

Abstract
QST is a standardized measure of the genetic differentiation of a quantitative trait among populations. The distribution of QST's for neutral traits can be predicted from the FST for neutral marker loci. To test for the neutral differentiation of a quantitative trait among populations, it is necessary to ask whether the QST of that trait is in the tail of the probability distribution of neutral traits. This neutral distribution can be estimated using the Lewontin–Krakauer distribution and the FST from a relatively small number of marker loci. We develop a simulation method to test whether the QST of a given trait is consistent with the null hypothesis of selective neutrality over space. The method is most powerful with small mean FST, strong selection, and a large number (>10) of measured populations. The power and type I error rate of the new method are far superior to the traditional method of comparing QST and FST.
IN 1993, Spitze (1993) and Prout and Barker (1993) introduced QST, a quantitative genetic analog of Wright's FST. Just as FST gives a standardized measure of the genetic differentiation among populations for a genetic locus, QST measures the amount of genetic variance among populations relative to the total genetic variance. In the years since, QST has been frequently used to test for the effects of spatially divergent (or less commonly, spatially uniform) selection (see reviews in Lynch et al. 1999; Merilä and Crnokrak 2001; McKay and Latta 2002; Howe et al. 2003; Leinonen et al. 2008; Whitlock 2008). In principle, the average QST of a neutral additive quantitative trait is expected to be equal to the mean value of FST for neutral genetic loci. FST can be readily measured on commonly available genetic markers, and QST can be measured as well with an appropriate breeding design in a common-garden setting. As a result, QST promises to be an index of the effect of selection on the quantitative trait. If QST is higher than FST, then this is taken as evidence of spatially divergent selection on the trait. If QST is much smaller than FST, then this has been taken as evidence of spatially uniform stabilizing selection, which makes the trait diverge less than expected by chance.
The comparison with FST is essential to rule out genetic drift as an alternative mechanism for phenotypic divergence among populations. Because finite populations may diverge genetically in the absence of selection, divergence must be greater than expected by drift alone if we are to conclusively demonstrate that divergent selection has played a role in genetic differentiation among populations. Therefore it has become common practice to use FST of putatively neutral markers as a control for the effects of genetic drift and to compare observed QST values for traits to these neutral FST values.
These comparisons follow two separate methods, to address related but distinct questions. First, many studies of quantitative genetic differentiation measure the QST of many traits and the FST of many loci, followed by a comparison of the mean QST to the mean FST. Such a comparison may judge whether the conditions are suitable in that species for local adaptation, that is, whether selective differences between populations are large enough relative to gene flow to allow adaptive differentiation (Whitlock 2008). We do not consider this sort of comparison in this article.
The other type of comparison asks whether the QST of a single trait is greater than expected by drift, as measured by FST. This type of comparison is most common, but it is statistically difficult. Unfortunately, as emphasized in a recent review by Whitlock (2008), there is great variation in the expected FST among neutral loci and among the QST of different neutral traits (see Figure 1). The majority of this variation results from evolutionary differences between loci and not sampling error in the observations. Rogers and Harpending (1983) imply that the distribution of QST of a single neutral trait should be approximately equivalent to that for FST of a single neutral locus, and this has been confirmed by simulation for traits determined by additive loci compared to biallelic marker loci (Whitlock 2008). The two distributions are similar, but there is great heterogeneity among traits or loci. As a result, to show that selection is acting on a trait, it is necessary to show that the value of QST has a low probability of being observed given the distribution of neutral QST.
Figure 1.—

The distribution of FST for neutral loci and QST for neutral quantitative traits. The histograms show the results of simulations of a set of 10 local populations each of 100 individuals, connected by 5% migration following island model assumptions. The solid line shows the distribution predicted by the Lewontin–Krakauer distribution. The distribution of QST for neutral traits is very similar to the distribution of FST for single neutral loci, as can be seen by their mutual good fit to the Lewontin–Krakauer distribution (Figure modified from Whitlock 2008).
Comparing QST to the distribution inferred from FST is difficult for two reasons. First, typical data sets rarely include enough loci to directly infer the distribution of FST without extra inferential steps. In our approach, we use the distribution of QST predicted from the mean FST and the χ2 distribution by Lewontin and Krakauer (1973) to bridge this gap. Whitlock (2008) has shown that this distribution is appropriate for nearly all realistic situations for traits determined by additive genetic effects. Second, QST for a trait is rarely measured with high precision, so the position of a given estimated QST value in the distribution cannot be known without error.
