transepithelial migration is dependent on these receptors. The results of these experiments indicate that siRNA-mediated knockdown of both PAR-1 and -2 prevents the decrease in TER induced by PMN contact with epithelial cells and significantly blocks the rate of PMN transepithelial migration. Because our results suggest that multiple proteases may play a role in regulating the decrease in TER induced by PMN contact, it is not surprising that knockdown of both PAR-1 and PAR-2 was necessary to attenuate the effects of this redundancy. Indeed, transmigrating PMNs express a multitude of proteases that may potentially activate PARs on epithelial surfaces. In turn, PAR activation may increase epithelial permeability through myosin L chain-dependent actin-myosin ring contraction and facilitate PMN transepithelial migration through the paracellular space. However, the possibility of indirect effects whereby PMN contact with the epithelium could induce the expression of epithelial factors involved in PAR activation cannot be excluded on the basis of the results in this study. Future investigations will determine the identity of the proteases responsible for epithelial PAR activation. Although our results indicate that epithelial PAR-1 and -2 may regulate PMN transepithelial migration, combined knockdown of these receptors failed to completely inhibit this effect, thus indicating that other mechanisms exist in regulating transepithelial migration. It is possible that induction of PAR signaling by subepithelial PMNs may serve to fine-tune PMN transepithelial migration by widening the paracellular space via myosin L chain contraction and unmask counterreceptors that may be engaged to facilitate PMN transmigration. Alternatively, incomplete knockdown of epithelial PAR-1 and -2 may have permitted PMNs to transmigrate across the epithelium. Further studies that develop the use of epithelial monolayers stably transfected with shRNA against PAR-1 and -2 may help address this issue.
In summary, our report indicates that basolateral activation of epithelial PAR-1 by subepithelial PMNs increases epithelial permeability and facilitates PMN transmigration through the paracellular space. We speculate that this novel mechanism may play a role in the pathogenesis of mucosal inflammatory disorders characterized by PMN infiltration and associated epithelial damage. Further characterization of PAR-1- and -2-mediated PMN transepithelial migration may prove to be useful in generating potential therapeutic strategies in controlling pathological inflammatory conditions.
Acknowledgments
The authors thank Dr. Susan Voss for expert tissue culture assistance and Eric Severson for technical advice on siRNA experiments. The rabbit polyclonal Ab raised against PAR-2 (B5/A5) was a gift from Dr. Morley Hollenberg.
Footnotes
This study was supported by research fellowships from the Canadian Association of Gastroenterology, Canadian Institutes of Health Research, Axcan Pharma, and the Crohn’s and Colitis Foundation of America (to A.C.C.); by grants from the Alberta Heritage Foundation for Medical Research, and the Canadian Institutes of Health Research, INSERM-Avenir, and the Foundation Bettencourt-Schueller (to N.V.); National Institutes of Health Grants DK59888 (to A.N.), DK079392, and DK72564 (to C.A.P.), and a Digestive Diseases Minicenter Grant DK064399 (epithelial cell culture core and microscopy core support).
- PMN
- polymorphonuclear neutrophil
- PAR
- protease-activated receptor
- MLCK
- myosin L chain kinase
- AAPV
- N-(methoxysuccinyl)-alanylalanylprolylvalyl chloromethyl ketone
- AEBSF
- 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride
- TER
- transepithelial electrical resistance
- siRNA
- small interfering RNA
Disclosures
The authors have no financial conflict of interest.
