

Abstract
Genomic Selection (GS) is a newly developed tool for the estimation of breeding values for quantitative traits through the use of dense markers covering the whole genome. For a successful application of GS, accuracy of the prediction of genomewide breeding value (GW-EBV) is a key issue to consider. Here we investigated the accuracy and possible bias of GW-EBV prediction, using real bovine SNP genotyping (18,991 SNPs) and phenotypic data of 500 Norwegian Red bulls. The study was performed on milk yield, fat yield, protein yield, first lactation mastitis traits, and calving ease. Three methods, best linear unbiased prediction (G-BLUP), Bayesian statistics (BayesB), and a mixture model approach (MIXTURE), were used to estimate marker effects, and their accuracy and bias were estimated by using cross-validation. The accuracies of the GW-EBV prediction were found to vary widely between 0.12 and 0.62. G-BLUP gave overall the highest accuracy. We observed a strong relationship between the accuracy of the prediction and the heritability of the trait. GW-EBV prediction for production traits with high heritability achieved higher accuracy and also lower bias than health traits with low heritability. To achieve a similar accuracy for the health traits probably more records will be needed.
GENOMIC selection (GS) is a new technology that is expected to revolutionize animal breeding. It is distinct from traditional selection methods where phenotype and pedigree information is combined to predict breeding values and where at least one source is necessary for a prediction. Estimation of GS breeding value is based on the estimation of marker effects covering the whole genome and combines these estimates with the marker genotypes to obtain breeding value estimates. Given a sufficiently dense genomewide marker map, all the genetic variance is expected to be explained by the markers, and all quantitative trait loci (QTL) are in linkage disequilibrium (LD) with at least one marker (Calus et al. 2008). This allows GS to predict genomewide estimates of breeding values (GW-EBV) without the need of phenotyping the selection candidates. A potential cost reduction of up to 90% can be achieved for a breeding program by GS (Schaeffer 2006), because only a moderate number of individuals are required to have both known marker genotypes and phenotypes. These individuals form a reference data set for the estimation of GW-EBV. The knowledge obtained from the reference data set can be applied to the calculation of GW-EBV for the selection candidates on the basis of their marker genotypes, with an accuracy that is found in the validation of the prediction (Goddard and Hayes 2007).
For a successful application of GS, based on a reference data set, to a usually much larger population of selection candidates without phenotypic records, accuracy of the prediction is a key issue to consider (Goddard and Hayes 2009). Since GS was first proposed by Meuwissen et al. (2001), many research works using simulated data have been performed on this issue (Calus and Veerkamp 2007; Habier et al. 2007; Kolbehdari et al. 2007; Calus et al. 2008; Solberg et al. 2008). The recent availability of genomewide dense SNP marker maps has made GS with real data feasible. Studies of the accuracy of genomic predictions have emerged in some animal species, including mice (Lee et al. 2008; Legarra et al. 2008), chickens (Gonzalez-Recio et al. 2009), and cattle (Hayes et al. 2009), and in plant species [for example, barley (Zhong et al. 2009)]. For GS applied to dairy cattle, accuracies for the GW-EBV have been reported in North American Holstein (VanRaden et al. 2009), Australian Holstein–Friesian (Hayes et al. 2009), and New Zealand Holstein–Friesian and Jersey dairy cattle (Harris et al. 2008).
In the present work we applied GS to Norwegian Red dairy cattle to investigate the accuracy and possible bias of GW-EBV prediction for the phenotypes of milk production, clinical mastitis, and calving ease, by using real bovine genotyping data. Three methods, best linear unbiased prediction, Bayesian statistics, and a mixture model approach were used in the study, and their accuracies and biases of the GW-EBV were compared. To estimate the accuracy and bias of the GW-EBV the approach of cross-validation was employed, making use of estimates of breeding value from the Norwegian Red dairy cattle breeding scheme.