To test the null hypothesis that the spatial distribution of a particular trait is not affected by selection, we wish to compare the observed 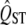 of that trait (marked with a hat to indicate it is an estimate) to the distribution of QST expected for neutral traits. Unfortunately, calculating the distribution of QST for neutral traits is not straightforward, because the estimate of QST for a particular trait is variable for several reasons. The estimate of QST is subject to measurement error, caused by the finite samples of families and individuals in the quantitative genetic experiment. These cause error in the estimate of the additive genetic variance within populations (VA,within) and the genetic variance among populations (VG,among), which translate into error of the estimate of QST. In addition, there is another source of variation in QST among neutral traits, caused by the idiosyncrasies of the evolutionary process in each local population in the study. The true value of QST for the set of populations being studied can vary tremendously around its expectation, even for neutral traits, because by chance a finite set of populations may drift in a similar direction (Whitlock 2008). As a result, measurements of QST can vary because of both statistical and evolutionary variation.
of that trait (marked with a hat to indicate it is an estimate) to the distribution of QST expected for neutral traits. Unfortunately, calculating the distribution of QST for neutral traits is not straightforward, because the estimate of QST for a particular trait is variable for several reasons. The estimate of QST is subject to measurement error, caused by the finite samples of families and individuals in the quantitative genetic experiment. These cause error in the estimate of the additive genetic variance within populations (VA,within) and the genetic variance among populations (VG,among), which translate into error of the estimate of QST. In addition, there is another source of variation in QST among neutral traits, caused by the idiosyncrasies of the evolutionary process in each local population in the study. The true value of QST for the set of populations being studied can vary tremendously around its expectation, even for neutral traits, because by chance a finite set of populations may drift in a similar direction (Whitlock 2008). As a result, measurements of QST can vary because of both statistical and evolutionary variation.
Fortunately, these two sources of variation are fairly well understood individually. The sampling error for the estimates of the variance components can be estimated from standard approaches, and this variation can be well approximated using information from the mean squares of the analysis of the breeding experiment (O'Hara and Merilä 2005). The variation in neutral QST that results from heterogeneity of evolutionary history can be approximated by the Lewontin–Krakauer distribution (Lewontin and Krakauer 1973), if information is available on the mean QST of neutral traits (Whitlock 2008). This approximation does not depend on the demographic details of the populations in question (Whitlock 2008), but the effects of deviations from assumptions of additive gene effect have not yet been tested. The mean of the distribution of values of QST for neutral traits is usually not known, but fortunately the mean of the distribution of FST of neutral loci is expected to be approximately equal to the mean QST of neutral traits (Spitze 1993), and this does not depend on demographic details (Whitlock 1999). Therefore the mean FST measured from a series of genetic markers thought to be selectively neutral can be combined with the Lewontin–Krakauer distribution to predict the distribution of true neutral QST across the range of possible evolutionary trajectories.
Given that the mean value of 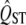 of neutral traits is expected to equal the mean FST of neutral markers under certain assumptions (discussed later), we will use
of neutral traits is expected to equal the mean FST of neutral markers under certain assumptions (discussed later), we will use 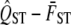 as a test statistic and compare the observed quantity to the zero value proposed by the null hypothesis. We will use a traditional hypothesis testing approach, which means that we need to specify the sampling distribution of
as a test statistic and compare the observed quantity to the zero value proposed by the null hypothesis. We will use a traditional hypothesis testing approach, which means that we need to specify the sampling distribution of 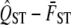 under the assumption of neutrality. Traditionally, the sampling distribution of
under the assumption of neutrality. Traditionally, the sampling distribution of 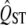 is inferred from the data on the trait itself, for example, using bootstrapping to infer the sampling distribution. This is appropriate when calculating a confidence interval for QST but is a biased measure of the sampling variance of neutral QST. The variance of the sampling distribution of
is inferred from the data on the trait itself, for example, using bootstrapping to infer the sampling distribution. This is appropriate when calculating a confidence interval for QST but is a biased measure of the sampling variance of neutral QST. The variance of the sampling distribution of 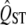 varies with its expected value; larger values of true QST have more variable sampling distributions than traits with smaller true QST. This association between QST and its sampling error is quite strong, as shown in Figure 2. As a result, if the sampling properties of neutral
varies with its expected value; larger values of true QST have more variable sampling distributions than traits with smaller true QST. This association between QST and its sampling error is quite strong, as shown in Figure 2. As a result, if the sampling properties of neutral  are inferred from a trait with high QST, the estimate of the variance of the null distribution will be too high, and the hypothesis test comparing
are inferred from a trait with high QST, the estimate of the variance of the null distribution will be too high, and the hypothesis test comparing 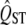 to FST will be conservative. On the other hand, if a low QST is used to estimate the variance of the null distribution, the estimated error will be too small, and the test will reject true null hypotheses too often.
to FST will be conservative. On the other hand, if a low QST is used to estimate the variance of the null distribution, the estimated error will be too small, and the test will reject true null hypotheses too often.
Figure 2.—

The width of the estimated sampling distribution of 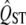 varies with mean QST. The solid line shows the sampling distribution of QST when the true mean QST value is 0.05. The dotted line shows the sampling distribution that would be estimated for QST from a trait that by chance was at the first percentile of this distribution, and the dashed line shows the sampling distribution that would be inferred from a value taken at the 99th percentile. If the QST of a trait differs from the expectation by chance, then the width of the sampling distribution will also be estimated with substantial error. In particular, the error variance of
varies with mean QST. The solid line shows the sampling distribution of QST when the true mean QST value is 0.05. The dotted line shows the sampling distribution that would be estimated for QST from a trait that by chance was at the first percentile of this distribution, and the dashed line shows the sampling distribution that would be inferred from a value taken at the 99th percentile. If the QST of a trait differs from the expectation by chance, then the width of the sampling distribution will also be estimated with substantial error. In particular, the error variance of 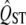 is overestimated with QST estimates that are too high and underestimated for small QST values.
is overestimated with QST estimates that are too high and underestimated for small QST values.
