

Abstract
During mucosal inflammation, a complex array of proinflammatory and protective mechanisms regulates inflammation and severity of injury. Secretion of anti-inflammatory mediators is a mechanism that is critical in controlling inflammatory responses and promoting epithelial restitution and barrier recovery. AnxA1 is a potent anti-inflammatory protein that has been implicated to play a critical immune regulatory role in models of inflammation. Although AnxA1 has been shown to be secreted in intestinal mucosal tissues during inflammation, its potential role in modulating the injury/inflammatory response is not understood. In this study, we demonstrate that AnxA1-deficient animals exhibit increased susceptibility to dextran sulfate sodium (DSS)-induced colitis with greater clinical morbidity and histopathologic mucosal injury. Furthermore, impaired recovery following withdrawal of DSS administration was observed in AnxA1 (−/−) animals compared with wild-type (WT) control mice that was independent of inflammatory cell infiltration. Since AnxA1 exerts its anti-inflammatory properties through stimulation of ALX/FPRL-1, we explored the role of this receptor-ligand interaction in regulating DSS-induced colitis. Interestingly, treatment with an ALX/FPRL-1 agonist, 15-epi-lipoxin A4 reversed the enhanced sensitivity of AnxA1 (−/−) mice to DSS colitis. In contrast, 15-epilipoxin A4 did not significantly improve the severity of disease in WT animals. Additionally, differential expression of ALX/FPLR-1 in control and DSS-treated WT and AnxA1-deficient animals suggested a potential role for AnxA1 in regulating ALX/FPRL-1 expression under pathophysiological conditions. Together, these results support a role of endogenous AnxA1 in the protective and reparative properties of the intestinal mucosal epithelium.
Inflammatory conditions of the gastrointestinal tract are a significant cause of morbidity and mortality worldwide. Conditions such as infectious colitis and inflammatory bowel disease induce erosion and ulceration of mucosal tissues and impaired gastrointestinal function. During the onset of mucosal injury and inflammation, a complex array of inflammatory signaling is initiated involving PG and cytokine production (1). Subsets of cytokines, including IL-8, IFN-γ, and TNF-α, impair intestinal epithelial function and lead to the recruitment of inflammatory cells to sites of injury (2). For example, IFN-γ and TNF-α have been shown to impair intestinal epithelial ion transport and induce disassembly of intercellular junctions and apoptosis (3-5). IL-8 is a potent recruiter of leukocytes (6). However, concurrent activation of protective mechanisms serves to regulate the degree of inflammation and severity of injury. Elaboration of a variety of factors, including IL-11 and inducible NO synthase, is critical to the defensive mechanism of the intestinal mucosa to sustain functional integrity (1, 7-9).
AnxA1 is a calcium-dependent phospholipid binding protein originally reported to be induced by glucocorticoids and inhibit phospholipase activity (10, 11). AnxA1 has subsequently been shown to regulate diverse cellular functions in a variety of cell types and exhibits profound inhibitory actions on leukocyte transmigration and activation (12, 13). A protective and anti-inflammatory role of AnxA1 has been demonstrated in models of endotoxemia, peritonitis, arthritis, as well as cerebral and myocardial ischemia (13-19). In vitro studies from our group have implicated a role for AnxA1 in stimulating migration of a model intestinal epithelial cell line important for regenerative mucosal responses (20). Consistent with these findings, recent studies have identified a role for AnxA1 in promoting healing of indomethacin-induced gastric ulcers (21).
In addition to its phospholipase inhibitory actions, protective and anti-inflammatory properties of AnxA1 have been shown to be mediated by signaling through formyl peptide receptors (FPR).4 In particular, the protective effect of AnxA1 in models of myocardial ischemia has been shown to be sensitive to antagonism of FPR (18). Additionally, the inhibition of leukocyte function and promotion of gastric mucosal wound healing has been ascribed to stimulation of ALX/FPRL-1 by AnxA1 (22, 23). ALX/FPRL-1 is expressed in intestinal epithelial cells where it has been shown to regulate anti-inflammatory signaling (24, 25). Interestingly, secretion of AnxA1 has been identified in inflamed intestinal mucosaltissues (26, 27); however, the functional significance of this event has yet to be determined. We therefore sought to determine the role of AnxA1 in modulating intestinal mucosal injury and inflammation. Using AnxA1 knockout animals, we demonstrate that lack of AnxA1 expression results in an increased susceptibility to acute dextran sulfate sodium (DSS)-induced colitis as well as impaired recovery following DSS withdrawal. Additionally, our findings also implicate a critical role for ALX/FPRL-1 in mediating the protective effects afforded by AnxA1 during the inflammatory process.
Materials and Methods
Animals
Wild-type (WT) female BALB/c mice were purchased from The Jackson Laboratory. AnxA1 null mice were generated as previously described (28). Animals were maintained on a 12-h light/12-h dark cycle under pathogen-free conditions. The mice had ad libitum access to a standard diet and water until reaching the desired age (8–10 wk) and/or weight (20–25 g). All procedures using animals were reviewed and approved by the Emory University Institutional Animal Care and Use Committee and were performed according to the criteria outlined by the National Institutes of Health.
Induction of colitis
Seven percent (w/v) DSS (molecular mass, 36–50 kDa; MP Biomedicals) was dissolved in purified water and administered to mice for 7 days (29). For rescue studies with ALX/FPRL-1 stimulation, we used 15-epi-lipoxin A4 ((5S,6R,15R)-5,6,15-trihydroxy-7,9,13-trans-11-cis-eicosatetraenoic acid; Calbiochem) which was injected i.p. (0.4 μg/animal) daily during the course of DSS administration.
Western blotting
Mucosal tissues manually stripped from mouse colons using stereomicroscopy, or cell monolayers were placed in lysis buffer (100 mM KCl, 3 mM NaCl, 3.5 mM MgCl2, 10 mM HEPES (pH 7.4), and 1% Triton X-100) containing protease and phosphatase inhibitors (Sigma-Aldrich). Lysates were dounced, and insoluble material removed by centrifugation (16,000×g/10 min/4°C). Cleared lysates were normalized for protein concentration, mixed with reducing Laemelli sample buffer (containing 20 mM DTT final concentration), and subject to SDS-PAGE. Proteins were transferred to nitrocellulose membranes and immunoblotted using rabbit anti-annexin 1 and actin Abs.
Immunofluorescence
Colonic tissue samples for immunofluorescence were embedded in O.C.T. (Sakura) and cryosectioned (5 μm thick). Tissue sections were then fixed in 3.4% paraformaldehyde for 20 min at room temperature and washed with HBSS+. Sections were then permeabilized with 1% Triton X-100 in HBSS+, washed, then blocked in HBSS+ containing 3% BSA for 1 h at room temperature. After incubation for 1 h with primary Abs in 3% BSA blocking buffer, sections were washed, incubated for 1 h with Alexa dye-conjugated secondary Abs, washed, and then incubated with To-Pro-3 iodide to visualize nuclei. The sections were mounted in p-phenylene-diamine and analyzed using a Zeiss LSM510 laser scanning confocal microscope (Zeiss). Integrated pixel intensity analysis was performed using the Zeiss LSM 510 (V 4.03) image analysis software. Images for this analysis were taken at identical detector gain settings.
