

Abstract
Lung cancer is the leading cause of cancer deaths accounting for more deaths than breast, colon and prostate cancer combined. The Rb/p16 tumor suppressive pathway is deregulated in most cancers. Loss of p16 occurs more frequently than Rb loss suggesting that p16 suppresses cancer by regulating Rb as well as the related proteins, p107 and p130. However, direct evidence demonstrating that p130 or p107 cooperate with Rb to suppress epithelial cancers associated with p16 loss is currently lacking. Moreover, the roles of p130 and p107 in lung cancer are not clear. In the present studies, Rb ablation was targeted to the lung epithelium in wild type, p107 or p130 null mice to determine unique and overlapping Rb family functions critical in tumor suppression. Rb ablation during development resulted in marked epithelial abnormalities despite p107 up-regulation. In contrast, p130 and p107 were not required during development but had distinct functions in the Rb deficient epithelium: p107 was required to suppress proliferation, whereas a novel pro-apoptotic function was identified for p130. Adult Rb ablated lungs lacked the epithelial phenotype seen at birth and showed compensatory p107 up-regulation and p16 induction in epithelial cell lineages that share phenotypic characteristics with human non-small cell lung cancers (NSCLC) that frequently show p16 loss. Importantly, Rb/p107, but not Rb/p130, deficient lungs developed tumors resembling NSCLC. Taken together, these studies identify distinct Rb family functions critical in controlling epithelial cell growth, and provide direct evidence that p107 cooperates with Rb to protect against a common adult cancer.
Keywords: Rb, p107, p130, lung cancer
INTRODUCTION
Carcinomas arise from transformed epithelial cells and account for >80% of adult malignancies. Lung cancer is the leading cause of cancer deaths with five year survival rates of 10-15%. This poor prognosis is due to lack of effective screening modalities resulting in patients presenting with advanced disease that is not responsive to current therapies. Understanding the molecular basis of lung cancer is fundamental to developing novel detection and therapeutic strategies. Clinicopathologic data strongly support a prominent role for the Rb/p16 pathway in the etiology of lung cancer (1). Patients with germline Rb mutations are at increased risk for lung cancer and the Rb/p16 pathway is deregulated in most, if not all, lung cancers providing convincing evidence that loss of Rb/p16 pathway function is essential in the genesis of this malignancy (2, 3).
The Rb/p16 pathway is classically viewed as a linear pathway wherein p16 positively regulates Rb which in turn suppresses cell proliferation by inhibiting expression of E2F target genes (4, 5). Rb is inactivated by cyclin D/cyclin dependent kinase (cdk) 4/6 dependent phosphorylation leading to cell cycle progression. The tumor suppressor p16 inhibits cyclin D-cdk4/6 activity thereby enhancing Rb activity and suppressing cell growth. Consistent with p16 and Rb functioning in a linear pathway, Rb mutations and p16 inactivation generally do not occur in the same tumor and Rb null cells are insensitive to p16 induced growth arrest (6-8). However, p16 also positively regulates the Rb related proteins, p107 and p130. In addition, p107 and p130 deficient cells are insensitive to p16 induced growth arrest (6) suggesting that p107 and p130 have growth suppressive functions that may be important in protecting against cancer.
Rb, p130 and p107 share extensive overlapping functions in cells in culture. All three proteins inhibit E2F responsive promoters, recruit chromatin remodeling enzymes, actively repress transcription, and induce growth arrest when overexpressed (9, 10). However, Rb, p130 and p107 clearly have distinct functions in vivo. Rb−/− mice die during mid-gestation whereas p107−/− and p130−/− mice develop normally in the same genetic backgrounds (11-16). While Rb family proteins can functionally compensate for one another during embryogenesis and cooperate in suppression of retinoblastoma and sarcomas (9, 17), the roles of p107 and p130 in suppressing common epithelial derived cancers is not clearly defined.
Lung cancers are divided into non-small cell (NSCLC) and small cell (SCLC) lung cancer based upon distinct clinical and pathologic features. Loss of p16 is detected in 30-70% of NSCLC whereas Rb mutations are detected in >90% of SCLC (1). Mouse models demonstrate that Rb cooperates with p53 to suppress SCLC (18). The role of p107 in lung cancer is largely unexplored and the contribution of p130 to lung tumorigenesis is under debate. Supporting a tumor suppressive role for p130 are reports of p130 mutations in a SCLC cell line and primary lung tumors, and studies demonstrating that p130 overexpression in a lung cancer cell line with relatively low p130 levels suppresses cell growth (19, 20). Challenging a significant role for p130 in lung cancer, however, are other studies reporting functional p130 expression in most, if not all, lung tumor cell lines and primary tumors leading to the conclusion that p130 mutation is a rare event in lung cancer (21, 22).
In order to identify unique and overlapping Rb family functions critical in lung epithelial regulation and tumor suppression, we developed mouse models with conditional Rb ablation targeted to the lung epithelium. The current studies demonstrate that p107, but not p130, cooperates with Rb to suppress lung tumorigenesis. Rb was specifically required to negatively regulate pulmonary neuroendocrine cells that share characteristics with SCLC but was not essential in non-neuroendocrine cell lineages that phenotypically resemble NSCLC. Rb ablation resulted in p107 up-regulation during development and postnatal induction of p16 providing mechanisms for cellular compensation after Rb loss. Interestingly, p107 and p130 had distinct roles in the Rb deficient epithelium p107 was critical for suppressing proliferation whereas p130 induced apoptosis. Combined Rb/p107, but not Rb/p130, loss resulted in lung tumors resembling NSCLC. Together these studies identify a novel pro-apototic function for p130 and demonstrate that p107 cooperates with Rb to suppress a common cancer.