We address this problem by using FST from putatively neutral maker loci in combination with estimates of the additive genetic variance within populations to predict the sampling variance that would be expected for the QST of a neutral trait. We show that the power and type I error rate of this test are greatly superior to traditional methods.
METHODS
Testing neutrality:
To generate the null distribution of  , we use a parametric simulation approach. To calculate a
, we use a parametric simulation approach. To calculate a  value from data, we need estimates of three quantities:
value from data, we need estimates of three quantities:  , VA,within, and VG,among. To calculate the null distribution, we simulate random sampling for each of these quantities under the assumption that the null hypothesis that QST equals
, VA,within, and VG,among. To calculate the null distribution, we simulate random sampling for each of these quantities under the assumption that the null hypothesis that QST equals 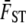 is true. We calculate
is true. We calculate 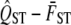 from the simulated values, and after repeating this 1000 times, we generate the sampling distribution of
from the simulated values, and after repeating this 1000 times, we generate the sampling distribution of 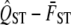 assuming the null hypothesis.
assuming the null hypothesis.
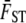 is calculated from marker loci; we use the Weir and Cockerham (1984) method in our test calculations. To simulate the sampling error in estimates of
is calculated from marker loci; we use the Weir and Cockerham (1984) method in our test calculations. To simulate the sampling error in estimates of  , for each replicate simulation we randomly sample with replacement from the marker loci until the number of loci in the simulated data set equals the number of loci in the real data set. Mean FST is calculated from these sampled loci using the method of Weir and Cockerham (1984), and the observed value of their θ is used as the simulated
, for each replicate simulation we randomly sample with replacement from the marker loci until the number of loci in the simulated data set equals the number of loci in the real data set. Mean FST is calculated from these sampled loci using the method of Weir and Cockerham (1984), and the observed value of their θ is used as the simulated  value.
value.
VA,within is calculated from a quantitative genetic breeding design. There are several suitable experimental designs for such estimates. In this article we assume that the additive genetic variance is estimated by a half-sib design, but the approach could easily be modified for other designs. VA,within can be estimated from four times the variance among sires; and to estimate the variance among sires we need the mean squares of sires (MSsires) and the mean squares of dams (MSdams). To simulate estimates of VA,within, we use an approach analogous to a parametric bootstrap (O'Hara and Merilä 2005). As tested by O'Hara and Merilä (2005),  and
and  should be χ2 distributed, where d.f. represents the degrees of freedom associated with a particular level and the overbar indicates the true value of the mean square. Therefore by multiplying the estimated
should be χ2 distributed, where d.f. represents the degrees of freedom associated with a particular level and the overbar indicates the true value of the mean square. Therefore by multiplying the estimated  . times a random number from a χ2 distribution for each of sires and dams we can simulate the sampling distribution of these quantities and therefore of VA,within. This procedure is implemented exactly as the parametric bootstrap in O'Hara and Merilä (2005), except to avoid a strong source of bias we do not constrain variance component estimates to be positive.
. times a random number from a χ2 distribution for each of sires and dams we can simulate the sampling distribution of these quantities and therefore of VA,within. This procedure is implemented exactly as the parametric bootstrap in O'Hara and Merilä (2005), except to avoid a strong source of bias we do not constrain variance component estimates to be positive.
VG,among is calculated from the variance among populations in the mean value of the trait when the organisms are grown in a common environment. The novel aspect of our design comes from how the sampling of VG,among is simulated. As mentioned in the introduction, the sampling variance for VG,among is correlated with the true value of VG,among, and therefore if the null hypothesis is true but VG,among incorrectly appears high by sampling error, the estimate of its sampling distribution will also be estimated poorly. If we were only estimating the value of QST itself, this would pose no real problems, but because we are trying to compare QST to the neutral expectation, it can be a real source of bias in the calculations. Our solution is to simulate the sampling distribution of VG,among assuming that the null hypothesis is true. We therefore calculate the value of VG,among that would be expected given the observed  and VA,within. Given that QST is defined as
and VA,within. Given that QST is defined as  and that for neutral traits and neutral loci the average values of QST and FST are approximately equal, we can find the expected value of VG,among under neutrality to be
and that for neutral traits and neutral loci the average values of QST and FST are approximately equal, we can find the expected value of VG,among under neutrality to be
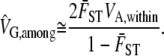 |
To simulate the sampling distribution around this expectation, we again assumed that the distribution of trait means among populations follows a normal distribution and multiply  times a random number drawn from a χ2 distribution with degrees of freedom equal to the number of populations (numpops) minus one. This sampling procedure is the same as assumed by the Lewontin–Krakauer distribution shown to work well to approximate the distribution of QST under a variety of demographic circumstances (Whitlock 2008). Simulating the sampling error in this way is identical to the approach taken by O'Hara and Merilä (2005) in their parametric bootstrapping, except for using the expected value of VG,among calculated from FST instead of the observed VG,among.
times a random number drawn from a χ2 distribution with degrees of freedom equal to the number of populations (numpops) minus one. This sampling procedure is the same as assumed by the Lewontin–Krakauer distribution shown to work well to approximate the distribution of QST under a variety of demographic circumstances (Whitlock 2008). Simulating the sampling error in this way is identical to the approach taken by O'Hara and Merilä (2005) in their parametric bootstrapping, except for using the expected value of VG,among calculated from FST instead of the observed VG,among.