References
- 1.Kumar NB, Nostrant TT, Appelman HD. The histopathologic spectrum of acute self-limited colitis (acute infectious-type colitis) Am. J. Surg. Pathol. 1982;6:523–529. doi: 10.1097/00000478-198209000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Parkos CA, Delp C, Arnaout MA, Madara JL. Neutrophil migration across a cultured intestinal epithelium: dependence on a CD11b/CD18-mediated event and enhanced efficiency in physiological direction. J. Clin. Invest. 1991;88:1605–1612. doi: 10.1172/JCI115473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nusrat A, Parkos CA, Liang TW, Carnes DK, Madara JL. Neutrophil migration across model intestinal epithelia: monolayer disruption and subsequent events in epithelial repair. Gastroenterology. 1997;113:1489–1500. doi: 10.1053/gast.1997.v113.pm9352851. [DOI] [PubMed] [Google Scholar]
- 4.Nash S, Stafford J, Madara JL. Effects of polymorphonuclear leukocyte transmigration on the barrier function of cultured intestinal epithelial monolayers. J. Clin. Invest. 1987;80:1104–1113. doi: 10.1172/JCI113167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parsons PE, Sugahara K, Cott GR, Mason RJ, Henson PM. The effect of neutrophil migration of prolonged neutrophil contact on epithelial permeability. Am. J. Pathol. 1987;129:302–312. [PMC free article] [PubMed] [Google Scholar]
- 6.Edens HA, Levi BP, Jaye DL, Walsh S, Reaves TA, Turner JR, Nusrat A, Parkos CA. Neutrophil transepithelial migration: evidence for sequential, contact-dependent signaling events and enhanced paracellular permeability independent of transjunctional migration. J. Immunol. 2002;169:476–486. doi: 10.4049/jimmunol.169.1.476. [DOI] [PubMed] [Google Scholar]
- 7.Owen CA, Campbell MA, Sannes PL, Boukedes SS, Campbell EJ. Cell surface-bound elastase and cathepsin G on human neutrophils: a novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J. Cell Biol. 1995;131:775–789. doi: 10.1083/jcb.131.3.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sengelov H, Kjeldsen L, Borregaard N. Control of exocytosis in early neutrophil activation. J. Immunol. 1993;150:1535–1543. [PubMed] [Google Scholar]
- 9.Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. J. Biol. Chem. 2000;275:6819–6823. doi: 10.1074/jbc.275.10.6819. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki T, Moraes TJ, Vachon E, Ginzberg HH, Huang TT, Matthay MA, Hollenberg MD, Marshall J, McCulloch CA, Abreu MT, et al. Proteinase-activated receptor-1 mediates elastase-induced apoptosis of human lung epithelial cells. Am. J. Respir. Cell. Mol. Biol. 2005;33:231–247. doi: 10.1165/rcmb.2005-0109OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uehara A, Muramoto K, Takada H, Sugawara S. Neutrophil serine proteinases activate human nonepithelial cells to produce inflammatory cytokines through protease-activated receptor 2. J. Immunol. 2003;170:5690–5696. doi: 10.4049/jimmunol.170.11.5690. [DOI] [PubMed] [Google Scholar]
- 12.Uehara A, Sugawara S, Muramoto K, Takada H. Activation of human oral epithelial cells by neutrophil proteinase 3 through protease-activated receptor-2. J. Immunol. 2002;169:4594–4603. doi: 10.4049/jimmunol.169.8.4594. [DOI] [PubMed] [Google Scholar]
- 13.Chin AC, Vergnolle N, MacNaughton WK, Wallace JL, Hollenberg MD, Buret AG. Proteinase-activated receptor 1 activation induces epithelial apoptosis and increases intestinal permeability. Proc. Natl. Acad. Sci. USA. 2003;100:11104–11109. doi: 10.1073/pnas.1831452100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacob C, Yang PC, Darmoul D, Amadesi S, Saito T, Cottrell GS, Coelho AM, Singh P, Grady EF, Perdue M, Bunnett NW. Mast cell tryptase controls paracellular permeability of the intestine: role of protease-activated receptor 2 and β-arrestins. J. Biol. Chem. 2005;280:31936–31948. doi: 10.1074/jbc.M506338200. [DOI] [PubMed] [Google Scholar]