MATERIALS AND METHODS
Mouse strains and genotyping
SPC-rtTA (23) and tetCre (24) transgenic mice were mated to RbLoxP/LoxP (24) or Rb+/− (B6.129S2-Rb1tm1Tyj, Jackson Laboratory) mice. Double transgenic/RbLoxP/LoxP mice were mated to p130−/− and p107−/− mice to generate mice with combined Rb family deficient lung epithelium (15). Gestational age was assigned by vaginal plug date designated embryonic day (E) 0.5. Dams were treated with doxycycline (Sigma) throughout gestation with treatment discontinued at birth (24). Genotypes were determined by PCR analysis using established primers (24).
Histology, immunohistochemistry, TUNEL analysis and ß-galactosidase staining
Lung tissue was fixed in 10% neutral buffered formalin and paraffin embedded. Sections were stained with hematoxylin and eosin for histological analysis. Immunohistochemistry was performed using Vectastain Elite ABC, M.O.M. Immunodetection, and DAB Substrate Kits (Vector Laboratories). Methanol/hydrogen peroxide pretreatment, microwave/10 mM citrate antigen retrieval and serum block were performed. Antibodies were incubated at room temperature for 45 minutes (p16) or 4°C overnight at the following dilutions: Ki67 (1:50; BD PharMingen), CGRP (1:10,000; Sigma), proSPC (09337, 1:2000; Jeffrey Whitsett, Cincinnati, OH), CCSP (1:20,000; Steve Brody, St. Louis, MO), and p16 (M156, 1:100, Santa Cruz Biotechnology). TUNEL analysis was performed using ApoTag Peroxidase Detection Kit (Chemicon International). Slides were counterstained with hematoxylin or nuclear fast red. Immunofluorescence was performed by co-staining sections with Ki67 (1:100) and proSPC (GP993, 1:500; Jeffrey Whitsett) followed by incubation with Alexa 594 anti-guinea pig and Alexa 488 anti-mouse IgG1 secondary antibodies (1:200; Invitrogen) for one hour at room temperature and coverslipping with Vectashield with DAPI (Vector Laboratories). Staining was visualized using a Zeiss Axioplan 2 Imaging fluorescent microscope and counts were obtained using Metamorph Version 7.5.0.0. Proliferation was quantified by determining the percentage of conducting airway or SPC positive epithelial cells with nuclear Ki67 staining. Apoptosis was quantified by determining the percentage of conducting airway epithelial cells that were TUNEL positive, showed morphologic features of apoptosis and remained attached to the basement membrane. TUNEL positive cells within airway lumens were not counted to avoid including necrotic cells. Counts reflect analysis of 200-450 epithelial cells and at least two lung lobes per mouse. Statistical analysis was performed with SigmaStat Version 3.10 using parametric (Ki67, SPC/Ki67) or nonparametric Kruskal-Wallis (TUNEL) one way ANOVA followed by multiple comparisons using the Student-Newman-Keuls method. Whole mount and frozen section ß-galactosidase staining was performed as described (24).
Western blot analysis
Lungs were sonicated in lysis buffer supplemented with protease inhibitors (Pierce Biotechnology) and 100 μg total lung protein were resolved by SDS-polyacrylamide electrophoresis under reducing conditions. The 3T3-L1 cell lysate was used as a positive p16 control (Santa Cruz Biotechnology). Proteins were transferred to nitrocellulose membranes and probed for p107 (C-18, 1:1000), p130 (C-20, 1:1000), and p16 (M156, 1:1000) (Santa Cruz Biotechnology), and tubulin (DM 1A; 1:2500) and ß-actin (20-33, 1:1000) (Sigma). Antibody binding was detected by incubation with horseradish peroxidase conjugated secondary antibodies followed by chemiluminescence (ECL Plus, Amersham Biosciences).
Quantitative real-time RT-PCR
RNA was isolated using Qiagen RNeasy Mini Kit and treated with DNAse (Qiagen). Total RNA was used to generate cDNA using SuperScript III RT (Invitrogen) and real time RT-PCR was performed using the TaqMan® Real-Time PCR Gene Expression System (Applied Biosystems). All samples were performed in triplicate and data was analyzed using 7300 System Software and ß-actin as the internal control. Statistically significant differences were determined by unpaired Student t-tests assuming equal variance.
Primary type II cell isolation
Primary type II cell cultures were generated from lungs of 5-6 week old mice as described (25). Cell viability measured by trypan blue dye exclusion was consistently >90%. SPC staining was performed by fixing cell cytospins in 4% paraformaldehyde for 15 minutes, blocking in 4% donkey serum and incubating with a proSPC antibody (GP993, 1:500). Staining was detected and visualized as described for tissue sections. Type II cell purity was consistently >85% as assessed morphologically after staining with hematoxylin or immunofluorescent staining for SPC.