For a given hypothesis test using a specific data set, we generate 1000 simulated estimates of 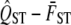 . For each simulation,
. For each simulation, 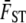 , VA,within, and VG,among are randomly drawn as specified above, and
, VA,within, and VG,among are randomly drawn as specified above, and 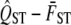 is calculated from these simulated values. The distribution of these 1000 simulated values is the null distribution of the hypothesis test. Therefore by comparing the quantile of the observed value of
is calculated from these simulated values. The distribution of these 1000 simulated values is the null distribution of the hypothesis test. Therefore by comparing the quantile of the observed value of 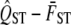 to the simulated distribution, we may determine the P-value of the hypothesis test of neutrality.
to the simulated distribution, we may determine the P-value of the hypothesis test of neutrality.
Supporting information, File S1 includes an R program to implement this procedure.
Simulations:
We tested the method using simulations conducted with the population genetics simulation software Nemo (guillaume and Rougemont 2006) updated to include quantitative traits. Neutral marker loci were simulated with 100 biallelic loci, with mutation rates of 10−5 in either direction. One hundred loci potentially affected the quantitative traits. Mutation was based on an infinite allele model, where the allelic effect of an allele was, if mutated, changed by a factor randomly selected from a Gaussian distribution with genomic mutational variance equal to 0.001. Mutation rates for the quantitative trait loci were set at 10−5. Each of 20 local populations had an effective population size of 500 diploid individuals, and the migration rate among populations varied from m = 0.05 to m = 0.001 to produce different FST values, ranging from approximately FST = 0.01 to FST = 0.3. Measurements were taken on the populations after 50,000 generations (or 25,000 generations for the neutral cases), allowing the populations to reach an approximate equilibrium before sampling. The 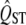 of 10,000 traits was simulated for the neutral traits and 100 for each set of parameters with selection.
of 10,000 traits was simulated for the neutral traits and 100 for each set of parameters with selection.
In addition to the island model calculations that make the bulk of the simulation tests, we also simulated a one-dimensional, circular stepping-stone model with 60 local populations. Simulations with FST = 0.04 were performed, corresponding to a migration rate of 0.12. Migration occurred only between adjacent (left and right) populations in the stepping-stone model, and at most, every third population was sampled for FST and the QST calculations, as suggested by Beaumont and Nichols (1996) and Whitlock (2008). For the heterogeneous selection cases, the populations were alternatively assigned to habitats in groups of five.
In some simulations, the quantitative trait was selectively neutral, to allow tests of the type I error rates of the method. In other simulations, the quantitative trait was subjected to either uniform stabilizing selection (for which all local populations had the same optimum with Gaussian selection with VS = 5) or heterogeneous selection (for which the selective optimum for half of the local populations was different from the optimum in the other half of the populations.) The strength of selection for the heterogeneous environment case was calculated such that a perfectly adapted individual on one environment would have a 5 or 50% reduction in fitness in the other selective environment in the island or stepping-stone model, respectively. The parameters of the selection functions were VS = 5, and the difference between the habitat optimum phenotypes was 0.716 in the island model, and 2.63 in the stepping-stone model. There was no environmental effect added to the genotypic values of the quantitative trait loci (VE = 0).
For each simulation,  was calculated from a simulated half-sib breeding design. In the default configuration, samples were taken from 20 populations, and for each population five sires were mated to five dams each. These numbers were varied to better understand the power of the approach. Five offspring from each dam were measured, and from the results
was calculated from a simulated half-sib breeding design. In the default configuration, samples were taken from 20 populations, and for each population five sires were mated to five dams each. These numbers were varied to better understand the power of the approach. Five offspring from each dam were measured, and from the results  was calculated from the population and sire effects using an analysis of variance.
was calculated from the population and sire effects using an analysis of variance.
For all parameter combinations, we tested the null hypothesis of neutrality using the new method and with the best method previously available, the parametric bootstrap approach from O'Hara and Merilä (2005). We refer to this latter approach as the “traditional approach” throughout.