MATERIALS AND METHODS
Genotypic and phenotypic data:
There were 500 Norwegian Red bulls selected for this study with 466 sons of 34 sires, with no son also being a sire. Sons had been progeny tested between 2001 and 2006, and sires tested before 2001. The numbers of sons chosen for years 2006, 2005, 2004, 2003, 2002, and 2001 are 36, 44, 98, 98, 100, and 90, respectively. All sons and sires were genotyped at CIGENE (www.cigene.no), using the 25K MIP-SNP chip array from Affymetrix (San Diego). All data from an individual SNP were deleted if (a) pedigree information exposed >2.5% non-Mendelian sire–offspring inheritance patterns, (b) its genotype probabilities significantly deviated from the Hardy–Weinberg proportions (P < 0.01%), (c) across samples >25% genotypes were missing, or (d) its minor allele frequency (MAF) <2.5%. A total of 18,991 SNPs remained after filtering. The phenotypic data of all 500 bulls used for the study are daughter-yield deviations (DYDs) (Wiggans et al. 1992) for the traits: metric tons of milk yield, kilograms of milk fat yield, kilograms of milk protein yield, calving ease, and clinical mastitis (cm). Clinical mastitis was considered in three traits defined by period of first lactation: cm1, 15–30 days in milk; cm2, 31–120 days in milk; and cm3, 121–305 days in milk. The data were based on the average of 1038 daughters, varying from 111 to 21,391, and were available for all bulls for all traits. Accuracy of DYD for a trait is defined as the average of the accuracies of the DYDs of 500 bulls for the trait and was obtained together with DYD from BoviBank Ltd. (www.bovibank.no). The accuracies of DYDs are listed in Table 1. The genotype data and phenotype data of all 500 bulls constitute the complete data used in the present work.
TABLE 1.
Heritability and accuracy of DYD for studied traits
| Trait | h2 | Accuracy |
|---|---|---|
| Milk yield | 0.277 | 0.972 |
| Fat yield | 0.235 | 0.968 |
| Protein yield | 0.213 | 0.965 |
| cm1 | 0.030 | 0.803 |
| cm2 | 0.008 | 0.583 |
| cm3 | 0.015 | 0.668 |
| Calving ease |
0.030 |
0.812 |
Training data:
The training data sets were each obtained by masking the phenotype, i.e., setting the phenotype “unknown,” for a defined number of individuals. The individuals whose phenotype was masked were selected in two different ways. The first way was through random selection: here 100 individuals at a time were randomly selected, without replacement, to produce five nonoverlapping training data sets; i.e., every phenotype was masked precisely once in the training data sets. In this article this way is referred to as “20% random masking.” The second way was to select individuals on the basis of their year of progeny testing: seven training data sets were obtained for years 2006, 2005, 2004, 2003, 2002, 2001, and before 2001. The number of phenotype-masked individuals in a training data set for a year of testing is the number of bulls selected for this year. We called this way of selection “cohort masking.” The nonmasked data were analyzed by the models described in the “data analysis” section to predict the masked phenotypes. This resulted in each bull having a predicted phenotype from each masking method and this was compared to the realized DYD. The correlation coefficient between the predicted and realized DYDs was calculated and used as a measure of the accuracy of the GW-EBV predictions.
Additional training sets for production traits were generated to test the impact of increasing the random masking to 50% by randomly allocating each individual to one of two training sets. This implies that there were only 250 individuals with phenotypes in the training data set, instead of 400 as in 20% random masking. To obtain standard errors, for both 20% and 50% random masking the division into sets and all the subsequent analyses described in the Data analysis section were replicated six times.
