

Abstract
Rapid arterial O2 desaturation during apnea in the preterm infant has obvious clinical implications but to date no adequate explanation for why it exists. Understanding the factors influencing the rate of arterial O2 desaturation during apnea (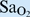 ) is complicated by the non-linear O2 dissociation curve, falling pulmonary O2 uptake, and by the fact that O2 desaturation is biphasic, exhibiting a rapid phase (stage 1) followed by a slower phase when severe desaturation develops (stage 2). Using a mathematical model incorporating pulmonary uptake dynamics, we found that elevated metabolic O2 consumption accelerates
) is complicated by the non-linear O2 dissociation curve, falling pulmonary O2 uptake, and by the fact that O2 desaturation is biphasic, exhibiting a rapid phase (stage 1) followed by a slower phase when severe desaturation develops (stage 2). Using a mathematical model incorporating pulmonary uptake dynamics, we found that elevated metabolic O2 consumption accelerates 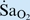 throughout the entire desaturation process. By contrast, the remaining factors have a restricted temporal influence: low pre-apneic alveolar
throughout the entire desaturation process. By contrast, the remaining factors have a restricted temporal influence: low pre-apneic alveolar 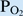 causes an early onset of desaturation, but thereafter has little impact; reduced lung volume, hemoglobin content or cardiac output, accelerates
causes an early onset of desaturation, but thereafter has little impact; reduced lung volume, hemoglobin content or cardiac output, accelerates 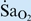 during stage 1, and finally, total blood O2 capacity (blood volume and hemoglobin content) alone determines
during stage 1, and finally, total blood O2 capacity (blood volume and hemoglobin content) alone determines 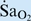 during stage 2. Preterm infants with elevated metabolic rate, respiratory depression, low lung volume, impaired cardiac reserve, anemia, or hypovolemia, are at risk for rapid and profound apneic hypoxemia. Our insights provide a basic physiological framework that may guide clinical interpretation and design of interventions for preventing sudden apneic hypoxemia.
during stage 2. Preterm infants with elevated metabolic rate, respiratory depression, low lung volume, impaired cardiac reserve, anemia, or hypovolemia, are at risk for rapid and profound apneic hypoxemia. Our insights provide a basic physiological framework that may guide clinical interpretation and design of interventions for preventing sudden apneic hypoxemia.
Author Summary
When breathing stops, the flow of O2 into and the flow of CO2 out of the body cease. Such an event, termed an apnea, can be especially dangerous in preterm infants in whom it can lead to a rapid decline in arterial O2 saturation, reaching rates of 3–8% per second, rapidly reducing O2 to a level that could lead to neurological damage. Despite extensive experimental research, we have a poor mechanistic understanding of the causes of rapidly developing hypoxemia. We describe a new mathematical model that allows examination of the importance of the major cardiorespiratory factors that are likely to influence the speed at which arterial hypoxemia worsens during apnea. We found that high metabolic rate as well as reduced pre-apneic ventilation, lung volume, cardiac output, hemoglobin content, blood O2 affinity, and blood volume accelerate the development of hypoxemia during apnea. Importantly, the cardiorespiratory factors that contribute to rapid hypoxemia are all pertinent to the preterm infant during early postnatal development. Thus the newborn is highly susceptible to rapid and severe desaturation, potentially explaining the propensity of preterm infants, particularly those with apnea, to neurological impairment.
Introduction
Apnea and its accompanying arterial O2 desaturation are common clinical complications in preterm infants, occurring in more than 50% of very low birth weight infants [1]. In preterm infants, apnea causes a reduction in heart rate [2] and cerebral perfusion [3], often requires mechanical ventilation, and is associated with neurodevelopmental impairment [4]. Apnea-related hypoxemia is of major concern in light of evidence that repetitive hypoxia in newborn animals results in irreversibly-altered carotid body function [5], raising the possibility of impaired ventilatory control, and causes neurocognitive and behavioural deficits [6]. Respiratory arrest and hypoxemia are also strongly implicated in sudden infant death syndrome (SIDS) [7],[8] where the speed at which hypoxemia develops is considered to be particularly dangerous.
In preterm infants, the rate of arterial O2 desaturation (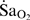 ) can be highly variable and rapid, with average rates as high as 4.3% s−1 during isolated apneas [9]. An earlier framework to describe
) can be highly variable and rapid, with average rates as high as 4.3% s−1 during isolated apneas [9]. An earlier framework to describe 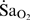 proposed that metabolic O2 consumption relative to alveolar volume determines the speed at which alveolar
proposed that metabolic O2 consumption relative to alveolar volume determines the speed at which alveolar 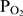 falls [10]; it was envisaged that
falls [10]; it was envisaged that 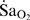 is then a function of falling
is then a function of falling 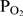 and the slope of the oxy-hemoglobin dissociation curve. However, such a model assumes that the rate of alveolar depletion of O2, denoted pulmonary O2 uptake (
and the slope of the oxy-hemoglobin dissociation curve. However, such a model assumes that the rate of alveolar depletion of O2, denoted pulmonary O2 uptake (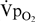 ), is equal to tissue O2 consumption during apnea (see Methods – Theory). Previous studies in adults have shown that
), is equal to tissue O2 consumption during apnea (see Methods – Theory). Previous studies in adults have shown that 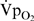 falls from metabolic consumption during apnea [11], and our previous modeling studies in lambs showed that the difference between
falls from metabolic consumption during apnea [11], and our previous modeling studies in lambs showed that the difference between 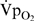 and metabolic O2 consumption has a crucial role in determining
and metabolic O2 consumption has a crucial role in determining 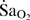 during recurrent apneas [12]. We found that apneic changes in
during recurrent apneas [12]. We found that apneic changes in 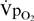 cause desaturation to occur in 2 stages. During stage 1, lung O2 stores are depleted, and
cause desaturation to occur in 2 stages. During stage 1, lung O2 stores are depleted, and 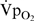 falls below metabolic consumption. During stage 2,
falls below metabolic consumption. During stage 2, 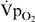 is close to zero, and tissue O2 needs are provided by depletion of blood O2 stores.
is close to zero, and tissue O2 needs are provided by depletion of blood O2 stores.
To date, no complete theoretical analysis of the factors influencing desaturation during apnea has been published. The only available study [13] has a number of critical limitations. First, the model incorporated a constraint of a fixed difference between 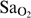 and mixed-venous saturation; thus dynamic changes in
and mixed-venous saturation; thus dynamic changes in 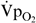 could not occur and their influence on
could not occur and their influence on 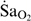 could not be examined. Second, no assessment was made of the impact of cardiorespiratory factors on the two stages of O2 desaturation. Third, in focusing on adults, the study did not examine profound desaturation to levels well below 60% as can often occur in preterm infants [9],[14].
could not be examined. Second, no assessment was made of the impact of cardiorespiratory factors on the two stages of O2 desaturation. Third, in focusing on adults, the study did not examine profound desaturation to levels well below 60% as can often occur in preterm infants [9],[14].
Accordingly, the aim of the current study was to quantify the importance of cardiorespiratory factors relevant to 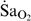 during apnea, with particular reference to the preterm infant. Using a model that permits variation of
during apnea, with particular reference to the preterm infant. Using a model that permits variation of 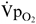 during apnea, we examine a number of factors known to influence
during apnea, we examine a number of factors known to influence 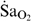 , such as lung volume [15], metabolic O2 consumption [16] and pre-apneic arterial oxygenation [17] as well as factors that are particularly pertinent for the developing newborn, including anemia, hypovolemia, reduced O2 affinity, and chronically and acutely reduced cardiac output. We use the results to develop a conceptual framework for the interpretation of mechanisms underlying rapid
, such as lung volume [15], metabolic O2 consumption [16] and pre-apneic arterial oxygenation [17] as well as factors that are particularly pertinent for the developing newborn, including anemia, hypovolemia, reduced O2 affinity, and chronically and acutely reduced cardiac output. We use the results to develop a conceptual framework for the interpretation of mechanisms underlying rapid 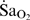 during apnea.
during apnea.
Results
Overview of the two-compartment model for gas exchange
To determine the independent influence of clinically relevant cardiorespiratory factors on 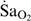 during a single isolated apnea, we used a two-compartment lung-body mathematical model which incorporated realistic blood O2 stores and gas exchange dynamics (Figure 1), as described in Methods – Mathematical model (a full list of symbols is provided in Table 1). We used published parameters for healthy preterm infants born at ∼30 wk gestational age (Table 2); the values represent measurements taken at approximately term equivalent age when surprisingly rapid desaturation has been observed [9]. We also derive analytic solutions for
during a single isolated apnea, we used a two-compartment lung-body mathematical model which incorporated realistic blood O2 stores and gas exchange dynamics (Figure 1), as described in Methods – Mathematical model (a full list of symbols is provided in Table 1). We used published parameters for healthy preterm infants born at ∼30 wk gestational age (Table 2); the values represent measurements taken at approximately term equivalent age when surprisingly rapid desaturation has been observed [9]. We also derive analytic solutions for 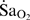 to quantify the importance of cardiorespiratory factors on
to quantify the importance of cardiorespiratory factors on 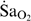 to obtain a detailed view of the arterial O2 desaturation process, as described in Methods – Theory.
to obtain a detailed view of the arterial O2 desaturation process, as described in Methods – Theory.
Figure 1. Model schematic representing O2 uptake, transport and consumption.

O2 stores are represented by the alveolar, arterial, and venous compartments. Two dynamically-independent levels of O2 uptake are denoted: pulmonary O2 uptake (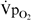 ) and metabolic consumption (
) and metabolic consumption (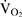 ). R-L shunt is also included. Ta is the arterial transit time. Symbols are described in Table 1.
). R-L shunt is also included. Ta is the arterial transit time. Symbols are described in Table 1.
Table 1. Mathematical symbols.
| Symbol | Description |

|
Arterial CO2 content |
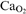
|
Arterial O2 content |
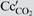
|
End-capillary arterial CO2 content |
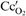
|
End-capillary arterial O2 content |
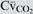
|
Mixed venous CO2 content |
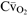
|
Mixed venous O2 content |
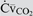
|
Rate of change in mixed venous CO2 content |
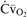
|
Rate of change in mixed venous O2 content |
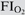
|
Fractional inspired O2 |
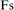
|
R-L pulmonary shunt fraction (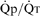 ) ) |
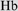
|
Hemoglobin content of blood |
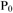
|
Barometric pressure, including conversion from STP to BTP, 863 mmHg |
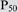
|
O2 partial pressure at 50% saturation |
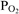
|
O2 partial pressure |

|
Arterial CO2 partial pressure |
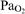
|
Arterial O2 partial pressure |

|
Alveolar CO2 partial pressure |
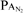
|
Alveolar N2 partial pressure |

|
Alveolar O2 partial pressure |
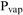
|
Alveolar water vapour partial pressure, 47 mmHg |
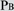
|
Barometric pressure, 760 mmHg |
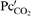
|
End-capillary CO2 partial pressure |
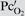
|
End-capillary O2 partial pressure |
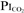
|
Inspired CO2 partial pressure |

|
Inspired N2 partial pressure |
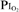
|
Inspired O2 partial pressure |
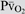
|
Mixed venous O2 partial pressure |
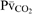
|
Mixed venous CO2 partial pressure |