Simulation results:
The simulations show that the new method has a more accurate type I error rate and more power than the traditional method. There is sufficient power to detect high QST when the 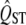 of a trait is severalfold greater than the mean FST and when large numbers of populations (10 or more) are included in the analysis. However, large numbers of marker loci are not necessary. On the other hand, it is difficult to reliably detect the signal of homogeneous selection; the power to discriminate significantly small QST values is low, even when the mean FST value is much higher than expected for most intraspecific comparisons.
of a trait is severalfold greater than the mean FST and when large numbers of populations (10 or more) are included in the analysis. However, large numbers of marker loci are not necessary. On the other hand, it is difficult to reliably detect the signal of homogeneous selection; the power to discriminate significantly small QST values is low, even when the mean FST value is much higher than expected for most intraspecific comparisons.
First, examine the cases where the null hypothesis is true; that is, when the trait is evolving without the influence of selection. The traditional method has an overall type I error rate that is a bit high overall (Table 1), but it is seen to be particularly poor when the type I errors are divided into the two tails. The type I error rate for the traditional method with low QST values is 7.0–7.8% (in contrast to the expected 2.5%), whereas the type I error rate is far too low for high values of QST compared to mean FST (0.41–0.44%). In all cases, the one-tailed error rates are different from the stated 2.5% with extremely small P-values (the largest being P = 4 × 10−59). In contrast, the new method has a much better type I error rate. The total error rate for the new method is always within the 95% confidence interval of the expected value of 5%, and the errors are more evenly divided into the two tails.
TABLE 1.
Type I error rates for the island model simulations based on the island model with 20 populations and 20 sires in the sample, for a two-sided test with α = 0.05
| Traditional method |
New method |
|||
|---|---|---|---|---|
| Migration rate | Left tail (low QST) | Right tail (high QST) | Left tail (low QST) | Right tail (high QST) |
| 0.001 | 0.0706 | 0.0042 | 0.0244 | 0.024 |
| 0.01 | 0.0700 | 0.0044 | 0.0257 | 0.026 |
| 0.05 |
0.0784 |
0.0041 |
0.0245 |
0.0293 |
With heterogeneous selection in the island model, the mean QST ranged from 0.026 to 0.564, depending on the amount of migration among populations (see Table 2). The power of the method depends in part on the relative value of the typical QST value in comparison to the mean FST. When QST is expected to be much greater than the mean FST, the method has substantial power (Figure 3). Importantly, the new method has much higher power to detect heterogeneous selection than the traditional method (Figure 3). With small sample sizes and low true differences between QST and FST, neither method is able to detect the effects of selection, and with extremely large samples both methods have high power. But for intermediate (and realistic) sample sizes with moderate QST values, the new method has substantially more power to detect heterogeneous selection than the traditional method. We also ran simulations of stronger selection (where an individual perfectly adapted to the other environment would have a 10% fitness reduction), where QST is higher. In these cases the power was very high for both methods, except for the cases when there were only two populations in the study. There again, the new method greatly outperformed the traditional method (results not shown).
TABLE 2.
Mean QST and FST values for different island model parameters
| Migration rate | Mean FST (neutral) | Mean QST (heterogeneous selection) | Mean QST (homogeneous selection) |
|---|---|---|---|
| 0.001 | 0.318 | 0.564 | 0.044 |
| 0.01 | 0.045 | 0.232 | 0.015 |
| 0.05 |
0.009 |
0.026 |
0.005 |
Figure 3.—

The power of the new approach (left graphs) compared to the traditional approach (right graphs), as a function of the number of populations included in the sample. Results are shown for the island model for three different migration rates. The populations experienced spatially heterogeneous selection; an individual that is perfectly adapted to one habitat will have a 5% reduction in fitness in the other habitat. Each habitat contains half of the populations. Each population was measured for 5 (top graphs) or 20 (bottom graphs) sires, each mated to five dams, with five offspring per dam for the  estimates, and FST was calculated from 10 loci. When FST is high (with low migration rates), it is more difficult to distinguish a high QST value caused by heterogeneous selection, and the power of the test is very weak if a small number of populations are measured in the study. The new simulation method has much better power than the traditional comparison of QST and FST.
estimates, and FST was calculated from 10 loci. When FST is high (with low migration rates), it is more difficult to distinguish a high QST value caused by heterogeneous selection, and the power of the test is very weak if a small number of populations are measured in the study. The new simulation method has much better power than the traditional comparison of QST and FST.
In contrast, under only rare circumstances was there much power to detect that the QST value of a trait was significantly smaller than expected under neutral differentiation (Figure 4). Even when the mean neutral FST is relatively high, the left tail of the distribution of neutral QST is still relatively dense for small values, making it difficult to separate a low QST from neutral expectations.
Figure 4.—

Power of QST to detect homogeneous selection. The trait experienced stabilizing selection in each population with a uniform optimum. Stabilizing selection was strong, with VS = 5. Sample sizes are the same as in the top panels in Figure 3.