RESULTS
Generation of mice with Rb ablation targeted throughout the lung epithelium
A conditional Rb knockout mouse model was generated by breeding double transgenic mice bearing 1) the reverse tetracycline responsive transactivator under control of the human surfactant protein C (SPC) promoter (SPC-rtTA) and 2) Cre recombinase under control of the tet operator (tetCre) into RbLoxP/LoxP or RbLoxP/− backgrounds (Fig. 1). A similar model using the rat Clara cell 10 kD/Clara cell secretory protein (CC10/CCSP) promoter was previously developed: however, the SPC promoter was used in the current studies because the SPC promoter more uniformly targets alveolar type II cells in the distal pulmonary epithelium thus providing the advantage of assessing Rb family function in the physiologically distinct conducting and respiratory regions of the lung. Rb ablation was induced during gestation by doxycycline administration to pregnant dams. Rb gene recombination was detected in double transgenic lungs from pups at birth but was not detected in littermate controls lacking one or both transgenes (Fig. 1). Cre mediated recombination was restricted to the lung epithelium and present in the vast majority of ciliated, Clara, and type II cells as assessed by ß-galactosidase staining on double transgenic lungs containing the ROSA26 reporter locus (26). These data confirm that gene recombination is confined to the lung epithelium and occurs in the vast majority of epithelial cells representing multiple cell lineages.
Figure 1. Epithelial hyperplasia and cell death seen in Rb ablated lungs at birth is not present in adult lungs.

A, SPC-rtTA+/−/tetCre+/− double transgenic mice were bred to RbLoxP/LoxP or RbLoxP/− mice. Doxycycline (ovals) activates the rtTA (arches) expressed specifically in lung epithelium leading to Cre expression and subsequent recombination at floxed Rb alleles. B, PCR analysis on lung DNA from day 1 pups. Rb recombination (RbRec) of floxed Rb alleles (RbLoxP) is only detected in lungs from mice containing both transgenes (SPC-rtTA, Cre). Mice were homozygous for the floxed Rb allele or heterozygous for the floxed and germline knockout (Rb−) alleles. C, Whole mount ß-galactosidase staining on lungs from day 1 mice pups containing the ROSA26 reporter locus demonstrates uniform staining in double transgenic lungs (SPC-rtTA+/Cre+) but not in controls lacking one or both transgenes (C). ß-galactosidase staining is restricted to the epithelium and present in Clara (arrow), ciliated (dashed arrow) and type II cells (arrowhead) in adult lungs. A subset of CGRP positive epithelial cells (brown, boxed area and open arrowhead) stain for ß-galactosidase (arrow in inset designates double positive cell). n ≥ 3 mice for each group. Original magnification: 200x (middle), 1000x (bottom). D, Hematoxylin and eosin stained lung sections show epithelial hypercellularity and apoptotic bodies (arrows) in Rb ablated E18.5 lungs (Pups) but not in Rb proficient lungs (Control) or Rb ablated lungs from 8 month old mice (Adults). Hypercellular neuroendocrine lesions are present in adult Rb ablated lungs (arrowhead). Original magnification: 400x (top row), 1000x (bottom row).
Rb loss results in neuroendocrine hyperplasia whereas non-neuroendocrine cells do not require Rb function
Rb deficient E18.5 lungs were grossly normal and showed branching morphogenesis; however morphologic analysis revealed profound epithelial abnormalities (Fig. 1). The epithelium lacked the normal pseudostratified organization and was comprised of hyperplastic cells with increased nuclear to cytoplasmic ratios and morphologic features of apoptosis. The phenotype was similar in the RbLoxP/LoxP and RbLoxP/− backgrounds and to the epithelial abnormalities previously reported after Rb ablation in a mouse model utilizing the CC10 promoter (24). Expression of rtTA was recently reported to be toxic to lung epithelial cells (27) however, single transgenic littermate controls and double transgenic mice with wild type Rb alleles lacked the epithelial abnormalities characteristic of Rb ablated lungs demonstrating that the phenotype was dependent upon Rb loss (data not shown). Thus, Rb has an essential role in regulating epithelial proliferation and survival during lung development.
Despite the marked lung abnormalities at birth, mice with Rb ablated lungs survived to adulthood and the adult lung epithelium lacked the overall epithelial hypercellularity and apoptosis noted at birth (Fig. 1). Interestingly, neuroendocrine cell hyperplasia was detected in 88% (14/16) of double transgenic adult mice examined between 5.5 to 14 months of age. Clusters of epithelial cells with high nuclear to cytoplasmic ratios and expressing the neuropeptide calcitonin gene related peptide (CGRP) protruded into airway lumens characteristic of neuroendocrine cell hyperplasia (28) (Fig. 1 and data not shown). Identical lesions were previously reported after Rb ablation in the lung epithelium using the CC10 promoter (24). Functional Cre activity was detected in a subset of pulmonary neuroendocrine cells in both the SPC and CC10 promoted models as assessed by colocalization of ß-galactosidase and CGRP staining (Fig. 1 and (24)). While neuroendocrine hyperplasia may represent a precursor for SCLC, progression to malignant tumors was not observed. Thus, the current studies together with previous findings in an independently derived mouse model identify a unique cell lineage specific function for Rb in pulmonary neuroendocrine cells. Moreover, these results demonstrate that non-neuroendocrine cell lineages compensate for Rb loss after birth resulting in resolution of the marked abnormalities incurred during development.