|
Rate of change in alveolar CO2 partial pressure |
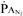
|
Rate of change in alveolar N2 partial pressure |
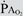
|
Rate of change in alveolar O2 partial pressure |
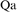
|
Arterial volume |
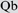
|
Blood volume |
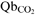
|
Blood volume for CO2 |
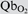
|
Blood volume for O2 |
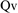
|
Venous (and tissue) volume |
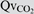
|
Venous (and tissue) volume for CO2 |
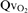
|
Venous (and tissue) volume for O2 |
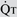
|
Cardiac output |
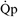
|
Pulmonary blood flow |
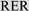
|
Respiratory exchange ratio (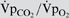 ) ) |
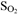
|
O2 saturation |

|
Arterial O2 saturation |
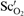
|
End-capillary arterial O2 saturation |
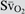
|
Mixed venous O2 saturation |
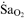
|
Rate of arterial O2 desaturation |

|
Average  from t = 0–10 s during apnea from t = 0–10 s during apnea |
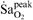
|
Peak instantaneous (‘linear’) 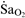 during apnea; stage 1 during apnea; stage 1 |
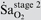
|
 during stage 2 during stage 2 |
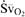
|
Rate of mixed-venous O2 desaturation |
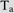
|
Arterial transit time |
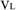
|
Lung volume |
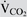
|
Metabolic CO2 production |
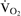
|
Metabolic O2 consumption |

|
Expired alveolar ventilation |
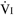
|
Inspired alveolar ventilation |
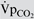
|
Pulmonary CO2 uptake (from capillary to alveoli) |
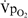
|
Pulmonary O2 uptake (from alveoli to capillary) |

|
Net pulmonary gas uptake from alveoli to capillary |
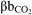
|
Capacitance co-efficient of blood for CO2 |

|
Capacitance co-efficient of blood for O2 relating changes in 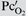 to changes in to changes in 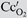
|
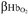
|
Capacitance co-efficient of hemoglobin for O2; slope of the O2-dissociation curve relating changes in 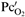 to changes in to changes in 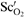
|
Table 2. Typical parameters for the preterm infant at term equivalent age.
| Parameter | Value | Reference/s |
Lung volume (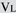 ) ) |
20 ml kg−1 | [31],[54] |
Metabolic O2 consumption (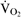 ) ) |
10 ml min−1 kg−1 | [55],[39],[40] |
Cardiac output (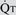 ) ) |
250 ml min−1 kg−1 | [56] |
| Hemoglobin content (Hb) | 8 g dl−1 | [22] |
| P50 | 24 mmHg | [22] |
Blood volume (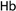 ) ) |
80 ml kg−1 | [57] |
P50 is the partial pressure at 50% saturation. 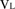 is taken from data on functional residual capacity. For all simulations unless otherwise stated: respiratory exchange ratio (RER) was assumed to be 0.8; shunt fraction (Fs) was adjusted to 8.7% to achieve a resting
is taken from data on functional residual capacity. For all simulations unless otherwise stated: respiratory exchange ratio (RER) was assumed to be 0.8; shunt fraction (Fs) was adjusted to 8.7% to achieve a resting 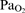 of 72 mmHg as is typical for normal healthy infants [58]; resting alveolar ventilation (
of 72 mmHg as is typical for normal healthy infants [58]; resting alveolar ventilation ( under normal conditions) was set to achieve resting
under normal conditions) was set to achieve resting 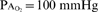 .
.
Pulmonary gas exchange dynamics during apnea
To examine changes in O2/CO2 exchange during apnea, a single apnea was imposed on the model. During apnea, changes in alveolar O2 and CO2 stores are not constant (Figure 2); importantly, alveolar 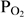 (
(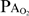 ) did not continue to fall at its initial rate as governed by metabolic O2 consumption (
) did not continue to fall at its initial rate as governed by metabolic O2 consumption (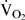 ), but instead the rate of fall in
), but instead the rate of fall in 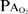 was reduced as it approached mixed venous
was reduced as it approached mixed venous 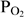 (
(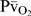 ), an observation also reflected in the falling
), an observation also reflected in the falling 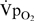 . As a result, two distinct phases for O2 depletion can be seen, which we refer to as stage 1 and stage 2 [12]. During stage 1,
. As a result, two distinct phases for O2 depletion can be seen, which we refer to as stage 1 and stage 2 [12]. During stage 1, 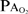 fell rapidly and
fell rapidly and 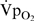 decreased and became dissociated from
decreased and became dissociated from 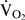 ; during stage 2, with
; during stage 2, with 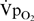 greatly reduced, both
greatly reduced, both 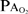 and
and 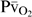 fell together at a reduced rate. The two distinct phases were also observed for alveolar and arterial
fell together at a reduced rate. The two distinct phases were also observed for alveolar and arterial 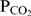 (
(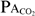 ,
, 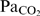 ) although stage 1 for CO2 was substantially shorter than that for O2. Such an effect results from the earlier fall in pulmonary CO2 uptake (
) although stage 1 for CO2 was substantially shorter than that for O2. Such an effect results from the earlier fall in pulmonary CO2 uptake (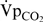 ) relative to the fall in
) relative to the fall in 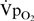 (Figure 2A) and is reflected in the reduction in respiratory exchange ratio (
(Figure 2A) and is reflected in the reduction in respiratory exchange ratio (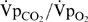 ) (Figure 2B). Consequently, a more rapid fall in
) (Figure 2B). Consequently, a more rapid fall in 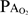 was observed compared with the rise in
was observed compared with the rise in 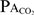 (see Methods – Derivation of equations), such that
(see Methods – Derivation of equations), such that 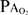 fell by 100 mmHg in the time
fell by 100 mmHg in the time 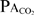 rose by just 14 mmHg (Figure 2C).
rose by just 14 mmHg (Figure 2C).
Figure 2. Pulmonary gas exchange during apnea.

(A) Rate of pulmonary O2/CO2 exchange. 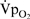 and
and 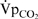 fall from resting levels during apnea. (B) Net alveolar-capillary gas uptake (
fall from resting levels during apnea. (B) Net alveolar-capillary gas uptake (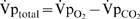 ) and respiratory exchange ratio (
) and respiratory exchange ratio (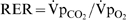 )during apnea. (C) Changes in alveolar, arterial and mixed venous
)during apnea. (C) Changes in alveolar, arterial and mixed venous 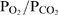 during apnea. Contrast the time-course in
during apnea. Contrast the time-course in 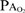 and
and 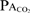 as they fall/rise towards
as they fall/rise towards 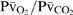 . (*) represents the fall in
. (*) represents the fall in 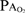 if
if 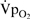 was assumed equal to
was assumed equal to 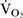 . S1 = stage 1; S2 = stage 2.
. S1 = stage 1; S2 = stage 2.
Time-course of 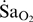 during apnea
during apnea
The time-course of 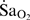 is complex (Figure 3), a consequence of the nonlinear O2-dissociation curve in combination with the fall in
is complex (Figure 3), a consequence of the nonlinear O2-dissociation curve in combination with the fall in 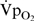 . At apnea onset,
. At apnea onset, 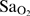 started to fall with a rate equivalent to that predicted by Equation 12, where
started to fall with a rate equivalent to that predicted by Equation 12, where 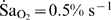 (Figure 3). During apnea, changes in the slope of the O2-dissociation curve (
(Figure 3). During apnea, changes in the slope of the O2-dissociation curve (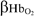 ) and
) and 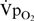 dominated the time-course of desaturation as hypoxemia progressed. As
dominated the time-course of desaturation as hypoxemia progressed. As 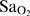 started to fall after apnea onset,
started to fall after apnea onset, 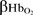 increased with little change in
increased with little change in 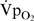 , resulting in a proportional increase in
, resulting in a proportional increase in 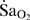 . However, as arterial hypoxemia developed, there was a concurrent decline in
. However, as arterial hypoxemia developed, there was a concurrent decline in 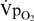 . As
. As 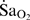 is directly proportional to the product
is directly proportional to the product 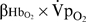 (Equation 11) it follows that during apnea, the peak
(Equation 11) it follows that during apnea, the peak 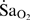 of 3.5% s−1 occurred when
of 3.5% s−1 occurred when 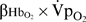 reached a maximum. This occurred when neither
reached a maximum. This occurred when neither 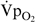 nor
nor 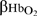 was at its maximum (both ∼50% of peak). Finally, with
was at its maximum (both ∼50% of peak). Finally, with 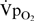 greatly reduced during stage 2,
greatly reduced during stage 2, 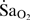 remained at a constant level (
remained at a constant level (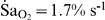 ), close to that predicted by Equation 13 (1.8% s−1).
), close to that predicted by Equation 13 (1.8% s−1).
Figure 3. The time course of 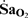 during apnea.
during apnea.