- 15.Cenac N, Chin AC, Garcia-Villar R, Salvador-Cartier C, Ferrier L, Vergnolle N, Buret AG, Fioramonti J, Bueno L. PAR2 activation alters colonic paracellular permeability in mice via IFN-γ-dependent and -independent pathways. J. Physiol. 2004;558:913–925. doi: 10.1113/jphysiol.2004.061721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol. Rev. 2004;84:579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- 17.Mandell KJ, McCall IC, Parkos CA. Involvement of the junctional adhesion molecule-1 (JAM1) homodimer interface in regulation of epithelial barrier function. J. Biol. Chem. 2004;279:16254–16262. doi: 10.1074/jbc.M309483200. [DOI] [PubMed] [Google Scholar]
- 18.Lee WY, Chin AC, Voss S, Parkos CA. In vitro neutrophil transepithelial migration. Methods Mol. Biol. 2006;341:205–215. doi: 10.1385/1-59745-113-4:205. [DOI] [PubMed] [Google Scholar]
- 19.Grisaru-Granovsky S, Salah Z, Maoz M, Pruss D, Beller U, Bar-Shavit R. Differential expression of protease activated receptor 1 (Par1) and pY397FAK in benign and malignant human ovarian tissue samples. Int. J. Cancer. 2005;113:372–378. doi: 10.1002/ijc.20607. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Zheng H, Hollenberg MD, Wijesuriya SJ, Ou X, Hauer-Jensen M. Up-regulation and activation of proteinase-activated receptor 2 in early and delayed radiation injury in the rat intestine: influence of biological activators of proteinase-activated receptor 2. Radiat. Res. 2003;160:524–535. doi: 10.1667/rr3080. [DOI] [PubMed] [Google Scholar]
- 21.Hein L, Ishii K, Coughlin SR, Kobilka BK. Intracellular targeting and trafficking of thrombin receptors: a novel mechanism for resensitization of a G protein-coupled receptor. J. Biol. Chem. 1994;269:27719–27726. [PubMed] [Google Scholar]
- 22.Dery O, Thoma MS, Wong H, Grady EF, Bunnett NW. Trafficking of proteinase-activated receptor-2 and β-arrestin-1 tagged with green fluorescent protein: β-arrestin-dependent endocytosis of a proteinase receptor. J. Biol. Chem. 1999;274:18524–18535. doi: 10.1074/jbc.274.26.18524. [DOI] [PubMed] [Google Scholar]
- 23.Ikebe M, Hartshorne DJ. Phosphorylation of smooth muscle myosin at two distinct sites by myosin light chain kinase. J. Biol. Chem. 1985;260:10027–10031. [PubMed] [Google Scholar]
- 24.Turner JR, Angle JM, Black ED, Joyal JL, Sacks DB, Madara JL. PKC-dependent regulation of transepithelial resistance: roles of MLC and MLC kinase. Am. J. Physiol. 1999;277:C554–C562. doi: 10.1152/ajpcell.1999.277.3.C554. [DOI] [PubMed] [Google Scholar]
- 25.Carden D, Xiao F, Moak C, Willis BH, Robinson-Jackson S, Alexander S. Neutrophil elastase promotes lung microvascular injury and proteolysis of endothelial cadherins. Am. J. Physiol. 1998;275:H385–H392. doi: 10.1152/ajpheart.1998.275.2.H385. [DOI] [PubMed] [Google Scholar]
- 26.Allport JR, Lim YC, Shipley JM, Senior RM, Shapiro SD, Matsuyoshi N, Vestweber D, Luscinskas FW. Neutrophils from MMP-9- or neutrophil elastase-deficient mice show no defect in transendothelial migration under flow in vitro. J. Leukocyte Biol. 2002;71:821–828. [PubMed] [Google Scholar]
- 27.Hermant B, Bibert S, Concord E, Dublet B, Weidenhaupt M, Vernet T, Gulino-Debrac D. Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J. Biol. Chem. 2003;278:14002–14012. doi: 10.1074/jbc.M300351200. [DOI] [PubMed] [Google Scholar]
- 28.Ginzberg HH, Cherapanov V, Dong Q, Cantin A, McCulloch CA, Shannon PT, Downey GP. Neutrophil-mediated epithelial injury during transmigration: role of elastase. Am. J. Physiol. 2001;281:G705–G717. doi: 10.1152/ajpgi.2001.281.3.G705. [DOI] [PubMed] [Google Scholar]