These preceding calculations are based on moderately large sample sizes for the quantitative genetic measurements but not very many (10) marker loci for the calculation of FST. Increasing the number of marker loci increases power, but not dramatically (Figure 5a). On the other hand, using more families per population to estimate 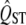 better has a beneficial effect (Figure 5b). However, the power of the analysis is critically dependent on the number of populations surveyed (Figure 3). The variance of the expected
better has a beneficial effect (Figure 5b). However, the power of the analysis is critically dependent on the number of populations surveyed (Figure 3). The variance of the expected 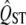 distribution reduces in proportion to the number of demes measured (Whitlock 2008), and the reliability of
distribution reduces in proportion to the number of demes measured (Whitlock 2008), and the reliability of  estimates increases strongly with number of demes (Goudet and Büchi 2006). Reliable inference about the neutrality of quantitative traits requires sampling of large numbers of populations. The estimation of both QST and FST depends critically on the estimate of the variance among populations, and the power of the estimate of this variance depends on the number of populations sampled. In studies with small numbers of populations, the
estimates increases strongly with number of demes (Goudet and Büchi 2006). Reliable inference about the neutrality of quantitative traits requires sampling of large numbers of populations. The estimation of both QST and FST depends critically on the estimate of the variance among populations, and the power of the estimate of this variance depends on the number of populations sampled. In studies with small numbers of populations, the 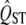 estimates were also quite biased for both methods (results not shown), explaining the apparently higher power for the smallest sample sizes.
estimates were also quite biased for both methods (results not shown), explaining the apparently higher power for the smallest sample sizes.
Figure 5.—

Power to detect heterogeneous selection as a function of (a) the number of marker loci examined and (b) the number of sires per population. All other sample sizes and parameters are the same as in Figures 3 and 4, with 20 (filled symbols) or 5 (open symbols) populations sampled. The power of the analysis is not much affected by the number of marker loci examined, but increasing the number of families per population can increase power.
Results under the stepping-stone model are quite similar. The mean QST for the stepping-stone simulations was 0.638 with selection and 0.0488 for the neutral case. The power of the analysis is largely dependent on the number of populations sampled (Figure 6) and varies in an equivalent way with the number of families and neutral loci sampled (results not shown).
Figure 6.—

The power of the simulation method applied to simulated data from a stepping-stone model. Sixty populations on a linear stepping stone were simulated with N = 500 and m = 0.12. FST averaged 0.04. In the heterogeneous selection case, each population experienced one of two selective environments, chosen at random for each population with equal probability. The resulting QST was approximately 0.6 on average. In the homogeneous selection case, the QST was ∼0.008. The method was applied using data from populations separated by at least two intervening populations, sampling 5 (solid lines) or 20 (dashed lines) populations.
DISCUSSION
The QST of neutral traits is potentially extremely variable from trait to trait, especially when the number of populations in the system (or in the study) is small. This distribution is approximately predictable with knowledge of the mean FST of neutral marker loci for the same populations (Whitlock 2008). A simple function of QST [equal to (numpops −1)QST/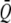 ST] is approximately distributed by a χ2 distribution with numpops − 1 degrees of freedom; this derives from the Lewontin–Krakauer distribution. Given that for traits determined by additively acting alleles the mean QST is approximately equal to the mean FST, the sampling distribution of neutral QST can be predicted.
ST] is approximately distributed by a χ2 distribution with numpops − 1 degrees of freedom; this derives from the Lewontin–Krakauer distribution. Given that for traits determined by additively acting alleles the mean QST is approximately equal to the mean FST, the sampling distribution of neutral QST can be predicted.
Most studies of QST explicitly compare  of a trait to FST, as a test of whether spatially heterogeneous or homogeneous selection affects the distribution of the trait. These studies use the observed properties of
of a trait to FST, as a test of whether spatially heterogeneous or homogeneous selection affects the distribution of the trait. These studies use the observed properties of 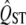 to predict its sampling distribution. However, when testing the null hypothesis of neutrality, we need to infer the sampling properties of
to predict its sampling distribution. However, when testing the null hypothesis of neutrality, we need to infer the sampling properties of 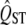 for neutral traits, not of traits with high or low expected QST's. The difference matters because the width of the sampling distribution of
for neutral traits, not of traits with high or low expected QST's. The difference matters because the width of the sampling distribution of 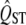 depends on its mean value (Figure 2).
depends on its mean value (Figure 2).
We have developed a new method to test for selective neutrality using the difference between 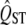 and mean FST. We account for the expected distribution of QST under neutrality using a distribution inferred from the mean FST. Compared to the traditional method, the new approach works extremely well. The traditional method, which infers the distribution of
and mean FST. We account for the expected distribution of QST under neutrality using a distribution inferred from the mean FST. Compared to the traditional method, the new approach works extremely well. The traditional method, which infers the distribution of 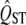 from the observed
from the observed 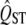 , has very poor false positive rates (type I error). High QST rejects the null hypothesis far too rarely, and low QST rejects the null hypothesis too often (Table 1). This is because the error variance is overestimated for high QST and underestimated for low QST (Figure 2). The type I error rate for our new method is close to the stated values, and it is symmetric in the upper and lower tails as is desirable.
, has very poor false positive rates (type I error). High QST rejects the null hypothesis far too rarely, and low QST rejects the null hypothesis too often (Table 1). This is because the error variance is overestimated for high QST and underestimated for low QST (Figure 2). The type I error rate for our new method is close to the stated values, and it is symmetric in the upper and lower tails as is desirable.