Assessment of inflammation in DSS-treated mice
Daily clinical assessment of DSS-treated animals included measurement of body weight, evaluation of stool consistency, and the presence of blood in the stools by a guaiac paper test (Hemoccult Sensa; Beckman Coulter). A validated clinical disease activity index (DAI) ranging from 0 to 4 was calculated using the following parameters: stool consistency, presence or absence of fecal blood, and percentage of weight loss (30). Mice were sacrificed at day 7, or 7 days following DSS withdrawal, and the colons were removed. The length and weight were measured (31) after exclusion of the cecum and before dividing the colon for histology and evaluation of myeloperoxidase (MPO) activity (32).
Tissue MPO activity
MPO activity was measured in tissue adjacent to that used for histology. Samples were rinsed with cold PBS, blotted dry, and immediately frozen in liquid nitrogen. They were stored at −80°C until assayed for MPO activity using the o-dianisidine method (33, 34).
Histology
For each animal, histological examination was performed on three samples of the distal colon. H&E-stained sections were generated from 5-mm segments of the distal colon. All histological quantification was performed in a blinded fashion using modified validated scoring systems (35-37). Five independent parameters were measured: 1) severity of inflammation (0–3: none, mild, moderate, severe); 2) depth of injury (0–3: none, mucosal, mucosal and submucosal, transmural); 3) crypt damage (0–4: none, basal one-third damaged, basal two-thirds damaged, only surface epithelium intact, entire crypt and epithelium lost); 4) submucosal edema (0–3: no changes, mild edema (the submucosa is <0.20-mm wide and accounts for <50% of the diameter of the entire intestinal wall), moderate edema (the submucosa is 21- to 45-mm wide and accounts for 50–80% of the diameter of the entire intestinal wall), profound edema (the submucosa is >0.46-mm wide and accounts for >80% of the diameter of the entire intestinal wall); and 5) polymorphonuclear granulocyte infiltration in the lamina propria (0 = <5 polymorphonuclear granulocyte; 1 = 5–20; 2 = 21–60; and 3 = 61–100). The scores of the parameters were multiplied by a factor reflecting the percentage of tissue involvement (×1: 0–25%, ×2: 26–50%, ×3: 51–75%, ×4: 76–100%), and these values were summed to obtain a maximum possible score of 68.
RT-PCR analysis
Total RNA extraction was performed on mucosal tissues isolated via scraping using TRIzol reagent (Invitrogen). Expression of murine FPR1 (NM_013521), FPR-rs2 (NM_008039), and ALX/FPRL-1 (NM_008042) was analyzed by real-time PCR using iQ SYBR mix (iCycler; Bio-Rad). Validated primers for the above genes of interest were obtained from Superarray Biosciences, and the following GAPDH primers were obtained from Integrated DNA Technologies: Forward- 5′-TGCACCACCAAC TGCTTAG-3′, reverse 5′-GATGCAGGGATGATGTTC-3′. Melt curve analysis was performed to verify specificity of reactions.
Statistical analysis
Results are expressed as the mean ± SEM. One-way ANOVA with post hoc testing or paired Student’s t tests were used to compare results from different experiments.
Results
Annexin A1 expression is increased in colonic mucosal tissues following DSS treatment
Increased expression and secretion of AnxA1 has been reported to occur in inflamed mucosal tissues in rodent models of colitis and in human ulcerative colitis (26, 27). Given the homeostatic and anti-inflammatory properties of AnxA1, it is likely that this increased expression might serve to counteract proinflammatory and injurious responses in the mucosa. However, experiments demonstrating the functional significance of such increased AnxA1 expression in the injured intestine are lacking. We therefore sought to examine the expression and role of AnxA1 in the DSS-induced acute colitis model. We first examined the total protein levels of AnxA1 in colonic mucosa in DSS-treated and nontreated WT BALB/c animals. As shown in Fig. 1A, western blot analysis of mucosal lysates revealed increased expression of AnxA1 in DSS-treated animals compared with nontreated controls. Based on densitometric analysis, AnxA1 levels were increased 1.5-fold in the DSS-treated animals (Fig. 1B; *, p-value <0.05). To control for the specificity of the anti-AnxA1 Abs, Western blot analysis of mucosal lysates was performed on AnxA1-deficient animals, which revealed no AnxA1 signal as expected (Fig. 1C). Thus, acute colitis induced by DSS results in increased levels of AnxA1 in the colonic mucosa.
FIGURE 1.

Increased expression of AnxA1 in colonic mucosal tissues following DSS treatment. Western blot analysis of mucosal lysates from nontreated and DSS-treated WT BALB/c animals (A) revealed increased AnxA1 protein. Based on densitometric analysis of Western blots from these studies (B), AnxA1 levels were increased 1.5-fold following DSS treatment (*, p-value <0.05). No AnxA1 expression was observed in mucosal lysates from AnxA1 (−/−) mice (C). Western blot for actin was used as loading control.
Annexin A1 expression is increased in intestinal epithelial cells following DSS treatment
To determine the cell types that express the protein and might contribute to the increased mucosal AnxA1 protein levels following DSS treatment, immunofluorescence studies were performed on frozen sections of WT BALB/c colonic mucosa. Both surface and crypt epithelial cells express AnxA1 (Fig. 2A, arrows). Mononuclear cells in the lamina propria also express AnxA1 (Fig. 2A, arrowheads). In surface enterocytes, AnxA1 was found along the apical and lateral membrane plasma membranes as well as in the cytoplasmic compartment (Fig. 2B). In contrast AnxA1 was identified predominantly in the cytoplasm of the crypt epithelium (Fig. 2C). Following DSS treatment, AnxA1 was up-regulated in both surface and crypt epithelial cells (Fig. 2, B-E; images were captured at identical detector gain settings). Based on integrated pixel intensity analysis, the epithelium exhibited a 1.6-fold increase in AnxA1 levels in DSS-treated animals compared with nontreated controls (Fig. 2F; *, p-value <0.05). Thus, AnxA1 is expressed in murine colonic epithelial cells and its expression is increased by DSS-induced injury. AnxA1 was also identified in infiltrating leukocytes in DSS-treated animals (15, 38). Therefore, the enhanced intestinal epithelial expression of AnxA1 and infiltration of inflammatory cells expressing AnxA1 contribute to the increase in total AnxA1 levels in mucosal tissues of DSS-treated animals.
FIGURE 2.

AnxA1 expression is increased in intestinal epithelial cells following DSS treatment. Immunofluorescence analysis of WT BALB/c mucosa revealed expression of AnxA1 in both crypt and surface intestinal epithelial cells (A; arrows) [Bar = 50 μm]. In surface enterocytes (B), AnxA1 localizes in the apical and lateral plasma membranes as well as in the cytoplasm, whereas in crypt epithelial cells it appears to be exclusively in the cytoplasm (C) [Bars = 20 μm]. Following DSS treatment, AnxA1 levels are increased in both surface and crypt epithelial cells compared with nontreated animals (B-E; images taken at identical detector gain settings) [Bars = 20 μm]. Integrated pixel intensity analysis (F) revealed a 1.7-fold increase in AnxA1 levels in intestinal epithelial cells after 7 days of DSS treatment (*, p-value <0.05). A secondary Ab control (G) and staining of AnxA1 (−/−) mucosa (H) demonstrated no tissue staining [Bars = 50 μm].