Data analysis:
Three models were used to estimate the marker effects: best linear unbiased prediction (G-BLUP), Bayesian statistics (BayesB), and MIXTURE. G-BLUP estimates the effects of the markers by best linear unbiased prediction (Henderson 1975), assuming that every marker explains an equal proportion of the total genetic variance. BayesB is described in detail by Meuwissen et al. (2001) and estimates the variance explained by every marker, using a prior distribution that assumes that this variance is small, denoted as σ2, with probability (1 − γB); i.e., the marker has virtually no effect or comes from an inverse-chi-square distribution with probability γB. The probability γB represents the probability that a marker has a substantial effect and was varied in the data analysis since it is generally unknown. Meuwissen et al. assumed σ2 = 0 for markers with small estimated effects, but by having σ2 slightly larger than zero, the model can be implemented as a Gibbs sampling algorithm, which has computational advantages. Here, the small variance σ2 was estimated from the data, i.e., from the genes with small effect. In the Gibbs chain σ2 was sampled from the conditional posterior, which is 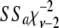 , where
, where 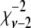 is a random deviate from the inverse-chi-square distribution with ν − 2 d.f.; ν is the number of SNPs with small effect in the current iteration of the chain;
is a random deviate from the inverse-chi-square distribution with ν − 2 d.f.; ν is the number of SNPs with small effect in the current iteration of the chain; 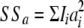 , where Ii is an indicator variable taking values 1 if SNPi belongs to the SNPs with small effects in the current iteration and Ii = 0 otherwise; and ai is the current solution of the effect of SNPi. As mentioned above, if SNPs were assumed to have substantial effect, they had an individual variance estimated. If this individual variance was <σ2, the SNP effect was removed from the set of SNPs with substantial effect, to ensure that the substantial effect SNPs always had higher variance than the small effect SNPs. This is similar to including a polygenic term in the BayesB model (Calus and Veerkamp 2007; Solberg et al. 2008), but where the correlation matrix of the polygenic effects is defined by the markers with small effect instead of by the pedigree.
, where Ii is an indicator variable taking values 1 if SNPi belongs to the SNPs with small effects in the current iteration and Ii = 0 otherwise; and ai is the current solution of the effect of SNPi. As mentioned above, if SNPs were assumed to have substantial effect, they had an individual variance estimated. If this individual variance was <σ2, the SNP effect was removed from the set of SNPs with substantial effect, to ensure that the substantial effect SNPs always had higher variance than the small effect SNPs. This is similar to including a polygenic term in the BayesB model (Calus and Veerkamp 2007; Solberg et al. 2008), but where the correlation matrix of the polygenic effects is defined by the markers with small effect instead of by the pedigree.
Since BayesB makes quite strong assumptions about the prior distribution of the marker effects, which may not be true in real data, we also used a model that attempts to estimate this prior distribution. For the latter, we made use of a property of mixtures of normal distributions, namely that they can be used to approximate any other (prior) distribution (Silverman 1996). In the MIXTURE model we assumed that the marker effects came from a mixture of two distributions: one distribution with large variance (accommodating large marker effects) and one with small variance (accommodating small marker effects). This model was also implemented in a Gibbs sampling algorithm as described by George and McCulloch (1996) except that the variance of the small SNP effects is estimated here. The distribution to which the marker belongs is sampled from the Bernoulli distribution, with parameter γM. The parameter γM, which reflects the proportion of the markers belonging to the large and the small variance distribution, is sampled using a noninformative Beta distribution as a prior. The variances of the two distributions underlying the mixture (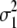 and
and 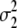 ) are estimated using a noninformative chi-square distribution (Sorenson and Gianola 2007).
) are estimated using a noninformative chi-square distribution (Sorenson and Gianola 2007).