p107 and p16 expression are induced in Rb ablated lungs
Previous studies showed that Rb loss can result in p107 and/or p130 induction, and that these family proteins can compensate for Rb loss in restricted cell lineages and environmental contexts (14, 15, 17, 29-31). p130 levels were not altered in Rb ablated lungs. In contrast, p107 was increased in Rb ablated day 1 and adult lungs demonstrating that p107 up-regulation occurred during development and was sustained into adulthood (Fig. 2). Given that Rb ablated lungs showed marked epithelial abnormalities at birth despite p107 up-regulation, investigations were aimed toward identifying effectors induced postnatally. p16 was explored as a potential candidate since Rb was shown to negatively regulate p16 in fibroblasts in culture (32). Indeed, p16 was induced in adult Rb ablated lungs but not in lungs from day 1 pups (Fig. 3). p16 was expressed in non-neuroendocrine progenitor cells lineages, namely Clara and type II cells, that share phenotypic features with NSCLC. Moreover, p16 was induced 10 fold in Rb ablated primary type II cell isolates as compared to Rb proficient controls (Fig. 3). Expression of p107 was also increased in Rb ablated type II cells whereas p130 levels were unchanged (Fig. 3). Taken together these data demonstrate that Rb loss results in p107 up-regulation during development followed by postnatal p16 induction in epithelial cell lineages that share phenotypic similarities with NSCLC.
Figure 2. p107, but not p130, is induced in Rb ablated lungs during development.
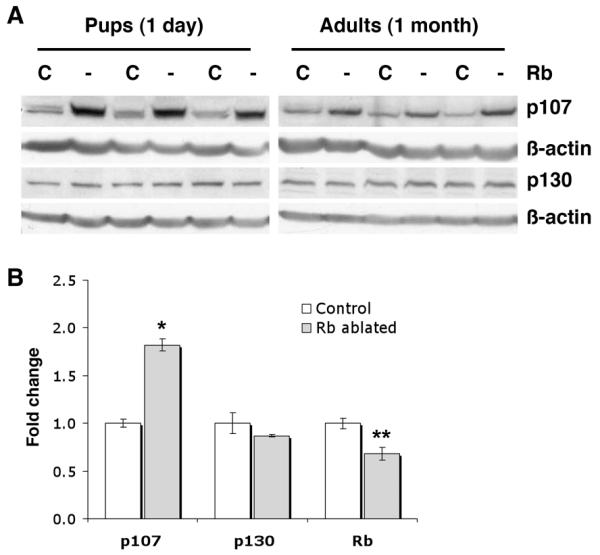
A, Western blot analyses show increased p107, but not p130, in Rb ablated (Rb -) day 1 and adult lungs as compared to Rb proficient littermate controls (Rb C). Lung lysates were evenly loaded as assessed by reprobing for β-actin. n=4-8 animals per group. B, Quantitative real-time RT-PCR shows increased p107, but not p130, in Rb ablated adult lungs (*p=0.0004, n=3 per group). Rb is significantly reduced in Rb deficient lungs (**p=0.023, n=3 per group). Graphs depict mRNA fold change relative to controls ± SE.
Figure 3. p16 is induced in Rb ablated lungs after birth and is expressed in Clara and type II cells.

A, Western blot analyses show increased p16 in Rb ablated (Rb -), and 1 and 6-9 month adult lungs as compared to Rb proficient controls (Rb C). p16 is not detected in Rb ablated or control day 1 lungs. Lung lysates from p16−/− mice (p16−/−) and 3T3-L1 cell lysates (p16 C) represent negative and positive controls, respectively. Lung lysates were evenly loaded as assessed by reprobing for β-actin or tubulin. n=4-9 per group. Quantitative real-time RT-PCR shows increased p16 in Rb ablated 1 month (*p=0.0017) and 6-9 month (**p=0.045) adult lungs. p16 was not detected in E18.5 lungs. n=3 per group. Graphs depict mRNA fold change relative to controls ± SE. B, Immunohistochemical analyses show p16 expression in Clara cells within conducting airways (left panel) and type II cells within the parenchyma (right panel) in both Rb ablated and Rb proficient (Control) 9 month adult lungs. No specific staining is present in p16−/− lungs confirming assay specificity. Higher power views (insets) of boxed areas show representative Clara, ciliated and type II cells. n=3 Rb ablated and control lungs. Original magnification: 1000x. C, Primary type II cell isolate cytospins stained with hematoxylin or stained for SPC and DAPI by immunofluorescence. PCR analysis shows Rb recombination (RbRec) of floxed Rb alleles (RbLoxP) in cells isolated from Rb ablated (+) but not Rb proficient control lungs (−). D, Quantitative real-time RT-PCR shows increased p107, but not p130, in Rb ablated type II cells as compared to Rb proficient controls (*p=0.038). Rb is significantly reduced in Rb ablated type II cells (**p=0.0001). p16 is increased in Rb ablated 1 month lungs (Whole Lung) and type II cells (*p≤0.002). n=3-4 per group. Graphs depict mRNA fold change relative to controls ± SE.