Annexin A1-deficient mice exhibit increased susceptibility to DSS-induced acute colitis
To determine the role of AnxA1 in regulating epithelial barrier function following acute colonic injury and inflammation, WT and AnxA1 (−/−) BALB/c mice (n ≥ 6) were treated with 7% DSS for 7 days and clinical assessments were performed. As shown in Fig. 3A, a more pronounced weight loss was observed in AnxA1 (−/−) mice within 3 days after DSS administration compared with DSS-treated WT animals (*p-value <0.05). AnxA1 (−/−) mice also displayed increased DAI scores beginning on day 1 of DSS treatment, which remained elevated throughout the course of DSS treatment compared with WT animals (Fig. 3B; *, p-value <0.05). Rectal bleeding scores were increased during DSS treatment in AnxA1 (−/−) mice compared with WT controls (Fig. 3C; *, p-value <0.05) and the colon length:weight ratio was ~18% higher in AnxA1 (−/−) animals than in WT mice following 7 days of DSS administration (Fig. 3D; *, p-value <0.05). No mortality was observed in WT or AnxA1 (−/−) mice during the 7 days of DSS treatment (data not shown). Histologic analysis revealed a greater degree of injury in AnxA1 (−/−) mice compared with WT animals following DSS treatment. As shown in Fig. 4A, representative photomicrographs of H&E-stained sections revealed a greater degree of epithelial injury, increased neutrophil infiltration, and inflammatory changes in submucosal tissues (arrows) of AnxA1 (−/−) mice compared with WT animals following 7 days of DSS treatment. Thus, DSS-treated AnxA1 (−/−) animals exhibited an increased histologic score of injury and MPO activity in mucosal tissues compared with controls (Fig. 4, B and C; *, p-value <0.05). Taken together, the above data demonstrate increased susceptibility of AnxA1 (−/−) mice to DSS treatment and support a protective role for AnxA1 in acute colonic injury.
FIGURE 3.

AnxA1-deficient mice exhibit increased susceptibility to DSS-induced acute colitis. AnxA1 (−/−) mice demonstrated a significantly higher weight loss compared with WT controls beginning at day 3 of DSS treatment (A; *, p-value <0.05, n = 7 mice). A sustained increase in the DAI in AnxA1 (−/−) mice compared with WT controls was observed on days 1–7 of DSS treatment (B; *, p-value <0.05, n = 7 mice). Rectal bleeding scores (C) were also increased in AnxA1 (−/−) animals compared with WT controls during DSS treatment (*, p-value <0.05, n = 7 mice). An increase in the colon weight/length ratio was observed in AnxA1 (−/−) mice compared with WT controls following 7 days of DSS administration (D; *, p-value <0.05, n = 7 mice).
FIGURE 4.
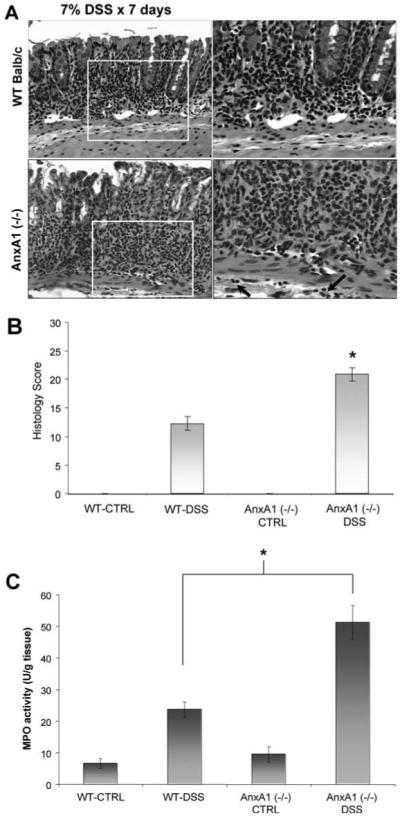
AnxA1-deficient mice have more severe histopathologic injury in DSS-induced colitis. Representative photomicrographs of H&E-stained histologic sections (A) demonstrate more severe colonic injury in AnxA1 (−/−) animals compared with WT controls following 7 days of DSS treatment. Note the increased degree of epithelial injury, increased leukocyte infiltration, and involvement of submucosal tissues in AnxA1 (−/−) mice (inset, arrows). Histologic scoring of colonic injury (B) is significantly higher than that of WT DSS-treated animals (*, p-value <0.05, n = 7 mice). Colonic MPO activity (C) is also significantly higher in AnxA1 (−/−) mice compared with WT BALB/c animals following DSS treatment (*, p-value <0.05, n = 7 mice).
AnxA1-deficient mice exhibit impaired clinical and histopathologic improvement following withdrawal of DSS treatment
To determine whether AnxA1 plays a role in reparative responses following colonic injury, AnxA1 (−/−) and WT mice were assessed over a course of DSS treatment for 7 days, at which time DSS administration was withdrawn and mice were followed for a subsequent 7 days. AnxA1 (−/−) mice exhibited a 25% mortality rate within the first 48 h following withdrawal of DSS, while no mortality was observed for WT animals (data not shown). All subsequent data shown was derived using only AnxA1 (−/−) animals that survived the entire course of the experiment. As shown in Fig. 5A, AnxA1 (−/−) animals exhibited increased weight loss compared with WT controls over the initial 7 days of DSS treatment. Following withdrawal of DSS, WT animals regained an average of 1.75% original body weight/day returning to a similar weight to that before DSS treatment. However, AnxA1 (−/−) mice exhibited continued weight loss for the first 3 days following DSS treatment and did not demonstrate any significant improvement in body weight over the 7-day period following DSS withdrawal (Fig. 5A; *, p-value <0.05). Similarly, WT animals demonstrated a significant improvement in DAI following DSS withdrawal with an average reduction of 0.3 points/day in DAI score over the 7-day recovery period. In contrast, AnxA1 (−/−) animals only began to show a reduction in the DAI score starting 5 days post DSS withdrawal and on the average an overall reduction of 0.2 points/day over the 7-day recovery period was observed (Fig. 5B; *, p-value <0.05). Histopathologic analysis revealed signs of improvement in mucosal injury in WT animals following 7 days of DSS withdrawal with a reduction in the degree of epithelial injury (Fig. 5C) and a reduction of 5.95 in the histologic scoring of injury (Fig. 5D). Interestingly, in AnxA1 (−/−) animals, a significant reduction in leukocyte infiltration into mucosal tissues was observed 7 days following DSS withdrawal. However, the degree of epithelial loss continued to increase (Fig. 5C), and, largely due to this loss, the histologic injury score increased by an average of 11 points (Fig. 5D). Note that in WT and AnxA1 (−/−) animals, abnormal mucosal architecture denoted by irregular spacing and loss of crypts was identified. Such crypt architectural abnormality and damage was significantly more pronounced in the AnxA1 (−/−) mice (Fig. 5C). MPO scores correlated with the histologic observations (Fig. 5E). However, AnxA1 (−/−) mice exhibited a reduction in MPO activity similar to that of WT animals following 7 days of recovery (average of 28 points; Fig. 5E, *, p-value <0.05). Taken together, the above data indicate an impaired ability of AnxA1 (−/−) animals to recover following DSS withdrawal and support a role for AnxA1 in mucosal reparative responses. These data also implicate an important role for AnxA1 in regulating the biologic responses of murine intestinal epithelial cells following acute injury given that AnxA1 (−/−) mice exhibited greater epithelial loss over the recovery period while the inflammatory cell infiltration decreased to levels similar to that of WT controls.
FIGURE 5.