Panel (A) shows the increase in the slope of the oxy-hemoglobin dissocation curve at the level of alveolar 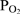 (
(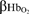 ), and the fall in pulmonary oxygen uptake (
), and the fall in pulmonary oxygen uptake (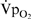 ) that occurs during apnea. Panel (B) shows that changes in the product
) that occurs during apnea. Panel (B) shows that changes in the product 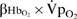 explain the time course of the instantaneous slope of arterial O2 desaturation (
explain the time course of the instantaneous slope of arterial O2 desaturation (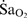 ) during apnea. Note that the peak
) during apnea. Note that the peak 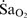 occurs when
occurs when 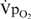 is substantially less than its resting value. Note also that the rate of fall of mixed-venous saturation (
is substantially less than its resting value. Note also that the rate of fall of mixed-venous saturation (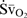 ) and
) and 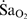 become equal and constant after 20 s.
become equal and constant after 20 s.
Factors influencing 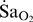
The following parameters were individually varied from their ‘normal’ values to quantify their influence on 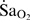 : resting
: resting 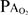 , lung volume (
, lung volume (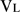 ), metabolic O2 consumption (
), metabolic O2 consumption (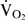 ), blood hemoglobin content (Hb), cardiac output (
), blood hemoglobin content (Hb), cardiac output (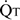 ), R-L shunt fraction (Fs), and the
), R-L shunt fraction (Fs), and the 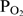 at 50% saturation (P50). All other parameters were kept constant to remove confounding effects, unless specified otherwise.
at 50% saturation (P50). All other parameters were kept constant to remove confounding effects, unless specified otherwise.
To quantify 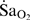 we used 3 different measures. First, since apnea is considered clinically significant if it lasts for >10 s and is accompanied by bradycardia or O2 desaturation [18], we calculated the average rate of fall in
we used 3 different measures. First, since apnea is considered clinically significant if it lasts for >10 s and is accompanied by bradycardia or O2 desaturation [18], we calculated the average rate of fall in 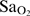 between apnea onset and 10 s later (
between apnea onset and 10 s later (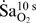 ); such a measure describes the immediacy of onset of desaturation and is analogous to the practical measurement of average
); such a measure describes the immediacy of onset of desaturation and is analogous to the practical measurement of average 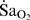 used in many clinical studies [9],[15],[19],[20]. Second, we determined the peak instantaneous
used in many clinical studies [9],[15],[19],[20]. Second, we determined the peak instantaneous 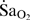 during apnea (
during apnea (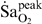 ), the value during the linear portion of arterial desaturation [10],[21] which we find is not confounded by resting
), the value during the linear portion of arterial desaturation [10],[21] which we find is not confounded by resting 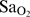 . Third, we report a measure of
. Third, we report a measure of 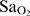 during stage 2 apnea (
during stage 2 apnea (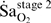 ). To quantify the sensitivity of
). To quantify the sensitivity of 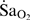 to changes in each cardiorespiratory factor, we defined the term impact ratio as the ratio of proportional increase in
to changes in each cardiorespiratory factor, we defined the term impact ratio as the ratio of proportional increase in 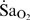 to a small increase from the normal value of each factor. For example, an impact ratio of 1 indicates a one-to-one increase in
to a small increase from the normal value of each factor. For example, an impact ratio of 1 indicates a one-to-one increase in 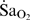 with an increase in the factor, and a negative ratio indicates an inverse relationship. The impact of each cardiorespiratory factor on
with an increase in the factor, and a negative ratio indicates an inverse relationship. The impact of each cardiorespiratory factor on 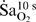 ,
, 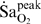 , and
, and 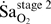 is summarised in Table 3.
is summarised in Table 3.
Table 3. Impact ratios describing the effect of cardiorespiratory factors on 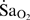 .
.
| Parameter alteration |
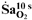
|
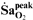
|
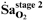
|
Resting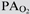
|
−3.97 | −0.35 | −0.01 |
| Lung volume (VL) | −2.24 | −0.82 | −0.09 |
| Blood volume (Qb) | −0.01 | −0.06 | −0.68 |
O2 consumption (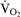 ) CC ) CC
|
+2.29 | +1.00 | +1.00 |
O2 consumption ( ) nCC ) nCC
|
+2.73 | +1.92 | +1.00 |
| Hemoglobin content (Hb) nCC | −0.38 | −1.00 | −0.89 |
| Hemoglobin content (Hb) CC | +0.01 | −0.10 | −0.89 |
| P50 | +1.37 | −0.68 | −0.11 |
Cardiac output (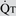 , resting) , resting) |
−0.39 | −0.90 | 0.00 |
Cardiac output (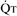 , transient) , transient) |
+1.45 | −0.06 | 0.00 |
| Shunt Fraction (Fs) | −0.01 | +0.01 | 0.00 |
Impact ratio is defined as the ratio of proportional increase in 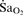 to the proportional increase in each factor, based on small changes around normal values. An impact ratio of 1 indicates a one-to-one increase in
to the proportional increase in each factor, based on small changes around normal values. An impact ratio of 1 indicates a one-to-one increase in 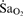 >with an increase in the factor, and a negative ratio indicates an inverse relationship. CC = cardiac compensated, nCC = cardiac uncompensated.
>with an increase in the factor, and a negative ratio indicates an inverse relationship. CC = cardiac compensated, nCC = cardiac uncompensated.
Resting 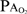
Changes in 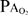 , achieved via reduced resting ventilation or increasing inspired O2 (
, achieved via reduced resting ventilation or increasing inspired O2 (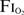 ), had a substantial effect on the onset of desaturation. Reduced pre-apneic
), had a substantial effect on the onset of desaturation. Reduced pre-apneic 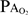 dramatically increased
dramatically increased 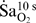 (Figure 4A), but had little effect on
(Figure 4A), but had little effect on 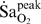 or
or 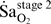 . In contrast, increasing pre-apneic
. In contrast, increasing pre-apneic 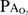 with the application of supplemental O2 achieved the opposite, essentially right-shifting or delaying the arterial desaturation curve, where one second of delay can be achieved by an increase in
with the application of supplemental O2 achieved the opposite, essentially right-shifting or delaying the arterial desaturation curve, where one second of delay can be achieved by an increase in 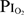 (
(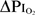 ) of ∼7 mmHg, or
) of ∼7 mmHg, or 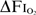 of ∼1% (see Methods – Derivation of equations). These results occurred despite only a minor influence being visible on resting
of ∼1% (see Methods – Derivation of equations). These results occurred despite only a minor influence being visible on resting 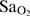 . For example, a reduction of
. For example, a reduction of 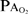 from 100 to 60 mmHg caused a 6% reduction in resting
from 100 to 60 mmHg caused a 6% reduction in resting 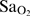 but at the same time led to a more than 2-fold elevation in
but at the same time led to a more than 2-fold elevation in 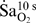 (Figure 4B). Additionally, a severe reduction in
(Figure 4B). Additionally, a severe reduction in 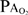 , to below 70 mmHg, was required to elevate
, to below 70 mmHg, was required to elevate 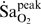 .
.
Figure 4. Impact of pre-apneic alveolar 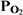 (ventilation, supplemental O2) on
(ventilation, supplemental O2) on 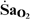 .
.

(A) Effect of three levels of alveolar 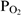 (
(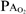 ), (i) 100 mmHg, (ii) 80 mmHg and (iii) 60 mmHg, on arterial (
), (i) 100 mmHg, (ii) 80 mmHg and (iii) 60 mmHg, on arterial (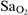 ) and mixed venous (
) and mixed venous (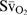 ) O2 desaturation during apnea. Note that arterial O2 desaturation is substantially right-shifted with increased
) O2 desaturation during apnea. Note that arterial O2 desaturation is substantially right-shifted with increased 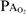 . (B) Sensitivity of
. (B) Sensitivity of 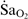 to changes in pre-apneic
to changes in pre-apneic 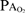 (
(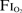 ). Note that reduced
). Note that reduced 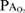 has a major impact on
has a major impact on 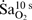 but little impact on
but little impact on 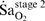 ; the influence on
; the influence on 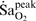 is small in the normal range but becomes stronger at low
is small in the normal range but becomes stronger at low 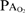 . n = ‘normal’ 'values; S1, stage 1 slope; S2, stage 2 slope.
. n = ‘normal’ 'values; S1, stage 1 slope; S2, stage 2 slope.
Lung volume (VL) and blood volume (Qb)
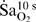 and
and 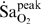 were inversely related to VL during stage 1 (Figure 5A, B), but changes in VL had no influence on
were inversely related to VL during stage 1 (Figure 5A, B), but changes in VL had no influence on 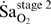 . In direct contrast, reduced Qb strongly increased
. In direct contrast, reduced Qb strongly increased 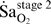 , but had no effect on stage 1 desaturation as reflected in no change in
, but had no effect on stage 1 desaturation as reflected in no change in 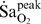 or
or 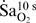 (Figure 5C, D).
(Figure 5C, D).
Figure 5. Impact of lung volume (VL) and blood volume (Qb) on 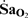 .
.

(A) Effect of three levels of VL, (i) 30, (ii) 20 and (iii) 10 ml kg−1, on arterial (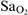 ) and mixed venous (
) and mixed venous (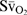 ) O2 desaturation during apnea. (B) Sensitivity of
) O2 desaturation during apnea. (B) Sensitivity of 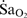 to changes in VL. Note that reduced VL has a strong impact on
to changes in VL. Note that reduced VL has a strong impact on 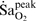 and
and 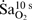 but no impact on
but no impact on 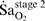 . (C) Effect of three levels of Qb, (iv) 120, (v) 80 and (iv) 40 ml kg−1, on
. (C) Effect of three levels of Qb, (iv) 120, (v) 80 and (iv) 40 ml kg−1, on 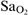 and
and 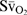 during apnea. (D) Sensitivity of
during apnea. (D) Sensitivity of 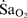 to changes in Qb. Note that reduced Qb has little impact on
to changes in Qb. Note that reduced Qb has little impact on 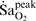 or
or 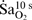 but has a large impact on
but has a large impact on 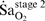 . n = ‘normal’ values; S1, stage 1 slope; S2, stage 2 slope.
. n = ‘normal’ values; S1, stage 1 slope; S2, stage 2 slope.
Metabolic O2 consumption (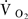 )
)
To examine the impact of changing 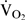 on
on 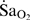 , independent of resting
, independent of resting 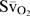 ,
, 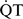 was adjusted to maintain resting
was adjusted to maintain resting 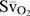 constant, where
constant, where 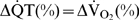 ; we refer to this procedure as ‘cardiac compensation’. Under this condition, elevated
; we refer to this procedure as ‘cardiac compensation’. Under this condition, elevated 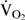 caused a directly proportional increase in
caused a directly proportional increase in 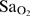 throughout stages 1 and 2 (Figure 6A, B). Without cardiac compensation, the effect of increased
throughout stages 1 and 2 (Figure 6A, B). Without cardiac compensation, the effect of increased 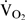 on
on 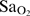 during stage 1 was magnified, as shown by the further increase in
during stage 1 was magnified, as shown by the further increase in 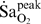 (Figure 6A, B).
(Figure 6A, B).
Figure 6. Impact of metabolic O2 consumption (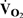 ) on
) on 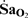 .
.

Panel (A) shows the effect of doubling 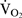 on arterial (
on arterial (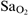 ) and mixed venous (
) and mixed venous (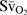 ) O2 during apnea; (i) 10 ml min−1kg−1, (ii) 20 ml min−1kg−1 with cardiac compensation (CC), and (iii) 20 ml min−1kg−1 with no CC (nCC). Note that with CC, increased
) O2 during apnea; (i) 10 ml min−1kg−1, (ii) 20 ml min−1kg−1 with cardiac compensation (CC), and (iii) 20 ml min−1kg−1 with no CC (nCC). Note that with CC, increased 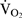 , from (i) to (ii), elevated
, from (i) to (ii), elevated 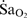 uniformly at all levels of
uniformly at all levels of 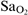 during both stages 1 and 2; note that the level of
during both stages 1 and 2; note that the level of 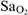 at the inflection point (shown by short black lines) is unchanged. With nCC (iii), increased
at the inflection point (shown by short black lines) is unchanged. With nCC (iii), increased 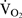 caused a reduced resting
caused a reduced resting 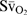 and lower
and lower 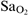 inflection, and greater
inflection, and greater 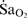 during stage 1, compared to (ii). (B) Sensitivity of
during stage 1, compared to (ii). (B) Sensitivity of 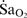 to changes in
to changes in 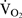 . Note that with increased
. Note that with increased 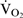 : a uniform increase in
: a uniform increase in 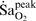 occurred with CC, and a more-than-proportional increase was seen with nCC;
occurred with CC, and a more-than-proportional increase was seen with nCC; 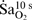 is elevated in both cases, but more so with nCC; a uniform increase in
is elevated in both cases, but more so with nCC; a uniform increase in 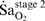 is shown regardless of CC. n = ‘normal’ values; S1, stage 1 slope; S2, stage 2 slope.
is shown regardless of CC. n = ‘normal’ values; S1, stage 1 slope; S2, stage 2 slope.
Hemoglobin content (Hb) and oxygen affinity (P50)
Reduced hemoglobin content (Hb) increased 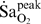 and
and 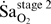 but had little effect on
but had little effect on 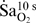 (Figure 7A, B). The increase in
(Figure 7A, B). The increase in 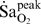 occurred with an increase in the peak of the product
occurred with an increase in the peak of the product 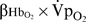 as
as 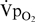 was higher at each level of
was higher at each level of 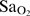 . The simulation was repeated with cardiac compensation for the reduction in hemoglobin content, where
. The simulation was repeated with cardiac compensation for the reduction in hemoglobin content, where 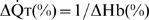 , to maintain constant resting
, to maintain constant resting 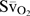 . Following such compensation, no changes in
. Following such compensation, no changes in 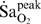 or
or 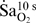 were observed but reduced Hb continued to increase
were observed but reduced Hb continued to increase 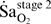 . In examining the influence of P50, P90 was adjusted in equal proportion on the basis of published data [22]. Increased P50 increased the immediate
. In examining the influence of P50, P90 was adjusted in equal proportion on the basis of published data [22]. Increased P50 increased the immediate 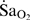 , increased
, increased 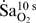 , decreased
, decreased 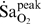 and had no effect on
and had no effect on 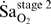 (Figure 7C, D).
(Figure 7C, D).
Figure 7. Impact of hemoglobin content (Hb) and O2 affinity (P50) on 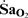 .
.