The new method is also more powerful than the traditional method for detecting spatially heterogeneous selection. Both the new and traditional methods work well when QST is much greater than FST and with data from many populations, and both fail with too few data (e.g., when the number of populations is two). However, in intermediate cases with moderate QST and moderately large sample sizes, the new method has much more power than the traditional approach. With homogeneous selection, the traditional method appears to have more power, but this is largely due to its inflated type I error rate. Positive results are not reliable for homogeneous selection and small numbers of populations.
Unfortunately, in some biologically interesting circumstances, there are a limited number of populations that exist in nature, and in these circumstances it is simply not possible to reliably show that even a large 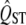 is different from the neutral expectation. This is especially true when the mean FST of neutral markers is also high. For example, some applications of the QST approach have been made comparing a pair of subspecies. In these cases, the mean FST is typically high (or the two populations would not have been given subspecific status) and the total number of such populations in nature is just two. In this case, there is little hope of finding significant evidence of selective differentiation via the QST approach. For example, when there are only two populations, the 97.5 percentile of the distribution of FST or QST is approximately five times the mean of the distribution, according to the Lewontin–Krakauer distribution. Even with no error in estimating QST, a trait would have to have a QST value five times as large as the mean FST to be significantly in the tail of the distribution, for the two-population case. QST is never estimated with such small error, so in practice the
is different from the neutral expectation. This is especially true when the mean FST of neutral markers is also high. For example, some applications of the QST approach have been made comparing a pair of subspecies. In these cases, the mean FST is typically high (or the two populations would not have been given subspecific status) and the total number of such populations in nature is just two. In this case, there is little hope of finding significant evidence of selective differentiation via the QST approach. For example, when there are only two populations, the 97.5 percentile of the distribution of FST or QST is approximately five times the mean of the distribution, according to the Lewontin–Krakauer distribution. Even with no error in estimating QST, a trait would have to have a QST value five times as large as the mean FST to be significantly in the tail of the distribution, for the two-population case. QST is never estimated with such small error, so in practice the 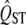 of the trait would have to be much larger than five times the mean FST to find statistical evidence of selection.
of the trait would have to be much larger than five times the mean FST to find statistical evidence of selection.
There is little power in typical data sets to test for spatially uniform stabilizing selection using 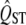 − FST comparisons. It has been suggested that small values of QST relative to FST may indicate strong stabilizing selection with the same optimum in all populations, because such selection would oppose genetic drift and maintain approximately the same mean in each local population. However, the distribution of neutral
− FST comparisons. It has been suggested that small values of QST relative to FST may indicate strong stabilizing selection with the same optimum in all populations, because such selection would oppose genetic drift and maintain approximately the same mean in each local population. However, the distribution of neutral 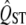 includes a dense left-hand tail in most intraspecific comparisons, because, with a small mean FST and a few populations sampled, a large number of loci with small FST (or neutral traits with small QST) are expected just by chance. Only with very strong selection and levels of FST that verge on interspecific values (FST = 0.2) have we found even moderate power to detect spatially uniform selection (Figure 4).
includes a dense left-hand tail in most intraspecific comparisons, because, with a small mean FST and a few populations sampled, a large number of loci with small FST (or neutral traits with small QST) are expected just by chance. Only with very strong selection and levels of FST that verge on interspecific values (FST = 0.2) have we found even moderate power to detect spatially uniform selection (Figure 4).
There are a few other caveats that need to be kept in mind when applying this method, in common with all interpretations of QST. It is crucial that FST and QST are both estimated without bias, and there are many sources of bias that affect most 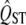 measures (Whitlock 2008). In particular, it is important that
measures (Whitlock 2008). In particular, it is important that 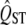 is estimated from a breeding design and not just from phenotypic data. Furthermore, it is essential that the study organisms are grown in a common garden to avoid conflating phenotypic plasticity with local adaptation.
is estimated from a breeding design and not just from phenotypic data. Furthermore, it is essential that the study organisms are grown in a common garden to avoid conflating phenotypic plasticity with local adaptation.
Importantly, the simulations conducted here all assumed that traits are determined by alleles that interact additively, both between and within loci. Dominance variance can under some circumstances cause mean QST to be greater than mean FST, even for neutral traits. There is controversy over whether the effects of dominance will typically lead to increased values of QST (Lopez-Fanjul et al. 2003, 2007; Goudet and Büchi 2006; Goudet and Martin 2007), but importantly the distribution of QST among neutral traits has not been investigated for traits affected by dominance or epistasis. Our ability to use the distribution predicted from the FST of marker loci depends on the distribution being similar for QST, and this has not been investigated for traits with dominance. This method, and indeed any comparison of QST and FST, requires stringent assumptions about the additive basis of the quantitative trait.
The method also relies on the assumption that we are able to identify neutral markers to use for FST to generate the null distribution. With a large number of marker loci, the chances may be high that at least some of the loci are affected by spatially heterogeneous selection. If such loci can be identified by a procedure such as fdist2 (Beaumont and Nichols 1996), then removing them from the analysis is probably best, although this may make the test less conservative. Alternatively, all marker loci could be left in the analysis, on the assumption that the loci affecting quantitative traits may sometimes differentiate by pleiotropic effects or by linkage to other selected loci. Keeping the full spectrum of marker loci potentially would control for these extraneous effects.