AnxA1-deficient mice exhibit impaired clinical and histopathologic improvement following withdrawal of DSS treatment. AnxA1 (−/−) mice exhibited a significant reduction in the rate of weight gain following withdrawal of DSS treatment compared with WT controls (A; *, p-value <0.05, n = 7 mice). Similarly, AnxA1 (−/−) animals showed an impairment in recovery following DSS withdrawal as determined by DAI (B; *, p-value <0.05, n = 7 mice). Representative photomicrographs of H&E-stained histologic sections (C) demonstrate more severe histopathologic injury following 7 days of DSS treatment in AnxA1 (−/−) animals compared with WT controls. After 7 days of recovery, WT animals appeared to exhibit slightly less epithelial injury and reduced inflammation. However, a mild degree of architectural irregularity as evidenced by minor irregular crypt spacing was identified. In contrast, while the inflammation in AnxA1 (−/−) animals was improved to a small extent following 7 days of recovery from DSS-induced colitis, significantly increased epithelial injury with the development of architectural irregularity was observed (C). Histologic scoring (D) of colonic injury revealed some improvement of histopathologic changes in WT animals, whereas an overall increase in injury was found in AnxA1 (−/−) animals (*, p-value <0.05, n = 7 mice). Measurement of MPO activity (E) did not reveal a statistically significant difference in leukocyte infiltration in WT animals between 7 days of DSS treatment and after 7 days of recovery. MPO activity in AnxA1-deficient animals improved to a degree similar to that of WT controls at day 7 of DSS treatment and was less than that of WT controls following a 7 day recovery period (*, p-value <0.05, n = 7 mice).
ALX/FPRL-1 stimulation decreases the susceptibility of AnxA1-deficient mice to DSS-induced colitis
Major anti-inflammatory effects of AnxA1 are mediated via activation of ALX/FPRL-1 (22, 23, 39). Thus, we sought to determine whether lack of ALX/FPRL-1 stimulation could contribute to the susceptibility of AnxA1-deficient animals to DSS-induced colitis. For these studies, WT and AnxA1 (−/−) BALB/c mice (n ≥ 6) were treated with 7% DSS for 7 days with or without daily i.p. injection of a metabolically stable ALX/FPRL-1 agonist, 15-epilipoxin A4 (0.4 μg/day). AnxA1 and lipoxin A4 analogues have been shown to induce similar anti-inflammatory and protective effects via activation of ALX/FPRL-1 (39-41). As shown in Fig. 6A, treatment of AnxA1 (−/−) animals with 15-epi-lipoxin A4 over the course of DSS administration results in similar weight loss to that of WT DSS-treated controls. Interestingly, no significant improvement in weight loss due to DSS treatment was observed in WT BALB/c animals. The DAI score for AnxA1 (−/−) animals was significantly higher than that of controls, and treatment with 15-epi-lipoxin A4 resulted in DAI scores similar to that of WT DSS-treated animals (Fig. 6B). No improvement in DAI was observed in WT animals treated with 15-epi-lipoxin A4 compared with WT animals treated with DSS alone. Finally, histopathologic analysis demonstrated a significant improvement in mucosal injury due to DSS administration in 15-epi-lipoxin A4-treated AnxA1 (−/−) animals compared with those receiving DSS alone (Fig. 6, C and D). In WT animals treated with DSS, concurrent 15-epi-lipoxin A4 treatment did not result in a statistically significant improvement in the histologic injury score (Fig. 6, C and D). Thus, in absence of AnxA1, ALX/FPRL-1 stimulation is effective in reducing the severity of DSS-induced disease analogous to that observed in WT controls. In the presence of AnxA1 (in WT mice), ALX/FPRL-1 stimulation with the current protocol and ligand did not improve the course of disease.
FIGURE 6.

ALX/FPRL-1 stimulation rescues the susceptibility of AnxA1-deficient mice to DSS-induced colitis. AnxA1 (−/−) animals demonstrated a more pronounced weight loss following DSS treatment that was restored to that of WT controls with ALX/FPRL-1 stimulation via administration of 15-epi-lipoxin A4 (denoted as L in the figure) (A; *, p-value <0.05, n = 7 mice). Conversely, WT animals did not show a reduction in weight loss due to ALX/FPRL-1 stimulation during DSS treatment. AnxA1 (−/−) mice exhibit an increased DAI over the course of DSS treatment which was restored similar to that of WT controls on days 4–7 with simultaneous ALX/FPRL-1 agonization via 15-epi-lipoxin A4 treatment (B; *, p-value <0.05, n = 7 mice). Treatment of WT animals with 15-epi-lipoxin A4 did not results in a consistent decrease in the DAI during DSS administration. Representative photomicrographs of H&E-stained histologic sections (C) and histologic scoring (D; *, p-value <0.05, n = 7 mice) demonstrate a similar degree of DSS-induced colonic injury in AnxA1-deficient animals treated with 15-epi-lipoxin A4 and WT controls (*, p-value <0.05). 15-epi-lipoxin A4 did not significantly improve the histologic injury induced by DSS in WT BALB/c mice.
AnxA1-deficient mice exhibit reduced expression of ALX/FPRL-1 during DSS-induced colitis
Given that treatment with 15-epi-lipoxin A4 rescued the severity of DSS-induced colitis in AnxA1 (−/−) animals and did not significantly influence disease in WT animals, we hypothesized that AnxA1 (−/−) animals may have differences in their expression of FPR family members compared with WT animals. We focused on expression of murine receptors most closely related to the human counterparts that interact with AnxA1 and lipoxin A4 analogues, namely mouse ALX/FPRL-1 (NM_008042), FPR-rs2 (NM_008039; the murine orthologue to human FPRL-1), and mouse FPR1 (NM_013521; the murine orthologue to human FPR) (42-46). Mucosal tissues from nontreated and DSS-treated WT and AnxA1 (−/−) animals were isolated by scraping and total RNA was extracted. Following cDNA synthesis, samples were subject to real time-PCR with SYBR green using Bio-Rad’s iCycler. In WT animals, expression of ALX/FPRL-1 is dramatically reduced (7-fold) following 7 days of DSS treatment (Fig. 7A; *, p-value <0.05). In AnxA1 (−/−) animals, baseline expression of ALX/FPRL-1 was reduced by an average of 27% compared with nontreated WT animals (Fig. 7A). Following DSS administration, AnxA1 (−/−) animals exhibited a greater degree of suppression of ALX/FPRL-1 expression compared with WT animals, averaging 4.7-fold less than DSS-treated WT animals (Fig. 7A). For FPR-rs2, no difference in expression was detected between control WT, DSS-treated WT, and control AnxA1 (−/−) animals (Fig. 7B). However, following 7 days of DSS treatment, FPR-rs-2 expression was dramatically reduced in AnxA1 (−/−) animals only (Fig. 7B; *, p-value <0.05). Expression of FPR-1 was similarly reduced in both WT and AnxA1 (−/−) following DSS treatment (Fig. 7C). Thus, AnxA1 (−/−) mice exhibit a reduction in ALX/FPRL-1, and in particular, FPR-rs2 expression following DSS treatment compared with WT animals.
FIGURE 7.