(A) Effect of three levels of Hb, (i) 12 g dl−1, (ii) 8 g dl−1 and (iii) 4 g dl−1, on arterial (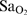 ) and mixed venous (
) and mixed venous (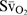 ) O2 desaturation during apnea. Note the fall in
) O2 desaturation during apnea. Note the fall in 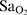 at the inflection point (shown by short black lines). Note also that the reduced Hb has little impact on desaturation above
at the inflection point (shown by short black lines). Note also that the reduced Hb has little impact on desaturation above 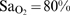 . (B) Sensitivity of
. (B) Sensitivity of 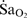 to changes in Hb. (C) Effect of three levels of P50, (iv) 18 mmHg, (v) 24 mmHg, and (vi) 36 mmHg, on
to changes in Hb. (C) Effect of three levels of P50, (iv) 18 mmHg, (v) 24 mmHg, and (vi) 36 mmHg, on 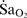 . (D) Sensitivity of
. (D) Sensitivity of 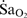 to changes in P50. n = ‘normal’ values; S1, stage 1 slope; S2, stage 2 slope.
to changes in P50. n = ‘normal’ values; S1, stage 1 slope; S2, stage 2 slope.
Cardiac output (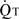 )
)
Reduced resting 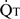 increased
increased 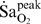 , but had little impact on
, but had little impact on 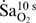 or
or 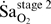 (Figure 8A, B). As with Hb, the increase in
(Figure 8A, B). As with Hb, the increase in 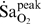 with reduced resting
with reduced resting  occurred with an increase in the peak of the product
occurred with an increase in the peak of the product 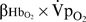 . To differentiate between the influence on
. To differentiate between the influence on 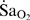 of an acute reduction in cardiac output, i.e. when bradycardia accompanies apnea, rather than a chronic reduction, we reduced cardiac output in a step-wise manner from the baseline value at the time of apnea onset. In constrast to the effect of reduced resting
of an acute reduction in cardiac output, i.e. when bradycardia accompanies apnea, rather than a chronic reduction, we reduced cardiac output in a step-wise manner from the baseline value at the time of apnea onset. In constrast to the effect of reduced resting 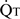 , a transient reduction in
, a transient reduction in 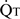 decreased
decreased 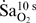 , but had a negligible impact on
, but had a negligible impact on 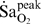 or
or 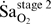 (Figure 8C, D).
(Figure 8C, D).
Figure 8. Impact of cardiac output (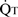 ) on
) on 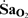 .
.

(A) Effect of three levels of resting 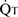 , (i) 375 ml min−1kg−1, (ii) 250 ml min−1kg−1, and (iii) 125 ml min−1kg−1, on arterial (
, (i) 375 ml min−1kg−1, (ii) 250 ml min−1kg−1, and (iii) 125 ml min−1kg−1, on arterial (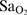 ) and mixed venous (
) and mixed venous (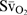 ) O2 during apnea. Note that reduced
) O2 during apnea. Note that reduced 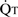 elevates
elevates 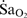 , associated with a reduction in resting
, associated with a reduction in resting 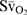 and reduction in
and reduction in 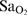 at the stage 1–2 transition or inflection point (shown by short black lines). (B) Sensitivity of
at the stage 1–2 transition or inflection point (shown by short black lines). (B) Sensitivity of 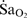 to changes in
to changes in 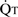 . Note the strong influence of
. Note the strong influence of 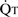 on
on 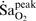 , but negligible effect on
, but negligible effect on 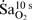 and
and 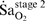 . (C) Simulations in (A) repeated for a step change in
. (C) Simulations in (A) repeated for a step change in 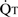 at apnea onset by (iv) +125 ml min−1kg−1 (e.g. tachycardia), (v) 0 ml min−1kg−1, and (vi) −125 ml min−1kg−1 (e.g. bradycardia), following resting
at apnea onset by (iv) +125 ml min−1kg−1 (e.g. tachycardia), (v) 0 ml min−1kg−1, and (vi) −125 ml min−1kg−1 (e.g. bradycardia), following resting 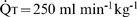 . Note that the transient effect of
. Note that the transient effect of 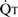 is opposite to the resting effect of
is opposite to the resting effect of 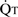 on arterial desaturation during apnea. (D) Sensitivity of
on arterial desaturation during apnea. (D) Sensitivity of 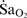 to acute changes in
to acute changes in 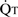 during apnea. Note the strong influence of a step-change in
during apnea. Note the strong influence of a step-change in 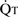 on
on 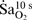 , but negligible effect on
, but negligible effect on 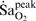 and
and 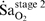 . n = ‘normal’ values.
. n = ‘normal’ values.
Resting R-L shunt fraction (Fs)
Increased Fs reduced resting 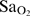 and
and 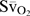 but had no effect on
but had no effect on 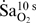 ,
, 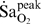 , or
, or 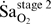 (Figure 9A, B).
(Figure 9A, B).
Figure 9. Impact of R-L shunt (Fs) on 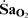 .
.