Finally, there are some specific issues with the new simulation method that limit its breadth of application. The method given here uses the Lewontin–Krakauer distribution to infer the distribution of neutral QST from mean FST. According to simulation results this should work fine for typical values of mean FST (less than ∼0.2). However, the Lewontin–Krakauer distribution is based on a χ2 distribution, and its right tail extends to positive infinity and is not constrained to be less than one. As a result, for large values of mean FST the probability of the right tail of this Lewontin–Krakauer distribution becomes an inaccurate representation of the true tail probability.
To use QST to test for selection, we have to compare an individual trait's  to the distribution of possible values of QST under neutrality. By doing so, we have developed a method that has much better type I error rates and higher power for detecting spatially heterogeneous selection than traditional approaches.
to the distribution of possible values of QST under neutrality. By doing so, we have developed a method that has much better type I error rates and higher power for detecting spatially heterogeneous selection than traditional approaches.
Acknowledgments
We thank Bob O'Hara for providing the R code for the parametric bootstrap, and Sally Otto, Jérôme Goudet, and an anonymous reviewer for extremely helpful comments on a previous version of this article. Jérôme Goudet pointed out that FST estimated from multiallelic loci have a different distribution, which helped us to clarify the use of the Lewontin-Krakauer distribution for QST. This research was supported by a Discovery Grant from the Natural Science and Engineering Research Council (Canada) (to M.C.W.) and a Swiss National Science Foundation grant PA00A3-115383 (to F.G.).
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.108.099812/DC1.
References
- Beaumont, M. A., and R. Nichols, 1996. Evaluating loci for use in the genetic analysis of populations structure. Proc. R. Soc. Lond. Ser. B 263 1619–1626. [Google Scholar]
- Goudet, J., and G. Martin, 2007. Under neutrality, QST ≤ FST when there is dominance in an island model. Genetics 176 1371–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet, J., and L. Büchi, 2006. The effects of dominance, regular inbreeding and sampling design on QST, an estimator of population differentiation for quantitative traits. Genetics 172 1337–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaume, F., and J. Rougemont, 2006. Nemo: an evolutionary and population genetics programming framework. Bioinformatics 22 2556–2557. [DOI] [PubMed] [Google Scholar]
- Howe, G. T., S. N. Aitken, D. B. Neale, K. D. Jermstad, N. C. Wheeler et al., 2003. From genotype to phenotype: unraveling the complexities of cold adaptation in forest trees. Can. J. Bot. 81 1247–1266. [Google Scholar]
- Leinonen, T., R. O'Hara, J. M. Cano and J. Merilä, 2008. Comparative studies of quantitative trait and neutral marker divergence: a meta-analysis. J. Evol. Biol. 21 1–17. [DOI] [PubMed] [Google Scholar]
- Lewontin, R. C., and J. Krakauer, 1973. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics 74 175–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Fanjul, C., A. Fernández and M. A. Toro, 2003. The effects of neutral nonadditive gene action on the quantitative index of population divergence. Genetics 164 1627–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Fanjul, C., A. Fernández and M. A. Toro, 2007. The effect of dominance on the use of the QST – FST contrast to detect natural selection on quantitative traits. Genetics 176 725–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., M. Pfrender, K. Spitze, N. Lehman, J. Hicks et al., 1999. The quantitative and molecular genetic architecture of a subdivided species. Evolution 53 100–110. [DOI] [PubMed] [Google Scholar]
- McKay, J. K., and R. G. Latta, 2002. Adaptive population divergence: markers, QTL and traits. Trends Ecol. Evol. 17 285–291. [Google Scholar]
- Merilä, J., and P. Crnokrak, 2001. Comparison of genetic differentiation at marker loci and quantitative traits. J. Evol. Biol. 14 892–903. [Google Scholar]
- O'Hara, R. B., and J. Merilä, 2005. Bias and precision in QST estimates: problems and some solutions. Genetics 171 1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prout, T., and J. S. F. Barker, 1993. F statistics in Drosophila buzzatii: selection, population size and inbreeding. Genetics 134 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, A. R., and H. C. Harpending, 1983. Population structure and quantitative characters. Genetics 105 985–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitze, K., 1993. Population structure in Daphnia obtusa: quantitative genetic and allozymic variation. Genetics 135 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B. S., and C. C. Cockerham, 1984. Estimating F-statistics for the analysis of population structure. Evolution 38 1358–1370. [DOI] [PubMed] [Google Scholar]
- Whitlock, M. C., 1999. Neutral additive genetic variance in a metapopulation. Genet. Res. 74 215–221. [DOI] [PubMed] [Google Scholar]
- Whitlock, M. C., 2008. Evolutionary inference from QST. Mol. Ecol. 17 1885–1896. [DOI] [PubMed] [Google Scholar]