AnxA1 (−/−) mice exhibit reduced ALX/FPRL-1 expression following DSS treatment. Total RNA was extracted from mucosal tissues from control and DSS-treated WT and AnxA1 (−/−) mice and subjected to RT-PCR analysis for ALX/FPRL-1, FPR-rs2, and FPR1. Although ALX/FPRL-1 levels were suppressed in WT type animals treated with DSS, a further diminishment of ALX/FPRL-1 mRNA was observed in AnxA1 (−/−) animals following DSS treatment (A; n = 3 animals). FPR-rs2 expression did not change following DSS administration in WT animals but was dramatically reduced in AnxA1 (−/−) mice following a 7 course of DSS treatment (B; n = 3 animals). A similar reduction in FPR1 expression was observed following DSS treatment in WT and AnxA1 (−/−) animals (C; n = 3 animals).
Discussion
AnxA1 has been shown to play protective or anti-inflammatory roles in a variety of disease models (13-15, 17, 47). However, relatively little is know about the role of AnxA1 in regulating injury and inflammation in the gastrointestinal tract. Recent studies have identified a role of AnxA1 in mediating the gastroprotective effects of dexamethasone against nonsteroidal anti-inflammatory drug-induced injury as well as in promoting repair of gastric ulcers (21, 48). In the lower gastrointestinal tract, studies using rodent models of colitis as well as analysis of human ulcerative colitis have identified increased expression and secretion of this anti-inflammatory mediator (26, 27). The present study was performed to examine the functional significance of AnxA1 expression during acute colonic injury and inflammation as well as in restitution of mucosal tissues.
AnxA1 is a functionally diverse anti-inflammatory protein that is expressed by a variety of cells types including epithelial cells (49-55). We identified AnxA1 expression in normal murine colonic epithelial cells along the crypt-surface axis. AnxA1 was found to localize to the cytoplasmic compartment of the crypt epithelium. Interestingly, AnxA1 was abundantly found along the apical and lateral plasma membrane domains in surface enterocytes in addition to the cytoplasm. The difference in localization between crypt epithelial cells and fully differentiated surface enterocytes may suggest that AnxA1 plays a role in regulating colonic epithelial cell differentiation and/or maturation. Such a role for AnxA1 has been observed in in vitro studies using model epithelial cell lines (53, 56). We did not observe any morphologic or overt functional differences in the colonic mucosa of WT and AnxA1 (−/−) animals. Thus, the homeostatic role of AnxA1 in the murine intestinal epithelium remains unknown and redundant systems could compensate for its deficiency. Alternatively, its protective function can become operative upon application of an insult.
Consistent with previous studies identifying increased expression of AnxA1 in trinitrobenzenesulfonic acid-induced colitis in rats (26), we observed significant up-regulation of AnxA1 expression in mucosal lysates and, more specifically, in the colonic epithelium following DSS administration. Furthermore, AnxA1-deficient animals exhibited significantly increased susceptibility to DSS-induced colonic injury and morbidity. These findings suggest a protective role for AnxA1 in acute colonic injury.
Although AnxA1 exhibits broad subcellular distribution and plays a role in a diversity of cellular functions, key anti-inflammatory actions of AnxA1 are mediated via activation of ALX/FPLR-1 in an autocrine/paracrine fashion. For example, externalized AnxA1 exhibits profound inhibitory actions on leukocyte activation and transmigration via interactions with ALX/FPRL-1 (12, 13, 57, 58). Furthermore, studies have shown that lipoxin A4-mediated ALX/FPRL-1 stimulation suppress innate inflammatory responses of intestinal epithelial cells and hence have implicated a protective role for ALX/FPLR-1 activation against DSS-induced colitis (24, 25, 59). We therefore hypothesized that one mechanism by which AnxA1 protects against DSS-induced colitis is via activation of ALX/FPRL-1. For these studies, we used metabolically stable 15-epi-lipoxin A4, which shares common anti-inflammatory actions with AnxA1 that are mediated through ALX/FPRL-1 (39-41). i.p. injection of 15-epi-lipoxin A4 throughout the course of DSS administration diminished the severity of disease of AnxA1-deficient animals to that of DSS-treated WT animals. We did not observe significant improvement in WT animals treated with 15-epi-lipoxin A4 during DSS treatment in contrast to previously published studies suggesting some attenuation of DSS-induced injury by lipoxin A4 (59). However, differences in the lipoxin A4 analog used, the method of lipoxin administration, as well animal strains may account for this discrepancy. It should be noted that in models of trinitrobenzenesulfonic acid-induced colitis, an oxidative resistant lipoxin A4 analog and a related endogenous lipid mediator, resolvin E1, have been shown to be protective in WT animals (60, 61). Our results support that treatment with the ALX/FPRL-1 agonist, 15-epi-lipoxin A4, reversed the enhanced susceptibility of AnxA1 (−/−) mice to DSS colitis.
In addition to increased susceptibility to DSS-induced colitis, AnxA1 (−/−) animals demonstrated impaired clinical and histopathologic recovery following withdrawal of DSS treatment. This finding suggests that AnxA1 plays an important role in promoting colonic mucosal wound healing. Previous in vitro studies from our group identified a role for AnxA1 in stimulating ALX/FPRL-1-mediated enhancement of epithelial cell migration through three-dimensional matrices (20). As epithelial cell migration is required for mucosal wound healing and regeneration, AnxA1-mediated activation of ALX/FPRL-1 may be an important mechanism in promoting intestinal mucosal wound healing. Consistent with this idea, studies have identified a role for AnxA1-mediated stimulation of ALX/FPRL-1 in promoting healing of indomethacin-induced gastric ulcers (21). In this report, however, AnxA1-deficient animals did not exhibit an increased susceptibility to the induction of ulcer formation in the stomach as we observed for DSS-induced injury.
The murine FPR family is more complex than that of humans and contains at least nine family members (42, 62). Murine receptors closely related to the human counterparts that interact with AnxA1 and lipoxin A4 have been described and include Lxa4r/FPRL-1 and FPR-rs2 (42-46, 63). FPR-rs2 is the orthologue to human ALX/FPRL-1, and mouse FPR1 is the orthologue to human FPR, which binds N-terminal-derived AnxA1 peptides (43, 44, 64). Since AnxA1 (−/−)-deficient animals benefited from lipoxin A4 treatment during DSS administration in contrast to WT animals, we explored expression of the above murine AnxA1 and lipoxin receptors in mucosal tissues in control and DSS-treated animals. Treatment of WT animals with DSS decreased the expression of ALX/FPRL-1 without influencing expression of FPR-rs2. However, the reduction of ALX/FPRL-1 in AnxA1 (−/−) animals was pronounced and these animals also demonstrated a decrease in FPR-rs2 expression. The reduction in the expression of these anti-inflammatory receptors, particularly FPR-rs2, could at least partially account for the increased susceptibility to and impaired recovery from DSS treatment. Additionally, these findings also suggest that AnxA1 may regulate the expression of FPRs since AnxA1 (−/−) animals exhibited reduced FPR-rs2 expression following DSS treatment that was not observed in WT animals.