(A) Effect of three levels of Fs, (i) 0%, (ii) 15%, and (iii) 30%, on arterial (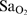 ) and mixed venous (
) and mixed venous (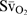 ) O2 during apnea. Note that resting R-L shunt fraction has a negligible impact on
) O2 during apnea. Note that resting R-L shunt fraction has a negligible impact on 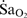 during apnea. (B) Sensitivity of
during apnea. (B) Sensitivity of 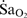 to changes in Fs. n = ‘normal’ values; S1, stage 1 slope; S2, stage 2 slope.
to changes in Fs. n = ‘normal’ values; S1, stage 1 slope; S2, stage 2 slope.
Discussion
Our model analysis of the rate of arterial O2 desaturation during apnea demonstrates that pre-apneic ventilation, lung volume, cardiac output, hemoglobin content and blood volume exert unique effects on 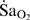 throughout the time-course of desaturation, while metabolic O2 consumption is uniformly influential throughout the process. Our analysis reveals that lung volume and the slope of the O2-dissociation curve are important early in the process, during what we refer to as stage 1 [12], but not stage 2. For the first time, our study reveals that reduced cardiac output and hemoglobin content, and as a consequence resting mixed-venous saturation, substantially accelerate peak
throughout the time-course of desaturation, while metabolic O2 consumption is uniformly influential throughout the process. Our analysis reveals that lung volume and the slope of the O2-dissociation curve are important early in the process, during what we refer to as stage 1 [12], but not stage 2. For the first time, our study reveals that reduced cardiac output and hemoglobin content, and as a consequence resting mixed-venous saturation, substantially accelerate peak 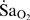 . Finally, low blood volume and hemoglobin content, and therefore a low total blood O2 capacity, increase the speed of desaturation, but only in stage 2. In addition to infants with elevated metabolic needs and low lung volume, those with anemia, cardiac dysfunction, or hypovolemia, which are common complications of prematurity, are at heightened risk of rapid and profound arterial desaturation during apnea.
. Finally, low blood volume and hemoglobin content, and therefore a low total blood O2 capacity, increase the speed of desaturation, but only in stage 2. In addition to infants with elevated metabolic needs and low lung volume, those with anemia, cardiac dysfunction, or hypovolemia, which are common complications of prematurity, are at heightened risk of rapid and profound arterial desaturation during apnea.
Methodological considerations
To evaluate the independent effects of cardiorespiratory factors on 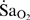 we used a two-compartment model, incorporating both alveolar and blood gas stores. The inclusion of a realistic blood store was crucial to reveal that changes in
we used a two-compartment model, incorporating both alveolar and blood gas stores. The inclusion of a realistic blood store was crucial to reveal that changes in 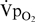 occur as a consequence of arterial and mixed-venous saturation falling asynchronously during apnea (Figure 3). Our approach allowed us to extend the previous framework based on the assumption of constant
occur as a consequence of arterial and mixed-venous saturation falling asynchronously during apnea (Figure 3). Our approach allowed us to extend the previous framework based on the assumption of constant 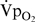 [23], which prevented the recognition that a steep O2-dissociation curve and low lung volume do not accelerate
[23], which prevented the recognition that a steep O2-dissociation curve and low lung volume do not accelerate 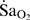 beyond stage 1. Furthermore, the varying
beyond stage 1. Furthermore, the varying 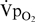 permitted recognition that cardiac output, hemoglobin content, and blood volume have a major influence on
permitted recognition that cardiac output, hemoglobin content, and blood volume have a major influence on 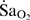 .
.
In the current study, the typical value of 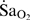 found using our model was 3.5% s−1 whereas Poets and Southall [9] using beat-by-beat oximetry in preterm infants reported a mean value for
found using our model was 3.5% s−1 whereas Poets and Southall [9] using beat-by-beat oximetry in preterm infants reported a mean value for 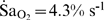 during isolated apneas. Reasons for our lower value may lie with our simplifying assumptions. Notably, we assumed a homogenous lung compartment and complete gas mixing and as such, the model incorporated neither limitation of alveolar-capillary diffusion nor an uneven ventilation-perfusion distribution, two factors that could cause an increase in
during isolated apneas. Reasons for our lower value may lie with our simplifying assumptions. Notably, we assumed a homogenous lung compartment and complete gas mixing and as such, the model incorporated neither limitation of alveolar-capillary diffusion nor an uneven ventilation-perfusion distribution, two factors that could cause an increase in 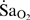 . In addition, we assumed a constant lung volume during apnea, equal to published values of functional residual capacity, whereas it is known that lung volume can fall during apnea [15],[24]; based on our data, a fall in lung volume to 15.5 ml min−1kg−1 immediately after apnea onset would achieve
. In addition, we assumed a constant lung volume during apnea, equal to published values of functional residual capacity, whereas it is known that lung volume can fall during apnea [15],[24]; based on our data, a fall in lung volume to 15.5 ml min−1kg−1 immediately after apnea onset would achieve 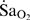 of 4.3% s−1 (Figure 5B).
of 4.3% s−1 (Figure 5B).
A final assumption implicit in our model is that all O2 transfer to the blood occurs via the pulmonary circulation. However, in very preterm infants there is evidence of percutaneous respiration in the first few days of life in both room air and with supplemental O2
[25]. With whole body exposure of 90% O2 to the newborn skin, it has been calculated that 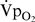 can be reduced by 8–10% [26], likely via an increased resting mixed-venous saturation; our study demonstrates that such an effect would decrease
can be reduced by 8–10% [26], likely via an increased resting mixed-venous saturation; our study demonstrates that such an effect would decrease 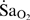 during apnea.
during apnea.
Pulmonary gas exchange dynamics during apnea
Our study is consistent with previous observations that 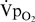 and
and 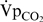 rapidly decline during apnea from their steady-state values [11], with
rapidly decline during apnea from their steady-state values [11], with 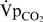 falling faster than
falling faster than 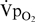 . The relatively low blood capacitance for O2 compared with that for CO2 results in the resting alveolar–mixed-venous partial pressure difference being ∼12-fold greater for O2 than for CO2. Consequently, when apnea begins ∼12 times more O2 than CO2 must diffuse across the lung to obliterate the alveolar–mixed-venous partial pressure difference. The slower fall in
. The relatively low blood capacitance for O2 compared with that for CO2 results in the resting alveolar–mixed-venous partial pressure difference being ∼12-fold greater for O2 than for CO2. Consequently, when apnea begins ∼12 times more O2 than CO2 must diffuse across the lung to obliterate the alveolar–mixed-venous partial pressure difference. The slower fall in 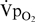 vs.
vs. 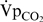 provides for a faster depletion of alveolar O2 vs. CO2 stores; such an effect results in complete desaturation of arterial blood in the time
provides for a faster depletion of alveolar O2 vs. CO2 stores; such an effect results in complete desaturation of arterial blood in the time 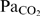 rises by just 14 mmHg. These findings lead us to conclude that short-term O2 homeostasis is more unstable than CO2 homeostasis and thus that the danger of isolated apneas in infants is likely to be mediated via hypoxemia rather than hypercapnia.
rises by just 14 mmHg. These findings lead us to conclude that short-term O2 homeostasis is more unstable than CO2 homeostasis and thus that the danger of isolated apneas in infants is likely to be mediated via hypoxemia rather than hypercapnia.
Factors influencing 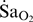
Our study provides for the first time a comprehensive analysis of the factors that determine arterial desaturation during apnea in preterm infants. We show that resting oxygenation in the form of alveolar 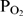 has the greatest influence on desaturation at apnea onset. When apnea begins at an increasingly lower alveolar
has the greatest influence on desaturation at apnea onset. When apnea begins at an increasingly lower alveolar 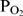 ,
, 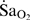 more quickly reaches its maximum because
more quickly reaches its maximum because 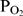 rapidly arrives at the steepest part of the O2-dissociation curve. This effect explains the inverse relationship between mean
rapidly arrives at the steepest part of the O2-dissociation curve. This effect explains the inverse relationship between mean 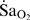 and pre-apneic
and pre-apneic 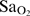 during apnea [17], but as we show the peak slope itself is negligibly affected by reduced resting
during apnea [17], but as we show the peak slope itself is negligibly affected by reduced resting 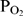 within the normal range.
within the normal range.
We demonstrate that 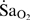 is inversely related to lung volume during stage 1 of apnea as a result of the greater reduction in alveolar
is inversely related to lung volume during stage 1 of apnea as a result of the greater reduction in alveolar 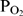 in poorly inflated lungs per unit of O2 transferred into the pulmonary capillaries. This analysis is consistent with the inverse correlation between
in poorly inflated lungs per unit of O2 transferred into the pulmonary capillaries. This analysis is consistent with the inverse correlation between 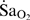 and lung volume [15], with the view that active upper airway closure maintains lung volume and slows
and lung volume [15], with the view that active upper airway closure maintains lung volume and slows 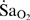 [27],[28], and with our recent report that the application of continuous positive airway pressure effectively slows
[27],[28], and with our recent report that the application of continuous positive airway pressure effectively slows 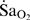 in lambs [29]. However, once stage 2 begins, the blood becomes the principal source of O2 and thus the only store which influences
in lambs [29]. However, once stage 2 begins, the blood becomes the principal source of O2 and thus the only store which influences 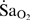 .
.
A novel finding from our study is that reduced resting mixed-venous saturation, caused by either a reduced cardiac output or reduced hemoglobin content, strongly elevates peak 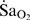 , independent of metabolic O2 consumption. We show that reduced resting mixed-venous saturation accelerates
, independent of metabolic O2 consumption. We show that reduced resting mixed-venous saturation accelerates 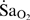 via an increase in the peak value of
via an increase in the peak value of 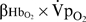 ; in other words, low mixed-venous saturation provides for a greater pulmonary O2 uptake even in the presence of a developing arterial hypoxemia, and thereby increases
; in other words, low mixed-venous saturation provides for a greater pulmonary O2 uptake even in the presence of a developing arterial hypoxemia, and thereby increases 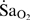 . A role for hemoglobin in determining
. A role for hemoglobin in determining 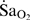 is consistent with the finding that elevated hemoglobin content in adults slows
is consistent with the finding that elevated hemoglobin content in adults slows 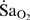 during apnea [21]. In contrast, blood transfusion to raise hemoglobin content in anemic preterm infants, a common clinical therapy, has little or no impact on the severity of apneic desaturation [30]. Our proposed explanation for the lack of benefit of raising hemoglobin content via transfusion is that it also reduces heart rate [30] and cardiac output. Thus, in the newborn, the rise in mixed-venous saturation expected after transfusion is counteracted by a tendency for mixed-venous saturation to fall as a result of reduced cardiac output. An investigation that failed to find an effect of cardiac output on
during apnea [21]. In contrast, blood transfusion to raise hemoglobin content in anemic preterm infants, a common clinical therapy, has little or no impact on the severity of apneic desaturation [30]. Our proposed explanation for the lack of benefit of raising hemoglobin content via transfusion is that it also reduces heart rate [30] and cardiac output. Thus, in the newborn, the rise in mixed-venous saturation expected after transfusion is counteracted by a tendency for mixed-venous saturation to fall as a result of reduced cardiac output. An investigation that failed to find an effect of cardiac output on 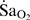 [23] did not account for our finding that pre-apneic and transient changes in cardiac output have opposing influence on
[23] did not account for our finding that pre-apneic and transient changes in cardiac output have opposing influence on 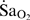 . Importantly, we find that a transient fall in cardiac output, characteristic of bradycardia during apnea in preterm infants [2], conserves alveolar O2 via reduced
. Importantly, we find that a transient fall in cardiac output, characteristic of bradycardia during apnea in preterm infants [2], conserves alveolar O2 via reduced 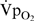 and thus reduces
and thus reduces 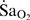 (see Equations 10 and 11). Consistent with this finding, apneic bradycardia prevents a rapid fall in
(see Equations 10 and 11). Consistent with this finding, apneic bradycardia prevents a rapid fall in 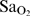 in adults [21].
in adults [21].
We found that each of the factors examined exerts a unique and therefore recognisable influence on the time course of the desaturation process (Figure 10). Low alveolar 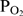 can be recognised by a left-shift of the desaturation trajectory so that desaturation begins sooner following the onset of apnea. A steep desaturation slope in the early phase of stage 1 points to a low ratio of lung volume to metabolic O2 consumption. In the late phase of stage 1, when desaturation proceeds in a linear fashion, a low resting mixed-venous saturation accelerates
can be recognised by a left-shift of the desaturation trajectory so that desaturation begins sooner following the onset of apnea. A steep desaturation slope in the early phase of stage 1 points to a low ratio of lung volume to metabolic O2 consumption. In the late phase of stage 1, when desaturation proceeds in a linear fashion, a low resting mixed-venous saturation accelerates 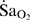 and leaves the fingerprint of a low inflection point in arterial O2 desaturation; low resting mixed-venous saturation reflects low cardiac output or hemoglobin content with respect to O2 consumption. Lastly rapid
and leaves the fingerprint of a low inflection point in arterial O2 desaturation; low resting mixed-venous saturation reflects low cardiac output or hemoglobin content with respect to O2 consumption. Lastly rapid 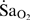 during stage 2 signifies a low total blood O2 capacity with respect to O2 consumption which would point to either low blood volume or anemia. The presence of a constant R-L shunt, while having no influence on
during stage 2 signifies a low total blood O2 capacity with respect to O2 consumption which would point to either low blood volume or anemia. The presence of a constant R-L shunt, while having no influence on 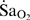 , causes a parallel downwards shift in the desaturation trajectory. The unique impact of different factors on the desaturation curve may be used to guide preventive clinical intervention.
, causes a parallel downwards shift in the desaturation trajectory. The unique impact of different factors on the desaturation curve may be used to guide preventive clinical intervention.
Figure 10. Conceptual framework depicting the temporal sequence of influence of the key cardiorespiratory factors on 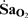 .
.