Epithelial loss of Rb and p107, but not p130, causes neonatal lethality
Combined Rb family deficient lungs were obtained by generating double transgenic/RbLoxP/LoxP mice in a p107−/− or p130−/− background. Mice screened at the time of weaning showed a marked reduction in the percentage of Rb ablated/p107−/− mice as compared to that expected by Mendelian genetics (Supplemental Fig. 1). Genotype analysis of E18.5 pups showed the expected numbers of Rb ablated/p107−/− pups indicating that combined Rb/p107 loss in the lung epithelium resulted in neonatal lethality (Supplemental Fig. 2). In contrast to p107, additional loss of p130 in Rb deficient lungs did not affect survival (Supplemental Fig. 3).
Surfactant protein B (SPB) and ABCA3 are expressed in the developing lung epithelium and required for neonatal survival (33, 34). SPB expression was decreased in Rb ablated/p107−/− lungs; however, SPB levels were similar in Rb/p107 and Rb deficient lungs providing evidence that neonatal death associated with combined Rb/p107 loss was not due solely to reduced SPB expression (Supplemental Fig. 4). ABCA3 levels were significantly reduced in Rb ablated/p107−/− lungs as compared to Rb ablation alone raising the possibility that decreased ABCA3 expression contributes to the neonatal death of pups with Rb/p107 deficient lungs. Of note, expression of other type II and Clara cell differentiation markers (namely SPC and CCSP) was also significantly reduced in Rb ablated/p107−/− lungs (Supplemental Fig. 4). Collectively these results demonstrate that loss of pocket protein function impairs lung epithelial cell differentiation, and that p107, but not p130, plays an essential role in compensating for Rb loss in the developing lung.
p107 and p130 have distinct roles in Rb deficient lung epithelium
Similar to Rb ablated lungs, Rb/p107 and Rb/p130 deficient lungs showed marked epithelial hypercellularity and cell death (Fig. 4). In contrast, p107−/− and p130−/− lungs were indistinguishable from Rb proficient controls demonstrating that Rb, but not p107 or p130, has a unique and critical role in lung development. Epithelial proliferation and apoptosis were significantly increased in Rb ablated lungs as compared to Rb proficient controls whereas p130 or p107 loss alone had no effect on epithelial proliferation or survival (Fig. 4). Interestingly, Rb ablated/p107−/− lungs showed increased proliferation as compared to Rb ablated lungs, but additional p107 loss had no effect on apoptosis (Fig. 4). Conversely, Rb ablated/p130−/− lungs showed decreased apoptosis but additional p130 loss had no effect on proliferation. Thus, pulmonary epithelial proliferation and survival are independently regulated, and p107 and p130 distinctly regulate these biologic processes.
Figure 4. Rb family proteins have distinct roles in regulating epithelial proliferation and survival.

A, Hematoxylin and eosin stained E18.5 lung sections show epithelial hyperplasia, dysplasia and apoptotic cell death in Rb ablated, Rb ablated/p130−/− and Rb ablated/p107−/− lungs. Lungs from p130−/− and p107−/− are indistinguishable from Rb family proficient lungs (Control). B, Immunohistochemical staining for the proliferation marker, Ki67 (arrow) shows an increase in proliferating epithelial cells in Rb ablated, Rb ablated/p130−/− and Rb ablated/p107−/− lungs as compared to Rb proficient controls (*p<0.001). Ki67 positive cells are also increased in Rb ablated/p107−/− as compared to Rb ablated lungs (**p<0.001). Proliferation in p130−/− and p107−/− lungs is similar to control Rb proficient lungs (p>0.67). C, TUNEL analysis shows an increase in apoptotic epithelial cells (arrow) in Rb ablated, Rb ablated/p130−/− and Rb ablated/p107−/−lungs as compared to Rb proficient controls (*p<0.05). Apoptotic cells are decreased in Rb ablated/p130−/− as compared to Rb ablated lungs (**p<0.05). Apoptosis in p130−/− and p107−/− lungs is similar to control Rb proficient lungs (p>0.05). Graphs depict percent Ki67 or TUNEL positive epithelial cells ± SE. n≥6 mice per group. Original magnification: 1000x.
Rb/p107 deficient lungs develop tumors with features of NSCLC
Adult mice with Rb, Rb/p130, and Rb/p107 deficient lungs along with Rb family proficient, p107−/− and p130−/− littermates lacking one or both transgenes were monitored for lung tumors. Due to neonatal lethality of Rb ablated/p107−/− pups, a cohort of adult double transgenic p107−/− mice with mosaic Rb/p107 deficient lungs were generated by taking advantage of the “leaky” Cre expression (and thus Rb ablation) that occurred in the absence of doxycycline treatment. Lungs from double transgenic mice not treated with doxycycline showed variable levels of Rb recombination (Fig. 5 and data not shown) allowing some mice with Rb ablated/p107−/− chimeric lungs to survive to adulthood. Interestingly, 67% (6/9) of these mice developed adenomas or adenocarcinomas by 5.5 to 15 months of age (Fig. 5 and Table 1). Rb recombination was detected in the tumors confirming that the tumors arose from Rb/p107 deficient epithelial cells (Fig. 5). In contrast to p107, p130 loss did not cooperate with Rb ablation in tumorigenesis. Adenocarcinomas were not detected in Rb/p130 deficient lungs or lungs deficient for a single Rb family member, and lung adenomas were only rarely detected in mice of other genotypes (Table 1).
Figure 5. Rb/p107 loss results in lung tumors with a type II cell phenotype.