Another mechanism by which AnxA1 may modulate inflammatory responses is via regulation of cytokine production. For example, studies have shown that AnxA1 negatively regulates the expression of the proinflammatory cytokine IL-6 (65). Additionally, AnxA1 has been shown to influence T cell differentiation and activation that influences the balance of cytokine production following stimulation. Differentiation of T cells in AnxA1 (−/−) mice has been shown to be skewed toward a Th2 phenotype following immunization with keyhole limpet hemocyanin or during allergic inflammation (66). We therefore examined cytokine profiles in control and DSS-treated WT and AnxA1 (−/−) animals. For these studies, serum samples were isolated and subjected to multiplex cytokine analysis at the National Institute of Allergy and Infectious Diseases Centers for Human Immunology and Biodefense Central Cytokine Core Facility under the direction of J. E. Connolly (Baylor Institute for Immunology Research, Dallas, TX; supported by funds from the Baylor Health Care System Foundation and National Institutes of Health 5U19AI057234 and U-19 5U19 AI062623). The levels of a variety of key cytokines including IL-1, IL-2, IFN-γ, and TNF-α were determined. We did not observe any significant differences in the cytokine profiles between WT and AnxA1 (−/−) DSS-treated and nontreated animals (data not shown). Therefore, the increased susceptibility of AnxA1 (−/−) mice to DSS colitis does not appear to be related to alterations in expression of the above key cytokines.
In summary, we have demonstrated that AnxA1 is expressed by murine intestinal epithelial cells and its expression is increased following DSS administration. Mice lacking AnxA1 exhibit increased susceptibility to DSS-induced mucosal injury, which results in an increase in associated morbidity. AnxA1-deficient mice also exhibit impaired clinical and histopathologic recovery following DSS administration. Furthermore, stimulation of ALX/FPRL-1 in AnxA1 (−/−) animals diminishes the severity of disease induced by DSS to that of WT animals but does not significantly improve the severity of disease in WT animals. Lastly, AnxA1 (−/−) mice exhibit reduced levels of murine lipoxin receptors following DSS treatment compared with WT animals. Taken together, these data support a protective role of AnxA1 following acute intestinal mucosal injury where it also facilitates epithelial barrier recovery. Our data also implicate a role for ALX/FPRL-1 activation in mediating the protective effects of AnxA1 against intestinal mucosal injury. Insight into the mechanisms by which AnxA1 protects against intestinal injury and promotes mucosal restitution may contribute to the development of therapies to prevent intestinal mucosal injury and enhance wound healing.
Acknowledgments
We thank Dr. E. Rodriguez-Boulan for the gift of SKCO-15 cells and Dr. S. Voss for cell culture assistance.
Footnotes
This work was supported by grants from the National Institutes of Health (DK 59888 and DK 55679 to A.N.), (R01-DK72564 and R01-DK61379 to C.A.P.), (K08 DK074706-01 to B.A.B.), DK64399 (National Institutes of Health Digestive Disease Research Center tissue culture and morphology cores), and T32 DK007771 (C.T.C. and E.P.); Crohn’s and Colitis Foundation of America fellowship award (P.N.D.); and the German Research Foundation (Deutsche Forschungsgemeinschaft-La 2359/1-1 to M.G.L.).
- FPR
- formyl peptide receptor
- DAI
- disease activity index
- DSS
- dextran sulfate sodium
- WT
- wild type
- MPO
- myeloperoxidase
Disclosures
The authors have no financial conflict of interest.
References
- 1.Dignass AU. Mechanisms and modulation of intestinal epithelial repair. Inflamm. Bowel Dis. 2001;7:68–77. doi: 10.1097/00054725-200102000-00014. [DOI] [PubMed] [Google Scholar]
- 2.Martin GR, Wallace JL. Gastrointestinal inflammation: a central component of mucosal defense and repair. Exp. Biol. Med. 2006;231:130–137. doi: 10.1177/153537020623100202. [DOI] [PubMed] [Google Scholar]
- 3.Bruewer M, Utech M, Ivanov AI, Hopkins AM, Parkos CA, Nusrat A. Interferon-γ induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. FASEB J. 2005;19:923–933. doi: 10.1096/fj.04-3260com. [DOI] [PubMed] [Google Scholar]
- 4.Markossian S, Kreydiyyeh SI. TNF-α down-regulates the Na+-K+ ATPase and the Na+-K+-2Cl-cotransporter in the rat colon via PGE2. Cytokine. 2005;30:319–327. doi: 10.1016/j.cyto.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, Parkos CA, Nusrat A. Mechanism of IFN-γ-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol. Biol. Cell. 2005;16:5040–5052. doi: 10.1091/mbc.E05-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kobayashi Y. The role of chemokines in neutrophil biology. Front. Biosci. 2008;13:2400–2407. doi: 10.2741/2853. [DOI] [PubMed] [Google Scholar]
- 7.Vallance BA, Dijkstra G, Qiu B, van der Waaij LA, van Goor H, Jansen PL, Mashimo H, Collins SM. Relative contributions of NOS isoforms during experimental colitis: endothelial-derived NOS maintains mucosal integrity. Am. J. Physiol. 2004;287:G865–G874. doi: 10.1152/ajpgi.00187.2004. [DOI] [PubMed] [Google Scholar]
- 8.Playford RJ, Ghosh S. Cytokines and growth factor modulators in intestinal inflammation and repair. J. Pathol. 2005;205:417–425. doi: 10.1002/path.1722. [DOI] [PubMed] [Google Scholar]
- 9.Naugler KM, Baer KA, Ropeleski MJ. Interleukin-11 antagonizes Fas ligand-mediated apoptosis in IEC-18 intestinal epithelial crypt cells: role of MEK1, 2 and Akt-dependent signaling. Am. J. Physiol. 2008;294:G728–G737. doi: 10.1152/ajpgi.00002.2007. [DOI] [PubMed] [Google Scholar]
- 10.Cirino G, Flower RJ. Human recombinant lipocortin 1 inhibits prostacyclin production by human umbilical artery in vitro. Prostaglandins. 1987;34:59–62. doi: 10.1016/0090-6980(87)90262-0. [DOI] [PubMed] [Google Scholar]
- 11.Goulding NJ, Godolphin JL, Sampson MB, Maddison PJ, Flower RJ. Hydrocortisone induces lipocortin 1 production by peripheral blood mononuclear cells in vivo in man. Biochem. Soc. Trans. 1990;18:306–307. doi: 10.1042/bst0180306. [DOI] [PubMed] [Google Scholar]
- 12.Chatterjee BE, Yona S, Rosignoli G, Young RE, Nourshargh S, Flower RJ, Perretti M. Annexin 1-deficient neutrophils exhibit enhanced transmigration in vivo and increased responsiveness in vitro. J. Leukocyte Biol. 2005;78:639–646. doi: 10.1189/jlb.0405206. [DOI] [PubMed] [Google Scholar]
- 13.Perretti M, Ahluwalia A, Harris JG, Goulding NJ, Flower RJ. Lipocortin-1 fragments inhibit neutrophil accumulation and neutrophil-dependent edema in the mouse: a qualitative comparison with an anti-CD11b monoclonal antibody. J. Immunol. 1993;151:4306–4314. [PubMed] [Google Scholar]