Note the regions of influence of lung volume (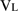 ), cardiac output (
), cardiac output (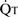 ) and blood volume (
) and blood volume (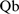 ), each with respect to metabolic O2 consumption (
), each with respect to metabolic O2 consumption (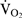 ). Hemoglobin content (Hb) influences the latter phase of stage 1 as well as stage 2. The impact of reduced
). Hemoglobin content (Hb) influences the latter phase of stage 1 as well as stage 2. The impact of reduced 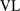 is limited to stage 1, and blood volume to stage 2. Reduced
is limited to stage 1, and blood volume to stage 2. Reduced 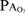 causes a leftward shift in the desaturation trajectory. Note that the point of inflection at the transition between stages reveals the resting
causes a leftward shift in the desaturation trajectory. Note that the point of inflection at the transition between stages reveals the resting 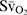 .
.
Clinical significance
We show theoretically that the lower lung volume [31] and higher metabolic O2 consumption [32] of preterm compared to term infants predisposes to a rapid onset and progression of desaturation during apnea. Two reports offer support for this view. First, rapid desaturation occurs in infants with low functional residual capacity [15], a finding that may help to explain the more frequent O2 desaturation events during active sleep [33] when functional residual capacity is reduced. Second, frequent desaturation is characteristic of preterm infants with bronchopulmonary dysplasia (BPD) [34] whose O2 consumption is 25% greater [35], and functional residual capacity is 25% less [36], than in preterm infants without BPD; Equations 11 and 12 predict that such differences increase both immediate and peak 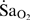 by ∼70%. In addition, hypoventilation and reduced resting
by ∼70%. In addition, hypoventilation and reduced resting 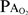 in infants with BPD, as inferred from elevated
in infants with BPD, as inferred from elevated 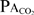 [37], further increase desaturation at apnea onset. Our finding that each rise of 1% in inspired O2 provides ∼1 s of delay (right-shift) in the onset of apneic desaturation (Equation 15) may guide the titration of supplemental O2 for the prevention of apneic hypoxemia while minimising the well known side-effects of long-term exposure to hyperoxia.
[37], further increase desaturation at apnea onset. Our finding that each rise of 1% in inspired O2 provides ∼1 s of delay (right-shift) in the onset of apneic desaturation (Equation 15) may guide the titration of supplemental O2 for the prevention of apneic hypoxemia while minimising the well known side-effects of long-term exposure to hyperoxia.
Our study has implications for the management of infants in clinical care. Metabolic O2 consumption can be elevated after feeding [38], with reduced ambient temperature [39], and via the adminstration of methylxanthines [40]. Despite the success of methylxanthines in reducing the frequency of apnea and bradycardia, such treatment has surprisingly little impact on hypoxemic episodes [41]; we suggest that the elevated O2 consumption and the absence of bradycardia are likely to increase 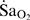 during those apneas that persist despite treatment. The severity of hypoxemic episodes is reduced by switching preterm infants from supine to prone [42], which may increase functional residual capacity [43] and improve diaphragm function, increase tidal volume and increase resting alveolar
during those apneas that persist despite treatment. The severity of hypoxemic episodes is reduced by switching preterm infants from supine to prone [42], which may increase functional residual capacity [43] and improve diaphragm function, increase tidal volume and increase resting alveolar 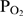 [44]. Our finding that low cardiac output leads to increased
[44]. Our finding that low cardiac output leads to increased 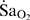 during apnea leads to the suggestion that judicious adjustment of inotropic support in infants with cardiac abnormalities could improve resting mixed-venous saturation and reduce apneic hypoxemia.
during apnea leads to the suggestion that judicious adjustment of inotropic support in infants with cardiac abnormalities could improve resting mixed-venous saturation and reduce apneic hypoxemia.
Hypoxemic events become less frequent between infancy and childhood, despite an unchanged apnea frequency [28], perhaps as a result of a fall in O2 consumption per body weight. However, before this occurs, infants experience a period of susceptibility to rapid desaturation during apnea as a result of a fall in hemoglobin content and O2 affinity [22] and a rise in O2 consumption [45]. The implications for SIDS are obvious in that these changes coincide with the peak incidence for SIDS at 2–3 months [46]. SIDS also occurs disproportionately in preterm infants [46], who manifest severe anemia [22] and greater O2 consumption. Infants resuscitated from apparent life threatening events have been found to have lower hemoglobin content [47], pointing to a potential role for rapid 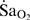 in the progression of such events. It is possible that the rapid development of apneic hypoxemia initiates prolonged hypoxic cardiorespiratory depression that in turn leads to SIDS.
in the progression of such events. It is possible that the rapid development of apneic hypoxemia initiates prolonged hypoxic cardiorespiratory depression that in turn leads to SIDS.
Conclusion
We have provided a mathematical framework for quantifying the relative importance of key cardiorespiratory factors on the rate of arterial O2 desaturation during apnea, with particular relevance to preterm infants. For the first time we have demonstrated that each of the factors examined has a signature influence on the trajectory of desaturation, providing quantitative insight into the causes of rapidly developing hypoxemia during apnea.
Methods
Mathematical model
Lung compartment
For the lung, a single homogeneous compartment is assumed based on the model of Grodins et al [48]. Each equation describing changes in the alveolar partial pressure of each gas (G) is based on the conservation of mass (specifically, the pressure–volume product) and is expressed in terms of inspired and expired alveolar ventilation and transfer of gases into the pulmonary capillary:
| (1) |
where 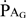 represents the rate of change of alveolar
represents the rate of change of alveolar 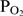 ,
, 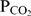 , and
, and 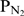 ;
; 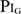 represents the inspired alveolar partial pressure of each gas G; P0 is atmospheric pressure converted from STP to BTP (863 mmHg);
represents the inspired alveolar partial pressure of each gas G; P0 is atmospheric pressure converted from STP to BTP (863 mmHg); 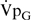 represents
represents 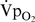 and
and 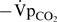 , pulmonary gas uptake (STPD) for O2 and CO2 (
, pulmonary gas uptake (STPD) for O2 and CO2 (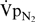 was neglected in this study for simplicity);
was neglected in this study for simplicity); 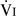 and
and 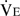 are inspired and expired alveolar ventilation (BTPS). Accounting for the difference in
are inspired and expired alveolar ventilation (BTPS). Accounting for the difference in 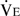 and
and 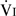 due to a net pulmonary gas uptake into the pulmonary blood, yields:
due to a net pulmonary gas uptake into the pulmonary blood, yields:
| (2) |
where 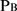 = barometric pressure (760 mmHg);
= barometric pressure (760 mmHg); 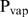 = water vapour pressure (47 mmHg);
= water vapour pressure (47 mmHg); 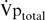 is the net pulmonary gas uptake,
is the net pulmonary gas uptake, 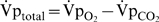 .
.
Since purely obstructive apneas are relatively rare in preterm infants [49], an unobstructed airway was chosen as the standard model in this study. In the current study it was assumed that lung volume did not fall during apnea, as in active sleep [24], when apneic desaturation events are most common [33]. With lung volume constant, conservation of mass requires that passive airflow into the unobstructed airway must occur in response to a net pulmonary gas uptake into the pulmonary blood [11]. To account for this effect, we can write:
| (3) |
Pilot simulations predicted that the volume of gas inflow during apnea is unlikely to exceed physiological deadspace. Thus, during apnea 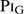 is taken as
is taken as 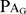 of the last exhaled breath prior to apnea onset.
of the last exhaled breath prior to apnea onset.
For the current study we assumed diffusion equilibrium at the pulmonary capillaries, such that 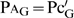 . Gas uptake is determined from the Fick equation; specifically, pulmonary blood flow (
. Gas uptake is determined from the Fick equation; specifically, pulmonary blood flow (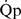 ), and the difference between end capillary (
), and the difference between end capillary (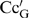 ) and mixed venous (
) and mixed venous (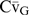 ) content:
) content:
| (4) |
Utilising equations for R-L shunt, arterial content of each gas G is determined from its end capillary (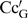 ) and mixed venous (
) and mixed venous (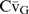 ) content, and pulmonary shunt fraction (
) content, and pulmonary shunt fraction (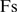 ):
):
| (5) |
Fs defines the ratio of pulmonary blood flow to cardiac output, such that Fs = 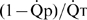 .
.
Body compartment
Assuming that the 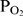 of the venous blood is equilibrated with the tissue
of the venous blood is equilibrated with the tissue 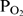 , the mass-balance equations are:
, the mass-balance equations are:
| (6) |
where 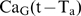 represents the gas content of O2 and CO2 in the arterioles; Ta is arterial transit time;
represents the gas content of O2 and CO2 in the arterioles; Ta is arterial transit time; 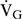 represents
represents 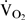 and
and 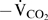 , the metabolic consumption of O2 and production of CO2;
, the metabolic consumption of O2 and production of CO2; 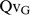 represents
represents 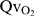 and
and 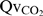 the combined venous/tissue volumes for O2 and CO2.
the combined venous/tissue volumes for O2 and CO2.
Blood O2 stores were partitioned by assigning blood volume (Qb) to arterial (25%) and venous (75%) compartments [50] and they were modelled assuming an entirely unmixed arterial compartment, and an entirely mixed and homogenous venous compartment. The arterial transit time (Ta) is constrained by the arterial volume (Qa) by the relationship 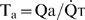 . The body compartment volume
. The body compartment volume 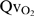 is taken as the venous volume.
is taken as the venous volume. 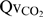 , the effective venous/tissue volume for CO2 is taken as the same value for QvO2, based on published data (see Methods – Derivation of equations). Physiologically this represents no additional contribution of a specific tissue reservoir for CO2 within the time frame of apnea.
, the effective venous/tissue volume for CO2 is taken as the same value for QvO2, based on published data (see Methods – Derivation of equations). Physiologically this represents no additional contribution of a specific tissue reservoir for CO2 within the time frame of apnea.
To characterise the O2-dissociation curve we used a modified form of the equation of Severinghaus [51]. We re-expressed the equation with respect to the partial pressure at 50% (P50) and at 90% (P90) saturation:
| (7) |
where 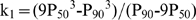 and
and 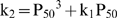 . Values for P50 (24.0 mmHg) and P90 (53.6 mmHg), were obtained from the data of Delivoria-Papadopoulos [22] for a 9–10 wk-old preterm infant. O2 content (
. Values for P50 (24.0 mmHg) and P90 (53.6 mmHg), were obtained from the data of Delivoria-Papadopoulos [22] for a 9–10 wk-old preterm infant. O2 content (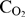 , ml ml−1) includes that bound to hemoglobin (Hb, g ml−1) and that dissolved in plasma:
, ml ml−1) includes that bound to hemoglobin (Hb, g ml−1) and that dissolved in plasma:
| (8) |
The relationship between CO2 content (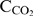 ) and
) and 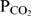 was assumed linear:
was assumed linear:
| (9) |
where 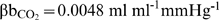 and
and 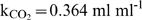 as adapted for STPD from Grodins et al. [52].
as adapted for STPD from Grodins et al. [52].
Simulations were performed using software written in MATLAB (The Mathworks; Natick, MA).
Theory
A general equation
In an earlier study we developed a general relationship that describes the factors influencing the magnitude of 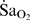 at any instant in time during apnea [12]:
at any instant in time during apnea [12]:
| (10) |
where 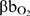 is the capacitance co-efficient of blood for O2. To specifically demonstrate the role of gas exchange, it is more useful to represent
is the capacitance co-efficient of blood for O2. To specifically demonstrate the role of gas exchange, it is more useful to represent 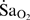 in terms of
in terms of 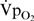 . Using Equation 1 for O2 under conditions of apnea (
. Using Equation 1 for O2 under conditions of apnea (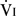 ,
,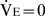 ), assuming
), assuming 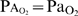 , and using
, and using 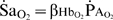 , reveals:
, reveals:
| (11) |
where 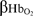 (% mmHg−1) is defined as the slope of the O2-dissociation curve, specifically regarding end-capillary
(% mmHg−1) is defined as the slope of the O2-dissociation curve, specifically regarding end-capillary 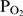 with respect to
with respect to 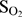 . It is clear from Equation 11 that
. It is clear from Equation 11 that 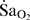 is directly proportional to the product
is directly proportional to the product 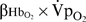 , which both vary substantially during apneic arterial desaturation. Although Equations 10 and 11 are useful conceptually, values for (
, which both vary substantially during apneic arterial desaturation. Although Equations 10 and 11 are useful conceptually, values for ( ) or
) or 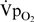 throughout apnea are unknown, and thus
throughout apnea are unknown, and thus 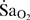 is not simple to predict explicitly.
is not simple to predict explicitly.
Special cases
The original framework to understand factors influencing 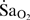 was based on the assumption that
was based on the assumption that 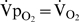 [10],[23] which does not hold true during apnea [11],[12]. However, such an assumption is valid prior to any substantial fall in
[10],[23] which does not hold true during apnea [11],[12]. However, such an assumption is valid prior to any substantial fall in 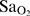 , and as therefore useful to explicitly describe
, and as therefore useful to explicitly describe 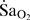 immediately upon apnea onset (
immediately upon apnea onset (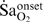 ):
):
| (12) |
Notably, Equation 12 demonstrates that for any level of 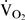 and
and 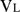 ,
, 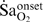 is intimately related to
is intimately related to 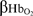 . Consequently,
. Consequently, 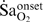 increases dramatically with reduced resting
increases dramatically with reduced resting 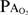 (Figure 11).
(Figure 11).
Figure 11. Relationship between the slope of the oxy-hemoglobin dissociation curve and alveolar 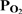 .
.