- 29.Dulon S, Cande C, Bunnett NW, Hollenberg MD, Chignard M, Pidard D. Proteinase-activated receptor-2 and human lung epithelial cells: disarming by neutrophil serine proteinases. Am. J. Respir. Cell Mol. Biol. 2003;28:339–346. doi: 10.1165/rcmb.4908. [DOI] [PubMed] [Google Scholar]
- 30.Tsuboi H, Naito Y, Katada K, Takagi T, Handa O, Kokura S, Ichikawa H, Yoshida N, Tsukada M, Yoshikawa T. Role of the thrombin/protease-activated receptor 1 pathway in intestinal ischemia-reperfusion injury in rats. Am. J. Physiol. 2007;292:G678–G683. doi: 10.1152/ajpgi.00361.2006. [DOI] [PubMed] [Google Scholar]
- 31.Kim JA, Choi SC, Yun KJ, Kim DK, Han MK, Seo GS, Yeom JJ, Kim TH, Nah YH, Lee YM. Expression of protease-activated receptor 2 in ulcerative colitis. Inflamm. Bowel Dis. 2003;9:224–229. doi: 10.1097/00054725-200307000-00002. [DOI] [PubMed] [Google Scholar]
- 32.Hamilton JR, Frauman AG, Cocks TM. Increased expression of protease-activated receptor-2 (PAR2) and PAR4 in human coronary artery by inflammatory stimuli unveils endothelium-dependent relaxations to PAR2 and PAR4 agonists. Circ. Res. 2001;89:92–98. doi: 10.1161/hh1301.092661. [DOI] [PubMed] [Google Scholar]
- 33.Ferrell WR, Lockhart JC, Kelso EB, Dunning L, Plevin R, Meek SE, Smith AJ, Hunter GD, McLean JS, McGarry F, et al. Essential role for proteinase-activated receptor-2 in arthritis. J. Clin. Invest. 2003;111:35–41. doi: 10.1172/JCI16913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buddenkotte J, Stroh C, Engels IH, Moormann C, Shpacovitch VM, Seeliger S, Vergnolle N, Vestweber D, Luger TA, Schulze-Osthoff K, Steinhoff M. Agonists of proteinase-activated receptor-2 stimulate upregulation of intercellular cell adhesion molecule-1 in primary human keratinocytes via activation of NF-κB. J. Invest. Dermatol. 2005;124:38–45. doi: 10.1111/j.0022-202X.2004.23539.x. [DOI] [PubMed] [Google Scholar]
- 35.Dattilio A, Vizzard MA. Up-regulation of protease activated receptors in bladder after cyclophosphamide induced cystitis and colocalization with capsaicin receptor (VR1) in bladder nerve fibers. J. Urol. 2005;173:635–639. doi: 10.1097/01.ju.0000143191.55468.1d. [DOI] [PubMed] [Google Scholar]
- 36.Babbin BA, Jesaitis AJ, Ivanov AI, Kelly D, Laukoetter M, Nava P, Parkos CA, Nusrat A. Formyl peptide receptor-1 activation enhances intestinal epithelial cell restitution through phosphatidylinositol 3-kinase-dependent activation of Rac1 and Cdc42. J. Immunol. 2007;179:8112–8121. doi: 10.4049/jimmunol.179.12.8112. [DOI] [PubMed] [Google Scholar]
- 37.Garcia JG, Verin AD, Schaphorst KL. Regulation of thrombin-mediated endothelial cell contraction and permeability. Semin. Thromb. Hemost. 1996;22:309–315. doi: 10.1055/s-2007-999025. [DOI] [PubMed] [Google Scholar]
- 38.Birukov KG, Csortos C, Marzilli L, Dudek S, Ma SF, Bresnick AR, Verin AD, Cotter RJ, Garcia JG. Differential regulation of alternatively spliced endothelial cell myosin light chain kinase isoforms by p60Src. J. Biol. Chem. 2001;276:8567–8573. doi: 10.1074/jbc.M005270200. [DOI] [PubMed] [Google Scholar]
- 39.Buresi MC, Buret AG, Hollenberg MD, MacNaughton WK. Activation of proteinase-activated receptor 1 stimulates epithelial chloride secretion through a unique MAP kinase- and cyclo-oxygenase-dependent pathway. FASEB J. 2002;16:1515–1525. doi: 10.1096/fj.02-0039com. [DOI] [PubMed] [Google Scholar]
- 40.Saito H, Minamiya Y, Kitamura M, Saito S, Enomoto K, Terada K, Ogawa J. Endothelial myosin light chain kinase regulates neutrophil migration across human umbilical vein endothelial cell monolayer. J. Immunol. 1998;161:1533–1540. [PubMed] [Google Scholar]