A, Hematoxylin and eosin stained lung sections (left) and immunohistochemical analysis (right) of lung tumors arising in double transgenic/p107−/− adult mice not treated with doxycycline show a solid adenoma (t, top left) and a papillary adenocarcinoma invading the airway (t, middle and bottom left and right panel) that are positive for SPC but not CCSP or CGRP. Non-neoplastic cells in the surrounding conducting airway show CCSP and CGRP positive cells (arrows) serving as internal controls. Original magnification: 100x (top and middle left), 400x (bottom left and SPC) 1000x (CCSP and CGRP). B, PCR analysis on DNA from non-neoplastic lung (lung) and lung tumors (tumor) obtained from double transgenic/p107−/− chimeric mice (+) and p107−/− control littermates lacking one or both transgenes (−) that are homozygous for the floxed Rb allele (RbLoxP) show enrichment for the Rb recombined allele (RbRec) in tumors as compared to non-neoplastic lung. Rb recombination is not detected in tail DNA or tissue from controls. C, Co-immunofluorescent staining for SPC (red) and Ki67 (green) shows increased type II cell proliferation in Rb ablated and Rb ablated/p107−/− E18.5 lungs as compared to Rb family proficient controls (*p<0.001). Type II cell proliferation was not increased in Rb ablated/p107−/− lungs as compared to Rb ablated lungs (p=0.07). Higher power views of boxed areas (insets) show representative Ki67 negative (Control), Ki67 positive (Rb ablated) and adjacent Ki67 positive and negative (Rb ablated/p107−/−) SPC positive type II cells. Yellow represents autofluorescent red blood cells. Graph depicts percent SPC positive type II cells with nuclear Ki67 staining ± SE. n=3-4 per group. br=bronchioles. Original magnification: 400x. D, Model for Rb family function in the lung epithelium. Rb is required to suppress epithelial proliferation and death during development despite p107 induction. p107 cooperates with Rb to induce cell cycle arrest whereas p130 promotes apoptosis in the developing Rb deficient lung epithelium. In the adult lung, Rb is required to negatively regulate neuroendocrine cells whereas non-neuroendocrine lineages compensate for Rb loss and show induction of p16 and p107. Rb and p107 cooperate to suppress development of lung tumors resembling NSCLC.
Table 1.
Rb/p107 loss results in lung tumors
| Resultant Genotype | Lung Tumors |
|---|---|
| Rb+/+ | 0/16 (0%) |
| Rb+/− | 0/14 (0%) |
| Rb−/− | 1/22 (5%) |
| p107−/− | 1/8 (13%) |
| p130−/− | 2/30 (7%) |
| Rb−/−;p130−/− | 0/28 (0%) |
| Rb−/−;p107−/− | 6/9 (67%) |
Lung tumors in Rb ablated/p107−/− chimeric lungs shared morphologic and phenotypic characteristics with human NSCLC. Tumors were comprised of epithelial cells with pleomorphic, hyperchromatic nuclei growing in solid nests or lining fibrovascular cores (Fig. 5). Focal necrosis and rare mitotic figures were identified. Tumors were classified as papillary adenocarcinoma, and papillary and solid adenomas (28). Tumors were positive for SPC but negative for Clara (CCSP) and neuroendocrine (CGRP) cell markers demonstrating a non-neuroendocrine type II cell phenotype (Fig. 5). Type II cell proliferation was increased in Rb ablated E18.5 lungs but additional loss of p107 was not associated with a further increase in Ki67 positive type II cells suggesting that tumorigenesis in Rb ablated/p107−/− lungs is not simply a result of increased type II cell proliferation (Fig. 5). Collectively these studies provide direct evidence that p107 cooperates with Rb in suppressing development of lung tumors that share phenotypic characteristics with human NSCLC.
DISCUSSION
The Rb/p16 pathway has a well established tumor suppressive role in human cancers. In the lung, p16 and Rb are preferentially lost in phenotypically distinct cancers providing a unique opportunity to explore Rb/p16 pathway function in carcinogenesis. We have developed novel mouse models that provide powerful systems to dissect how Rb interacts with other proteins to regulate epithelial biology and in turn impact development and tumorigenesis. p107 and p130 cooperate with Rb to suppress retinoblastoma, and Rb/p107 deficient chimeric mice develop sarcomas, cecal adenocarcinomas and rarely other tumors thus identifying p107 and p130 as tumor suppressors in restricted cell lineages (14, 16, 17). The current studies extend these observations by identifying distinct cell lineage specific pocket protein functions critical for suppressing epithelial cell growth and directly demonstrating that Rb and p107 have synergistic functions critical in suppressing a common adult cancer.
p130 has a novel pro-apoptotic function in Rb deficient epithelial cells in vivo
The present studies identify functional distinctions among the closely related Rb, p107 and p130 proteins in epithelial regulation in vivo. Rb was uniquely required for proper lung epithelial growth and survival during development. Interestingly, p107 was critical for suppressing proliferation whereas p130 cooperated with Rb in regulating apoptosis. Molecular mechanisms driving apoptosis in Rb family deficient tissues are highly cell context specific (35). Previously identified apoptotic mediators include p53, caspase 3, Apaf1, PTEN, E2F1 and E2F3. These studies identify p130 as a novel mediator of apoptosis in Rb deficient cells. This pro-apoptotic p130 function is in contrast to the role of p130 in suppressing neuronal cell apoptosis in culture (36). p130 depletion resulted in apoptosis in postmitotic neuronal PC12 cells but not in cycling cells providing evidence that cellular response to p130 loss is influenced by the proliferative state of the cell (36). Combined Rb and p130 loss was not sufficient for tumorigenesis but p130 could limit aberrant lung epithelial cell growth by inducing apoptosis. Consistent with this notion, p130 suppresses growth of oncogenic K-ras induced lung tumors in mice, and reduced or absent p130 expression in human lung cancers is associated with a poor prognosis (37, 38).