- 14.Relton JK, Strijbos PJ, O’Shaughnessy CT, Carey F, Forder RA, Tilders FJ, Rothwell NJ. Lipocortin-1 is an endogenous inhibitor of ischemic damage in the rat brain. J. Exp. Med. 1991;174:305–310. doi: 10.1084/jem.174.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Damazo AS, Yona S, D’Acquisto F, Flower RJ, Oliani SM, Perretti M. Critical protective role for annexin 1 gene expression in the endotoxemic murine microcirculation. Am. J. Pathol. 2005;166:1607–1617. doi: 10.1016/S0002-9440(10)62471-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Y, Leech M, Hutchinson P, Holdsworth SR, Morand EF. Antiinflammatory effect of lipocortin 1 in experimental arthritis. Inflammation. 1997;21:583–596. doi: 10.1023/a:1027330021479. [DOI] [PubMed] [Google Scholar]
- 17.D’Amico M, Di Filippo C, La M, Solito E, McLean PG, Flower RJ, Oliani SM, Perretti M. Lipocortin 1 reduces myocardial ischemia-reperfusion injury by affecting local leukocyte recruitment. FASEB J. 2000;14:1867–1869. doi: 10.1096/fj.99-0602fje. [DOI] [PubMed] [Google Scholar]
- 18.La M, D’Amico M, Bandiera S, Di Filippo C, Oliani SM, Gavins FN, Flower RJ, Perretti M. Annexin 1 peptides protect against experimental myocardial ischemia-reperfusion: analysis of their mechanism of action. FASEB J. 2001;15:2247–2256. doi: 10.1096/fj.01-0196com. [DOI] [PubMed] [Google Scholar]
- 19.La M, Tailor A, D’Amico M, Flower RJ, Perretti M. Analysis of the protection afforded by annexin 1 in ischaemia-reperfusion injury: focus on neutrophil recruitment. Eur. J. Pharmacol. 2001;429:263–278. doi: 10.1016/s0014-2999(01)01325-5. [DOI] [PubMed] [Google Scholar]
- 20.Babbin BA, Lee WY, Parkos CA, Winfree LM, Akyildiz A, Perretti M, Nusrat A. Annexin I regulates SKCO-15 cell invasion by signaling through formyl peptide receptors. J. Biol. Chem. 2006;281:19588–19599. doi: 10.1074/jbc.M513025200. [DOI] [PubMed] [Google Scholar]
- 21.Martin GR, Perretti M, Flower RJ, Wallace JL. Annexin-1 modulates repair of gastric mucosal injury. Am. J. Physiol. 2008;294:G764–G769. doi: 10.1152/ajpgi.00531.2007. [DOI] [PubMed] [Google Scholar]
- 22.Gavins FN, Yona S, Kamal AM, Flower RJ, Perretti M. Leukocyte antiadhesive actions of annexin 1: ALXR- and FPR-related anti-inflammatory mechanisms. Blood. 2003;101:4140–4147. doi: 10.1182/blood-2002-11-3411. [DOI] [PubMed] [Google Scholar]
- 23.Gavins FN, Sawmynaden P, Chatterjee BE, Perretti M. A twist in anti-inflammation: Annexin 1 acts via the lipoxin A(4) receptor. Prostaglandins Leukotrienes Essent. Fatty Acids. 2005;73:211–219. doi: 10.1016/j.plefa.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 24.Kucharzik T, Gewirtz AT, Merlin D, Madara JL, Williams IR. Lateral membrane LXA4 receptors mediate LXA4’s anti-inflammatory actions on intestinal epithelium. Am. J. Physiol. 2003;284:C888–C896. doi: 10.1152/ajpcell.00507.2001. [DOI] [PubMed] [Google Scholar]
- 25.Gronert K, Gewirtz A, Madara JL, Serhan CN. Identification of a human enterocyte lipoxin A4 receptor that is regulated by interleukin (IL)-13 and interferon γ and inhibits tumor necrosis factor α-induced IL-8 release. J. Exp. Med. 1998;187:1285–1294. doi: 10.1084/jem.187.8.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vergnolle N, Comera C, Bueno L. Annexin 1 is overexpressed and specifically secreted during experimentally induced colitis in rats. Eur. J. Biochem. 1995;232:603–610. doi: 10.1111/j.1432-1033.1995.tb20850.x. [DOI] [PubMed] [Google Scholar]
- 27.Vergnolle N, Pages P, Guimbaud R, Chaussade S, Bueno L, Escourrou J, Comera C. Annexin 1 is secreted in situ during ulcerative colitis in humans. Inflamm. Bowel Dis. 2004;10:584–592. doi: 10.1097/00054725-200409000-00013. [DOI] [PubMed] [Google Scholar]
- 28.Hannon R, Croxtall JD, Getting SJ, Roviezzo F, Yona S, Paul-Clark MJ, Gavins FN, Perretti M, Morris JF, Buckingham JC, Flower RJ. Aberrant inflammation and resistance to glucocorticoids in annexin 1−/− mouse. FASEB J. 2003;17:253–255. doi: 10.1096/fj.02-0239fje. [DOI] [PubMed] [Google Scholar]
- 29.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 30.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 31.Diaz-Granados N, Howe K, Lu J, McKay DM. Dextran sulfate sodium-induced colonic histopathology, but not altered epithelial ion transport, is reduced by inhibition of phosphodiesterase activity. Am. J. Pathol. 2000;156:2169–2177. doi: 10.1016/S0002-9440(10)65087-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krieglstein CF, Cerwinka WH, Sprague AG, Laroux FS, Grisham MB, Koteliansky VE, Senninger N, Granger DN, de Fougerolles AR. Collagen-binding integrin α1β1 regulates intestinal inflammation in experimental colitis. J. Clin. Invest. 2002;110:1773–1782. doi: 10.1172/JCI200215256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arndt H, Kubes P, Grisham MB, Gonzalez E, Granger DN. Granulocyte turnover in the feline intestine. Inflammation. 1992;16:549–559. doi: 10.1007/BF00918979. [DOI] [PubMed] [Google Scholar]
- 34.Krawisz JE, Sharon P, Stenson WF. Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity: assessment of inflammation in rat and hamster models. Gastroenterology. 1984;87:1344–1350. [PubMed] [Google Scholar]
- 35.Dieleman LA, Palmen MJ, Akol H, Bloemena E, Pena AS, Meuwissen SG, Van Rees EP. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin. Exp. Immunol. 1998;114:385–391. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barthel M, Hapfelmeier S, Quintanilla-Martinez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Russmann H, Hardt WD. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect. Immun. 2003;71:2839–2858. doi: 10.1128/IAI.71.5.2839-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J. Exp. Med. 2007;204:3067–3076. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Damazo AS, Yona S, Flower RJ, Perretti M, Oliani SM. Spatial and temporal profiles for anti-inflammatory gene expression in leukocytes during a resolving model of peritonitis. J. Immunol. 2006;176:4410–4418. doi: 10.4049/jimmunol.176.7.4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Souza DG, Fagundes CT, Amaral FA, Cisalpino D, Sousa LP, Vieira AT, Pinho V, Nicoli JR, Vieira LQ, Fierro IM, Teixeira MM. The required role of endogenously produced lipoxin A4 and annexin-1 for the production of IL-10 and inflammatory hyporesponsiveness in mice. J. Immunol. 2007;179:8533–8543. doi: 10.4049/jimmunol.179.12.8533. [DOI] [PubMed] [Google Scholar]