Note that reduced alveolar 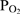 (
(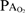 ) causes a substantial increase in the slope of the oxy-hemoglobin dissociation curve (
) causes a substantial increase in the slope of the oxy-hemoglobin dissociation curve (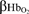 ; see inset) and in
; see inset) and in 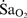 at apnea onset (
at apnea onset (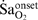 ; based on Equation 12).
; based on Equation 12).
Although no simple expression could be written to describe 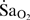 explicitly for stage 1, we derived an expression for
explicitly for stage 1, we derived an expression for 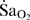 during stage 2 (see Methods – Derivation of equations), given by:
during stage 2 (see Methods – Derivation of equations), given by:
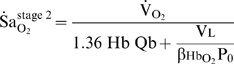 |
(13) |
Since the total blood O2 capacity (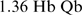 ) is much greater than
) is much greater than 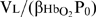 ,
, 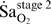 is determined principally by (
is determined principally by (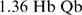 ) with negligible influence coming from lung volume (
) with negligible influence coming from lung volume (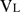 ) and the slope of the O2-dissociation curve (
) and the slope of the O2-dissociation curve (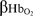 ), as well as
), as well as 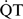 . Using the values for the preterm infant in Table 2 and maximum
. Using the values for the preterm infant in Table 2 and maximum 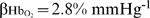 , Equation 13 predicts that
, Equation 13 predicts that 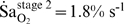 . The little remaining
. The little remaining 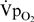 during stage 2 can be found by combining Equations 11 and 13:
during stage 2 can be found by combining Equations 11 and 13:
| (14) |
Equation 14 predicts that 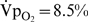 of its resting value during stage 2. Notably,
of its resting value during stage 2. Notably, 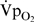 is increased by reducing Hb and Qb; the greater
is increased by reducing Hb and Qb; the greater 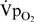 and thus a greater rate of alveolar O2 depletion with reduced blood O2 capacity (
and thus a greater rate of alveolar O2 depletion with reduced blood O2 capacity (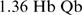 ) will increase
) will increase 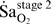 .
.
How can we reconcile that Equation 13 shows that 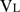 no longer influences
no longer influences 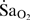 during stage 2, when the general equation (Equation 11) implies that reduced
during stage 2, when the general equation (Equation 11) implies that reduced 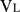 will accelerate
will accelerate 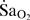 throughout apneic desaturation? Equation 14 reveals that during stage 2, elevated
throughout apneic desaturation? Equation 14 reveals that during stage 2, elevated 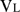 also acts to increase
also acts to increase 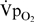 ; thus nearly entirely offsetting the direct influence on
; thus nearly entirely offsetting the direct influence on 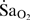 . The same applies for reduced
. The same applies for reduced 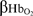 , which acts to elevate
, which acts to elevate 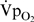 and therefore no longer accelerates
and therefore no longer accelerates 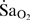 during stage 2.
during stage 2.
Derivation of equations
Here we derive the explicit equations used within the current study to encapsulate key relationships pertaining to gas exchange and arterial desaturation during apnea.
Stage 2 arterial O2 desaturation
This section details the derivation of an explicit equation to predict the rate of both arterial and venous desaturation during the severe desaturation of stage 2, a phase where 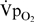 is substantially reduced below
is substantially reduced below 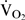 and both
and both 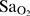 and
and 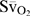 fall at the same rate. Ignoring dissolved plasma O2, consideration of Equation 1 for O2 and assuming
fall at the same rate. Ignoring dissolved plasma O2, consideration of Equation 1 for O2 and assuming 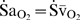 yields:
yields:
| (15) |
By substituting the following relationships into Equation 15: 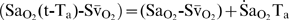 ;
; 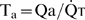 ; Qb = Qa+Qv;
; Qb = Qa+Qv; 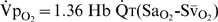 ; it can be shown that
; it can be shown that 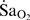 is directly proportional to the difference between
is directly proportional to the difference between 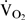 and
and 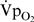 , where:
, where:
| (16) |
Combining Equations 11 and 16 yields Equation 13.
Estimation of effective blood volume for CO2
Using the same methodology as described above, the ratio of 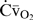 to
to 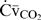 during stage 2 can be used to estimate the ratio of
during stage 2 can be used to estimate the ratio of 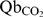 to
to 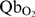 .
. 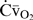 and
and 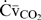 can be found using:
can be found using:
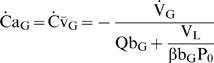 |
(17) |
where 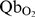 and
and 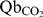 are the effective blood volumes for O2/CO2;
are the effective blood volumes for O2/CO2; 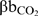 is the capacitance coefficient for CO2. Neglecting pulmonary gas exchange, combining Equation 17 for O2 and CO2 gives:
is the capacitance coefficient for CO2. Neglecting pulmonary gas exchange, combining Equation 17 for O2 and CO2 gives:
| (18) |
Equation 18 permitted the calculation of 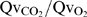 based on published data [53; their Figure 3] where during apnea the rate of rise in
based on published data [53; their Figure 3] where during apnea the rate of rise in 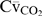 is very close to the rate of fall in the product of
is very close to the rate of fall in the product of 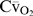 and the respiratory exchange ratio (RER); using
and the respiratory exchange ratio (RER); using 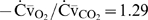 from their data, and assuming resting RER = 0.8, we find that
from their data, and assuming resting RER = 0.8, we find that 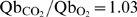 or approximately 1. Thus
or approximately 1. Thus 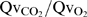 is assumed to be 1.
is assumed to be 1.
Stage 1 hypercapnia
Here we develop a relationship to describe the time-course of alveolar/arterial hypercapnia during stage 1 for CO2. Using Equation 1 for CO2, taking 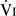 ,
,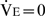 , gives the relationship
, gives the relationship 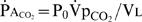 . Substituting the steady-state Fick equation,
. Substituting the steady-state Fick equation, 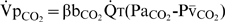 , assuming alveolar-arterial equilibrium (
, assuming alveolar-arterial equilibrium (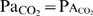 ), using
), using 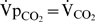 under resting conditions, assuming that
under resting conditions, assuming that 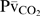 is constant, and solving for
is constant, and solving for 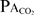 yields:
yields:
| (19) |
Calculating the rate of rise in 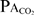 (
(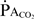 ) by taking the derivative gives:
) by taking the derivative gives:
| (20) |
Equations 19 and 20 describe the slowing of 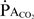 from the initial rate
from the initial rate 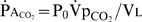 as
as 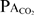 rises towards
rises towards 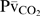 . Specifically, the time constant
. Specifically, the time constant 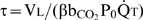 demonstrates that high
demonstrates that high 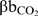 causes a rapid slowing of
causes a rapid slowing of 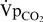 and hence of
and hence of 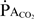 as the arterial value approaches venous value. Indeed, fitting an exponential curve to the
as the arterial value approaches venous value. Indeed, fitting an exponential curve to the 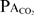 trace (Figure 2) during the first 5 s of apnea yielded a rapid time constant of 1.26 s, a value close to that predicted by
trace (Figure 2) during the first 5 s of apnea yielded a rapid time constant of 1.26 s, a value close to that predicted by 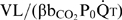 . The corollary is that the low value of
. The corollary is that the low value of 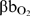 prevents the slowing of
prevents the slowing of 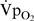 as desaturation progresses, giving rise to a rapid
as desaturation progresses, giving rise to a rapid 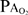 decline and thus rapid arterial desaturation. Likewise, further reducing
decline and thus rapid arterial desaturation. Likewise, further reducing 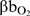 by lowering hemoglobin content potentiates such effect.
by lowering hemoglobin content potentiates such effect.
Impact of supplemental O2
The delay (right-shift) in arterial desaturation during apnea with increasing supplemental O2 (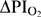 ) can be predicted explicitly. Using Equation 1 for O2 under the conditions of apnea, and assuming
) can be predicted explicitly. Using Equation 1 for O2 under the conditions of apnea, and assuming 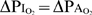 , the delay (
, the delay (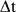 ) in arterial desaturation is given by:
) in arterial desaturation is given by:
| (21) |
Footnotes
The authors have declared that no competing interests exist.
We received no funding for this work.
References
- 1.Barrington K, Finer N. The natural history of the appearance of apnea of prematurity. Pediatr Res. 1991;29:372–375. doi: 10.1038/pr.1991.72500. [DOI] [PubMed] [Google Scholar]
- 2.Poets CF, Stebbens VA, Samuels MP, Southall DP. The relationship between bradycardia, apnea, and hypoxemia in preterm infants. Pediatr Res. 1993;34:144–147. doi: 10.1203/00006450-199308000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Perlman JM, Volpe JJ. Episodes of apnea and bradycardia in the preterm newborn: impact on cerebral circulation. Pediatrics. 1985;76:333–338. [PubMed] [Google Scholar]
- 4.Janvier A, Khairy M, Kokkotis A, Cormier C, Messmer D, et al. Apnea is associated with neurodevelopmental impairment in very low birth weight infants. J Perinatol. 2004;24:763–768. doi: 10.1038/sj.jp.7211182. [DOI] [PubMed] [Google Scholar]
- 5.Prabhakar NR, Peng YJ, Kumar GK, Pawar A. Altered carotid body function by intermittent hypoxia in neonates and adults: relevance to recurrent apneas. Respir Physiol Neurobiol. 2007;157:148–153. doi: 10.1016/j.resp.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 6.Row BW, Kheirandish L, Neville JJ, Gozal D. Impaired spatial learning and hyperactivity in developing rats exposed to intermittent hypoxia. Pediatr Res. 2002;52:449–453. doi: 10.1203/00006450-200209000-00024. [DOI] [PubMed] [Google Scholar]