Combined Rb and p130 loss does not lead to SCLC
Previous studies suggested that combined Rb and p130 loss was required for development of neuroendocrine hyperplasia, a presumed precursor for SCLC (17, 18). Given that combined Rb and p53 loss results in SCLC, it was further suggested that p130 and p53 may act in a similar tumor suppressive pathway and that p130 loss might substitute for loss of p53 in SCLC progression. The current studies demonstrate that neuroendocrine hyperplasia develops after Rb ablation alone and that additional loss of p130 does not lead to SCLC. Conditional Rb and p53 ablation in the current model results in neuroendocrine lung tumors resembling SCLC. Recombined Rb and p53 alleles are detected in the tumor cells providing evidence that cell autonomous Rb and p53 functions are required to suppress SCLC (data not shown). Together these data support a unique role for Rb in neuroendocrine cell regulation and directly demonstrate that p130 loss does not substitute for loss of p53 in development of SCLC.
In summary, the current studies demonstrate that Rb family proteins have cell type specific functions in the lung epithelium important for development and tumor suppression (see Fig. 5 for model). Rb plays a critical unique role in regulating epithelial proliferation and survival in the developing lung despite p107 induction. In contrast, p107 and p130 are not essential in the pulmonary epithelium but have distinct roles in Rb deficient cells: p107 induces cell cycle arrest whereas p130 promotes apoptosis. In the adult lung, Rb is required for negatively regulating the neuroendocrine cell lineage whereas p107 and p16 are induced in non-neuroendocrine cell types that compensate for Rb loss. Importantly, combined Rb/p107 loss results in tumors resembling NSCLC demonstrating that p107 and Rb have cooperative functions critical for suppressing lung cancer.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jeffrey Whitsett, Anton Berns and Tyler Jacks for mice, and Susan Wert and Paula Blair for expertise in immunohistochemistry.
Financial Support: This work was supported by grants from the American Cancer Society and NIH/NHLBI RO1 HL079193 to KAWB
REFERENCES
- 1.Minna JD, Roth JA, Gazdar AF. Focus on lung cancer. Cancer Cell. 2002;1:49–52. doi: 10.1016/s1535-6108(02)00027-2. [DOI] [PubMed] [Google Scholar]
- 2.Kleinerman RA, Tarone RE, Abramson DH, Seddon JM, Li FP, Tucker MA. Hereditary retinoblastoma and risk of lung cancer. J Natl Cancer Inst. 2000;92:2037–9. doi: 10.1093/jnci/92.24.2037. [DOI] [PubMed] [Google Scholar]
- 3.Sanders BM, Jay M, Draper GJ, Roberts EM. Non-ocular cancer in relatives of retinoblastoma patients. Br J Cancer. 1989;60:358–65. doi: 10.1038/bjc.1989.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 5.Knudsen ES, Knudsen KE. Retinoblastoma tumor suppressor: where cancer meets the cell cycle. Exp Biol Med (Maywood) 2006;231:1271–81. doi: 10.1177/153537020623100713. [DOI] [PubMed] [Google Scholar]
- 6.Bruce JL, Hurford RK, Jr., Classon M, Koh J, Dyson N. Requirements for cell cycle arrest by p16INK4a. Mol Cell. 2000;6:737–42. doi: 10.1016/s1097-2765(00)00072-1. [DOI] [PubMed] [Google Scholar]
- 7.Otterson GA, Kratzke RA, Coxon A, Kim YW, Kaye FJ. Absence of p16INK4 protein is restricted to the subset of lung cancer lines that retains wildtype RB. Oncogene. 1994;9:3375–8. [PubMed] [Google Scholar]
- 8.Shapiro GI, Edwards CD, Kobzik L, et al. Reciprocal Rb inactivation and p16INK4 expression in primary lung cancers and cell lines. Cancer Res. 1995;55:505–9. [PubMed] [Google Scholar]
- 9.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–82. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Classon M, Dyson N. p107 and p130: versatile proteins with interesting pockets. Exp Cell Res. 2001;264:135–47. doi: 10.1006/excr.2000.5135. [DOI] [PubMed] [Google Scholar]
- 11.Clarke AR, Maandag ER, van Roon M, et al. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–30. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- 12.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 13.Lee EY, Chang CY, Hu N, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–94. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 14.Lee MH, Williams BO, Mulligan G, et al. Targeted disruption of p107: functional overlap between p107 and Rb. Genes Dev. 1996;10:1621–32. doi: 10.1101/gad.10.13.1621. [DOI] [PubMed] [Google Scholar]
- 15.Cobrinik D, Lee MH, Hannon G, et al. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev. 1996;10:1633–44. doi: 10.1101/gad.10.13.1633. [DOI] [PubMed] [Google Scholar]