- 40.Perretti M, Chiang N, La M, Fierro IM, Marullo S, Getting SJ, Solito E, Serhan CN. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat. Med. 2002;8:1296–1302. doi: 10.1038/nm786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gavins FN, Kamal AM, D’Amico M, Oliani SM, Perretti M. Formyl-peptide receptor is not involved in the protection afforded by annexin 1 in murine acute myocardial infarct. FASEB J. 2005;19:100–102. doi: 10.1096/fj.04-2178fje. [DOI] [PubMed] [Google Scholar]
- 42.Chiang N, Takano T, Arita M, Watanabe S, Serhan CN. A novel rat lipoxin A4 receptor that is conserved in structure and function. Br. J. Pharmacol. 2003;139:89–98. doi: 10.1038/sj.bjp.0705220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao JL, Chen H, Filie JD, Kozak CA, Murphy PM. Differential expansion of the N-formylpeptide receptor gene cluster in human and mouse. Genomics. 1998;51:270–276. doi: 10.1006/geno.1998.5376. [DOI] [PubMed] [Google Scholar]
- 44.Gao JL, Murphy PM. Species and subtype variants of the N-formyl peptide chemotactic receptor reveal multiple important functional domains. J. Biol. Chem. 1993;268:25395–25401. [PubMed] [Google Scholar]
- 45.Gavins FN, Dalli J, Flower RJ, Granger DN, Perretti M. Activation of the annexin 1 counter-regulatory circuit affords protection in the mouse brain microcirculation. FASEB J. 2007;21:1751–1758. doi: 10.1096/fj.06-7842com. [DOI] [PubMed] [Google Scholar]
- 46.Takano T, Fiore S, Maddox JF, Brady HR, Petasis NA, Serhan CN. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: evidence for anti-inflammatory receptors. J. Exp. Med. 1997;185:1693–1704. doi: 10.1084/jem.185.9.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cuzzocrea S, Tailor A, Zingarelli B, Salzman AL, Flower RJ, Szabo C, Perretti M. Lipocortin 1 protects against splanchnic artery occlusion and reperfusion injury by affecting neutrophil migration. J. Immunol. 1997;159:5089–5097. [PubMed] [Google Scholar]
- 48.Zanardo RC, Perretti M, Wallace JL. Annexin-1 is an endogenous gastroprotective factor against indomethacin-induced damage. Am. J. Physiol. 2005;288:G481–G486. doi: 10.1152/ajpgi.00299.2004. [DOI] [PubMed] [Google Scholar]
- 49.Gerke V, Moss SE. Annexins and membrane dynamics. Biochim. Biophys. Acta. 1997;1357:129–154. doi: 10.1016/s0167-4889(97)00038-4. [DOI] [PubMed] [Google Scholar]
- 50.Gerke V, Moss SE. Annexins: from structure to function. Physiol. Rev. 2002;82:331–371. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- 51.Bai XF, Ni XG, Zhao P, Liu SM, Wang HX, Guo B, Zhou LP, Liu F, Zhang JS, Wang K, et al. Overexpression of annexin 1 in pancreatic cancer and its clinical significance. World J. Gastroenterol. 2004;10:1466–1470. doi: 10.3748/wjg.v10.i10.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Croxtall JD, Choudhury Q, Newman S, Flower RJ. Lipocortin 1 and the control of cPLA2 activity in A549 cells: glucocorticoids block EGF stimulation of cPLA2 phosphorylation. Biochem. Pharmacol. 1996;52:351–356. doi: 10.1016/0006-2952(95)02442-5. [DOI] [PubMed] [Google Scholar]
- 53.Solito E, de Coupade C, Parente L, Flower RJ, Russo-Marie F. Human annexin 1 is highly expressed during the differentiation of the epithelial cell line A 549: involvement of nuclear factor interleukin 6 in phorbol ester induction of annexin 1. Cell Growth Differ. 1998;9:327–336. [PubMed] [Google Scholar]
- 54.Wang Y, Serfass L, Roy MO, Wong J, Bonneau AM, Georges E. Annexin-I expression modulates drug resistance in tumor cells. Biochem. Biophys. Res. Commun. 2004;314:565–570. doi: 10.1016/j.bbrc.2003.12.117. [DOI] [PubMed] [Google Scholar]
- 55.William F, Haigler HT, Kraft AS. Lack of phosphorylation of lipocortin I in A431 epidermoid carcinoma cells treated with phorbol esters. Biochem. Biophys. Res. Commun. 1989;160:474–479. doi: 10.1016/0006-291x(89)92457-1. [DOI] [PubMed] [Google Scholar]
- 56.Violette SM, King I, Browning JL, Pepinsky RB, Wallner BP, Sartorelli AC. Role of lipocortin I in the glucocorticoid induction of the terminal differentiation of a human squamous carcinoma. J. Cell. Physiol. 1990;142:70–77. doi: 10.1002/jcp.1041420110. [DOI] [PubMed] [Google Scholar]
- 57.Perretti M, Flower RJ. Annexin 1 and the biology of the neutrophil. J. Leukocyte Biol. 2004;76:25–29. doi: 10.1189/jlb.1103552. [DOI] [PubMed] [Google Scholar]
- 58.Perretti M, Gavins FN. Annexin 1: an endogenous anti-inflammatory protein. News Physiol. Sci. 2003;18:60–64. doi: 10.1152/nips.01424.2002. [DOI] [PubMed] [Google Scholar]
- 59.Gewirtz AT, Collier-Hyams LS, Young AN, Kucharzik T, Guilford WJ, Parkinson JF, Williams IR, Neish AS, Madara JL. Lipoxin a4 analogs attenuate induction of intestinal epithelial proinflammatory gene expression and reduce the severity of dextran sodium sulfate-induced colitis. J. Immunol. 2002;168:5260–5267. doi: 10.4049/jimmunol.168.10.5260. [DOI] [PubMed] [Google Scholar]
- 60.Fiorucci S, Wallace JL, Mencarelli A, Distrutti E, Rizzo G, Farneti S, Morelli A, Tseng JL, Suramanyam B, Guilford WJ, Parkinson JF. A β-oxidation-resistant lipoxin A4 analog treats hapten-induced colitis by attenuating inflammation and immune dysfunction. Proc. Natl. Acad. Sci. USA. 2004;101:15736–15741. doi: 10.1073/pnas.0404722101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA, Blumberg RS, Serhan CN. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc. Natl. Acad. Sci. USA. 2005;102:7671–7676. doi: 10.1073/pnas.0409271102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chiang N, Serhan CN, Dahlen SE, Drazen JM, Hay DW, Rovati GE, Shimizu T, Yokomizo T, Brink C. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol. Rev. 2006;58:463–487. doi: 10.1124/pr.58.3.4. [DOI] [PubMed] [Google Scholar]
- 63.Vaughn MW, Proske RJ, Haviland DL. Identification, cloning, and functional characterization of a murine lipoxin A4 receptor homologue gene. J. Immunol. 2002;169:3363–3369. doi: 10.4049/jimmunol.169.6.3363. [DOI] [PubMed] [Google Scholar]
- 64.Ernst S, Lange C, Wilbers A, Goebeler V, Gerke V, Rescher U. An annexin 1 N-terminal peptide activates leukocytes by triggering different members of the formyl peptide receptor family. J. Immunol. 2004;172:7669–7676. doi: 10.4049/jimmunol.172.12.7669. [DOI] [PubMed] [Google Scholar]
- 65.Yang YH, Toh ML, Clyne CD, Leech M, Aeberli D, Xue J, Dacumos A, Sharma L, Morand EF. Annexin 1 negatively regulates IL-6 expression via effects on p38 MAPK and MAPK phosphatase-1. J. Immunol. 2006;177:8148–8153. doi: 10.4049/jimmunol.177.11.8148. [DOI] [PubMed] [Google Scholar]
- 66.D’Acquisto F, Paschalidis N, Sampaio AL, Merghani A, Flower RJ, Perretti M. Impaired T cell activation and increased Th2 lineage commitment in Annexin-1-deficient T cells. Eur. J. Immunol. 2007;37:3131–3142. doi: 10.1002/eji.200636792. [DOI] [PubMed] [Google Scholar]