- 7.Kato I, Groswasser J, Franco P, Scaillet S, Kelmanson I, et al. Developmental characteristics of apnea in infants who succumb to sudden infant death syndrome. Am J Respir Crit Care Med. 2001;164:1464–1469. doi: 10.1164/ajrccm.164.8.2009001. [DOI] [PubMed] [Google Scholar]
- 8.Naeye RL. Hypoxemia and the sudden infant death syndrome. Science. 1974;186:837–838. doi: 10.1126/science.186.4166.837. [DOI] [PubMed] [Google Scholar]
- 9.Poets CF, Southall DP. Patterns of oxygenation during periodic breathing in preterm infants. Early Hum Dev. 1991;26:1–12. doi: 10.1016/0378-3782(91)90038-5. [DOI] [PubMed] [Google Scholar]
- 10.Fletcher EC, Goodnight S, Miller T, Luckett RA, Rosborough J, et al. Atelectasis affects the rate of arterial desaturation during obstructive apnea. J Appl Physiol. 1990;69:1863–1868. doi: 10.1152/jappl.1990.69.5.1863. [DOI] [PubMed] [Google Scholar]
- 11.Lanphier EH, Rahn H. Alveolar gas exchange during breath holding with air. J Appl Physiol. 1963;18:478–482. doi: 10.1152/jappl.1963.18.3.478. [DOI] [PubMed] [Google Scholar]
- 12.Wilkinson MH, Berger PJ, Blanch N, Brodecky V. Effect of venous oxygenation on arterial desaturation rate during repetitive apneas in lambs. Respir Physiol. 1995;101:321–331. doi: 10.1016/0034-5687(95)00034-b. [DOI] [PubMed] [Google Scholar]
- 13.Farmery AD, Roe PG. A model to describe the rate of oxyhaemoglobin desaturation during apnoea. Br J Anaesth. 1996;76:284–291. doi: 10.1093/bja/76.2.284. [DOI] [PubMed] [Google Scholar]
- 14.Poets CF. Apparent life-threatening events and sudden infant death on a monitor. Paediatr Respir Rev. 2004;5(Suppl A):S383–386. doi: 10.1016/s1526-0542(04)90068-1. [DOI] [PubMed] [Google Scholar]
- 15.Poets CF, Rau GA, Neuber K, Gappa M, Seidenberg J. Determinants of lung volume in spontaneously breathing preterm infants. Am J Respir Crit Care Med. 1997;155:649–653. doi: 10.1164/ajrccm.155.2.9032208. [DOI] [PubMed] [Google Scholar]
- 16.Henderson-Smart DJ. Vulnerability to hypoxemia in the newborn. Sleep. 1980;3:331–342. doi: 10.1093/sleep/3.3-4.331. [DOI] [PubMed] [Google Scholar]
- 17.Strohl KP, Altose MD. Oxygen saturation during breath-holding and during apneas in sleep. Chest. 1984;85:181–186. doi: 10.1378/chest.85.2.181. [DOI] [PubMed] [Google Scholar]
- 18.Finer NN, Higgins R, Kattwinkel J, Martin RJ. Summary proceedings from the apnea-of-prematurity group. Pediatrics. 2006;117:S47–51. doi: 10.1542/peds.2005-0620H. [DOI] [PubMed] [Google Scholar]
- 19.Upton CJ, Milner AD, Stokes GM. Apnoea, bradycardia, and oxygen saturation in preterm infants. Arch Dis Child. 1991;66:381–385. doi: 10.1136/adc.66.4_spec_no.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams JA, Zabaleta IA, Sackner MA. Hypoxemic events in spontaneously breathing premature infants: etiologic basis. Pediatr Res. 1997;42:463–471. doi: 10.1203/00006450-199710000-00007. [DOI] [PubMed] [Google Scholar]
- 21.Stewart IB, Bulmer AC, Sharman JE, Ridgway L. Arterial oxygen desaturation kinetics during apnea. Med Sci Sports Exerc. 2005;37:1871–1876. doi: 10.1249/01.mss.0000176305.51360.7e. [DOI] [PubMed] [Google Scholar]
- 22.Delivoria-Papadopoulos M, Roncevic NP, Oski FA. Postnatal changes in oxygen transport of term, preterm, and sick infants: The role of red cell 2,3-Diphosphoglycerate and adult hemoglobin. Pediatric Research. 1971;5:235–245. [Google Scholar]
- 23.Fletcher EC, White SG, Munafo D, Miller CC, 3rd, Luckett R, et al. Effect of cardiac output reduction on rate of desaturation in obstructive apnea. Chest. 1991;99:452–456. doi: 10.1378/chest.99.2.452. [DOI] [PubMed] [Google Scholar]
- 24.Stark AR, Cohlan BA, Waggener TB, Frantz ID, 3rd, Kosch PC. Regulation of end-expiratory lung volume during sleep in premature infants. J Appl Physiol. 1987;62:1117–1123. doi: 10.1152/jappl.1987.62.3.1117. [DOI] [PubMed] [Google Scholar]
- 25.Cartlidge PH, Rutter N. Percutaneous respiration in the newborn infant. Effect of gestation and altered ambient oxygen concentration. Biol Neonate. 1987;52:301–306. doi: 10.1159/000242725. [DOI] [PubMed] [Google Scholar]
- 26.Cartlidge PH, Rutter N. Percutaneous respiration in the new-born infant. Effect of ambient oxygen concentration on pulmonary oxygen uptake. Biol Neonate. 1988;54:68–72. doi: 10.1159/000242826. [DOI] [PubMed] [Google Scholar]
- 27.Reix P, Arsenault J, Dome V, Fortier PH, Lafond JR, et al. Active glottal closure during central apneas limits oxygen desaturation in premature lambs. J Appl Physiol. 2003;94:1949–1954. doi: 10.1152/japplphysiol.00783.2002. [DOI] [PubMed] [Google Scholar]
- 28.Poets CF. Pathophysiology of apnea of prematurity. Implications from observational studies. In: Oommen MP, editor. Respiratory control and disorders in the newborn. New York: Marcel Dekker Inc; 2003. pp. 295–316. [Google Scholar]
- 29.Edwards BA, Sands SA, Feeney C, Skuza EM, Brodecky V, et al. Continuous positive airway pressure reduces loop gain and resolves periodic central apneas in the lamb. Respir Physiol Neurobiol. 2009 doi: 10.1016/j.resp.2009.07.006. doi: 10.1016/j.resp.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 30.Westkamp E, Soditt V, Adrian S, Bohnhorst B, Groneck P, et al. Blood transfusion in anemic infants with apnea of prematurity. Biol Neonate. 2002;82:228–232. doi: 10.1159/000065891. [DOI] [PubMed] [Google Scholar]
- 31.Hjalmarson O, Sandberg K. Abnormal lung function in healthy preterm infants. Am J Respir Crit Care Med. 2002;165:83–87. doi: 10.1164/ajrccm.165.1.2107093. [DOI] [PubMed] [Google Scholar]
- 32.Olhager E, Forsum E. Total energy expenditure, body composition and weight gain in moderately preterm and full-term infants at term postconceptional age. Acta Paediatr. 2003;92:1327–1334. doi: 10.1080/08035250310005396. [DOI] [PubMed] [Google Scholar]
- 33.Tourneux P, Leke A, Kongolo G, Cardot V, Degrugilliers L, et al. Relationship between functional residual capacity and oxygen desaturation during short central apneic events during sleep in “late preterm” infants. Pediatr Res. 2008;64:171–176. doi: 10.1203/PDR.0b013e318179951d. [DOI] [PubMed] [Google Scholar]
- 34.Sekar KC, Duke JC. Sleep apnea and hypoxemia in recently weaned premature infants with and without bronchopulmonary dysplasia. Pediatr Pulmonol. 1991;10:112–116. doi: 10.1002/ppul.1950100213. [DOI] [PubMed] [Google Scholar]
- 35.Weinstein MR, Oh W. Oxygen consumption in infants with bronchopulmonary dysplasia. J Pediatr. 1981;99:958–961. doi: 10.1016/s0022-3476(81)80032-7. [DOI] [PubMed] [Google Scholar]
- 36.Hjalmarson O, Sandberg KL. Lung function at term reflects severity of bronchopulmonary dysplasia. J Pediatr. 2005;146:86–90. doi: 10.1016/j.jpeds.2004.08.044. [DOI] [PubMed] [Google Scholar]
- 37.Kaempf JW, Campbell B, Brown A, Bowers K, Gallegos R, et al. PCO2 and room air saturation values in premature infants at risk for bronchopulmonary dysplasia. J Perinatol. 2007 doi: 10.1038/sj.jp.7211859. [DOI] [PubMed] [Google Scholar]
- 38.Dechert R, Wesley J, Schafer L, LaMond S, Beck T, et al. Comparison of oxygen consumption, carbon dioxide production, and resting energy expenditure in premature and full-term infants. J Pediatr Surg. 1985;20:792–798. doi: 10.1016/s0022-3468(85)80045-2. [DOI] [PubMed] [Google Scholar]
- 39.Hey EN. The relation between environmental temperature and oxygen consumption in the new-born baby. J Physiol. 1969;200:589–603. doi: 10.1113/jphysiol.1969.sp008710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bauer J, Maier K, Linderkamp O, Hentschel R. Effect of caffeine on oxygen consumption and metabolic rate in very low birth weight infants with idiopathic apnea. Pediatrics. 2001;107:660–663. doi: 10.1542/peds.107.4.660. [DOI] [PubMed] [Google Scholar]
- 41.Bucher HU, Duc G. Does caffeine prevent hypoxaemic episodes in premature infants? A randomized controlled trial. Eur J Pediatr. 1988;147:288–291. doi: 10.1007/BF00442697. [DOI] [PubMed] [Google Scholar]
- 42.McEvoy C, Mendoza ME, Bowling S, Hewlett V, Sardesai S, et al. Prone positioning decreases episodes of hypoxemia in extremely low birth weight infants (1000 grams or less) with chronic lung disease. J Pediatr. 1997;130:305–309. doi: 10.1016/s0022-3476(97)70360-3. [DOI] [PubMed] [Google Scholar]
- 43.Kassim Z, Donaldson N, Khetriwal B, Rao H, Sylvester K, et al. Sleeping position, oxygen saturation and lung volume in convalescent, prematurely born infants. Arch Dis Child Fetal Neonatal Ed. 2007;92:F347–350. doi: 10.1136/adc.2006.094078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wagaman MJ, Shutack JG, Moomjian AS, Schwartz JG, Shaffer TH, et al. Improved oxygenation and lung compliance with prone positioning of neonates. J Pediatr. 1979;94:787–791. doi: 10.1016/s0022-3476(79)80157-2. [DOI] [PubMed] [Google Scholar]
- 45.Hill JR, Rahimtulla KA. Heat balance and the metabolic rate of new-born babies in relation to environmental temperature; and the effect of age and of weight on basal metabolic rate. J Physiol. 1965;180:239–265. doi: 10.1113/jphysiol.1965.sp007701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blair PS, Sidebotham P, Berry PJ, Evans M, Fleming PJ. Major epidemiological changes in sudden infant death syndrome: a 20-year population-based study in the UK. Lancet. 2006;367:314–319. doi: 10.1016/S0140-6736(06)67968-3. [DOI] [PubMed] [Google Scholar]
- 47.Poets CF, Samuels MP, Wardrop CA, Picton-Jones E, Southall DP. Reduced haemoglobin levels in infants presenting with apparent life-threatening events–a retrospective investigation. Acta Paediatr. 1992;81:319–321. doi: 10.1111/j.1651-2227.1992.tb12234.x. [DOI] [PubMed] [Google Scholar]
- 48.Grodins FS, Buell J, Bart AJ. Mathematical analysis and digital simulation of the respiratory control system. J Appl Physiol. 1967;22:260–276. doi: 10.1152/jappl.1967.22.2.260. [DOI] [PubMed] [Google Scholar]
- 49.Milner AD, Greenough A. The role of the upper airway in neonatal apnoea. Semin Neonatol. 2004;9:213–219. doi: 10.1016/j.siny.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 50.Guyton AC. The systemic circulation. Textbook of medical physiology. 5 ed. Philapelphia: W. B. Saunders Company; 1976. pp. 237–249. [Google Scholar]
- 51.Severinghaus JW. Simple, accurate equations for human blood O2 dissociation computations. J Appl Physiol. 1979;46:599–602. doi: 10.1152/jappl.1979.46.3.599. [DOI] [PubMed] [Google Scholar]
- 52.Grodins FS, Gray JS, Schroeder KR, Norins AL, Jones RW. Respiratory responses to CO2 inhalation; a theoretical study of a nonlinear biological regulator. J Appl Physiol. 1954;7:283–308. doi: 10.1152/jappl.1954.7.3.283. [DOI] [PubMed] [Google Scholar]
- 53.Hong SK, Lin YC, Lally DA, Yim BJ, Kominami N, et al. Alveolar gas exchanges and cardiovascular functions during breath holding with air. J Appl Physiol. 1971;30:540–547. doi: 10.1152/jappl.1971.30.4.540. [DOI] [PubMed] [Google Scholar]
- 54.Hulskamp G, Hoo AF, Ljungberg H, Lum S, Pillow JJ, et al. Progressive decline in plethysmographic lung volumes in infants: physiology or technology? Am J Respir Crit Care Med. 2003;168:1003–1009. doi: 10.1164/rccm.200303-460OC. [DOI] [PubMed] [Google Scholar]
- 55.Hill JR, Robinson DC. Oxygen consumption in normally grown, small-for-dates and large-for-dates new-born infants. J Physiol. 1968;199:685–703. doi: 10.1113/jphysiol.1968.sp008673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walther FJ, Siassi B, Ramadan NA, Ananda AK, Wu PY. Pulsed Doppler determinations of cardiac output in neonates: normal standards for clinical use. Pediatrics. 1985;76:829–833. [PubMed] [Google Scholar]
- 57.Linderkamp O, Versmold HT, Riegel KP, Betke K. Estimation and prediction of blood volume in infants and children. Eur J Pediatr. 1977;125:227–234. doi: 10.1007/BF00493567. [DOI] [PubMed] [Google Scholar]
- 58.Koch G. Alveolar ventilation, diffusing capacity and the A-a PO2 difference in the newborn infant. Respir Physiol. 1968;4:168–192. doi: 10.1016/0034-5687(68)90050-9. [DOI] [PubMed] [Google Scholar]