- 16.Robanus-Maandag E, Dekker M, van der Valk M, et al. p107 is a suppressor of retinoblastoma development in pRb-deficient mice. Genes Dev. 1998;12:1599–609. doi: 10.1101/gad.12.11.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dannenberg JH, Schuijff L, Dekker M, van der Valk M, te Riele H. Tissue-specific tumor suppressor activity of retinoblastoma gene homologs p107 and p130. Genes Dev. 2004;18:2952–62. doi: 10.1101/gad.322004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–9. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- 19.Helin K, Holm K, Niebuhr A, et al. Loss of the retinoblastoma protein-related p130 protein in small cell lung carcinoma. Proc Natl Acad Sci U S A. 1997;94:6933–8. doi: 10.1073/pnas.94.13.6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Claudio PP, Howard CM, Pacilio C, et al. Mutations in the retinoblastoma-related gene RB2/p130 in lung tumors and suppression of tumor growth in vivo by retrovirus-mediated gene transfer. Cancer Res. 2000;60:372–82. [PubMed] [Google Scholar]
- 21.Jun H Xue, Gemma A, Hosoya Y, et al. Reduced transcription of the RB2/p130 gene in human lung cancer. Mol Carcinog. 2003;38:124–9. doi: 10.1002/mc.10152. [DOI] [PubMed] [Google Scholar]
- 22.Modi S, Kubo A, Oie H, Coxon AB, Rehmatulla A, Kaye FJ. Protein expression of the RB-related gene family and SV40 large T antigen in mesothelioma and lung cancer. Oncogene. 2000;19:4632–9. doi: 10.1038/sj.onc.1203815. [DOI] [PubMed] [Google Scholar]
- 23.Perl AK, Tichelaar JW, Whitsett JA. Conditional gene expression in the respiratory epithelium of the mouse. Transgenic Res. 2002;11:21–9. doi: 10.1023/a:1013986627504. [DOI] [PubMed] [Google Scholar]
- 24.Wikenheiser-Brokamp KA. Rb family proteins differentially regulate distinct cell lineages during epithelial development. Development. 2004;131:4299–310. doi: 10.1242/dev.01232. [DOI] [PubMed] [Google Scholar]
- 25.Rice WR, Conkright JJ, Na CL, Ikegami M, Shannon JM, Weaver TE. Maintenance of the mouse type II cell phenotype in vitro. Am J Physiol Lung Cell Mol Physiol. 2002;283:L256–64. doi: 10.1152/ajplung.00302.2001. [DOI] [PubMed] [Google Scholar]
- 26.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nature Genetics. 1999;21:70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 27.Morimoto M, Kopan R. rtTA toxicity limits the usefulness of the SP-C-rtTA transgenic mouse. Dev Biol. 2009;325:171–8. doi: 10.1016/j.ydbio.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nikitin AY, Alcaraz A, Anver MR, et al. Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res. 2004;64:2307–16. doi: 10.1158/0008-5472.can-03-3376. [DOI] [PubMed] [Google Scholar]
- 29.Guo J, Longshore S, Nair R, Warner BW. Retinoblastoma protein (pRb), but not p107 or p130, is required for maintenance of enterocyte quiescence and differentiation in small intestine. J Biol Chem. 2009;284:134–40. doi: 10.1074/jbc.M806133200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacLellan WR, Garcia A, Oh H, et al. Overlapping roles of pocket proteins in the myocardium are unmasked by germ line deletion of p130 plus heart-specific deletion of Rb. Mol Cell Biol. 2005;25:2486–97. doi: 10.1128/MCB.25.6.2486-2497.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayhew CN, Carter SL, Fox SR, et al. RB loss abrogates cell cycle control and genome integrity to promote liver tumorigenesis. Gastroenterology. 2007;133:976–84. doi: 10.1053/j.gastro.2007.06.025. [DOI] [PubMed] [Google Scholar]
- 32.Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev. 2007;21:49–54. doi: 10.1101/gad.1499407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fitzgerald ML, Xavier R, Haley KJ, et al. ABCA3 inactivation in mice causes respiratory failure, loss of pulmonary surfactant, and depletion of lung phosphatidylglycerol. J Lipid Res. 2007;48:621–32. doi: 10.1194/jlr.M600449-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Clark JC, Wert SE, Bachurski CJ, et al. Targeted disruption of the surfactant protein B gene disrupts surfactant homeostasis, causing respiratory failure in newborn mice. Proc Natl Acad Sci U S A. 1995;92:7794–8. doi: 10.1073/pnas.92.17.7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chau BN, Wang JY. Coordinated regulation of life and death by RB. Nat Rev Cancer. 2003;3:130–8. doi: 10.1038/nrc993. [DOI] [PubMed] [Google Scholar]
- 36.Liu DX, Nath N, Chellappan SP, Greene LA. Regulation of neuron survival and death by p130 and associated chromatin modifiers. Genes Dev. 2005;19:719–32. doi: 10.1101/gad.1296405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ho VM, Schaffer BE, Karnezis AN, Park KS, Sage J. The retinoblastoma gene Rb and its family member p130 suppress lung adenocarcinoma induced by oncogenic K-Ras. Oncogene. 2009;28:1393–9. doi: 10.1038/onc.2008.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caputi M, Groeger AM, Esposito V, et al. Loss of pRb2/p130 expression is associated with unfavorable clinical outcome in lung cancer. Clin Cancer Res. 2002;8:3850–6. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.