The model of analysis that was used by G-BLUP, BayesB, and MIXTURE was
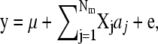 |
where y is a N × 1 vector of phenotypes (DYDs); Nm is the number of markers fitted; aj is the effect of the marker; Xj is a N × 1 vector denoting the genotype of the individuals for marker j, with Xij=0 if individual i is homozygous for the first allele at locus j, 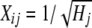 if heterozygous,
if heterozygous, 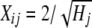 if individual i is homozygous for the second allele at locus j, and
if individual i is homozygous for the second allele at locus j, and 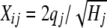 if the marker genotype is missing, where qj is the frequency of the second marker allele and Hj is the marker heterozygosity. The division by
if the marker genotype is missing, where qj is the frequency of the second marker allele and Hj is the marker heterozygosity. The division by 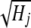 standardizes the variance of the marker genotype data to 1. The variance of aj is assumed to be Vs/Nm for G-BLUP, is estimated by BayesB, and in MIXTURE equals
standardizes the variance of the marker genotype data to 1. The variance of aj is assumed to be Vs/Nm for G-BLUP, is estimated by BayesB, and in MIXTURE equals 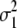 or
or 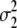 , depending on whether the marker effect is small or large and
, depending on whether the marker effect is small or large and 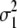 and
and 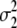 are both estimated. Since the traits are DYDs, Vs is the sire variance, which is one-quarter of the total genetic variance, and was obtained from Interbull (http://www-interbull.slu.se) together with the trait heritabilities (Table 1). Given the estimates of the marker effects and the marker genotypes, genetic values for the masked individuals are predicted as
are both estimated. Since the traits are DYDs, Vs is the sire variance, which is one-quarter of the total genetic variance, and was obtained from Interbull (http://www-interbull.slu.se) together with the trait heritabilities (Table 1). Given the estimates of the marker effects and the marker genotypes, genetic values for the masked individuals are predicted as
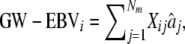 |
where Xij is the marker genotype of individual i for marker j coded the same as above, and 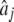 is the estimated effect of marker j. By adding the overall mean, μ, to the GW-EBVi, and assuming that the residual effect of the DYDs is on average 0, a predicted phenotype was obtained for every bull (whose phenotype was masked). Since every bull's phenotype was masked once in one of the training sets, a total of 500 predicted phenotypes were obtained for each model for a trait for a masking strategy. The correlation coefficient between the predicted and realized phenotypes was calculated and used as a measure of the accuracy of the GW-EBV predictions. The regression of the realized phenotypes on the predicted phenotypes is used as a measure of the bias of the GW-EBV, where a regression coefficient of 1 denotes no bias, <1 implies that extreme high (low) values of the GW-EBV over- (under)predict the realized phenotypes, and vice versa for a regression coefficient >1. These summary statistics were examined both overall and within each masked set.
is the estimated effect of marker j. By adding the overall mean, μ, to the GW-EBVi, and assuming that the residual effect of the DYDs is on average 0, a predicted phenotype was obtained for every bull (whose phenotype was masked). Since every bull's phenotype was masked once in one of the training sets, a total of 500 predicted phenotypes were obtained for each model for a trait for a masking strategy. The correlation coefficient between the predicted and realized phenotypes was calculated and used as a measure of the accuracy of the GW-EBV predictions. The regression of the realized phenotypes on the predicted phenotypes is used as a measure of the bias of the GW-EBV, where a regression coefficient of 1 denotes no bias, <1 implies that extreme high (low) values of the GW-EBV over- (under)predict the realized phenotypes, and vice versa for a regression coefficient >1. These summary statistics were examined both overall and within each masked set.
For BayesB calculation, the length of the Gibbs chain was 15,000 iterations and 5000 iterations were used for burn-in. For MIXTURE, the chain length was 12,000 iterations and burn-in was 8000 iterations. To ensure the convergence of the Gibbs chains used for BayesB and MIXTURE, 10 distinct chains were run for milk yield and the estimated accuracy was calculated from pooling the chains. It was found that the estimated accuracy did not change to three significant numbers after pooling 3 chains. This finding was tested for other production traits and health traits. Consequently all results presented for Gibbs analyses are the average of 3 distinct Gibbs chains.
RESULTS
Determination of the number of markers with effects:
The number of markers with a substantial effect in BayesB calculation determines the probability γB of a marker with substantial effect. In this study, this number is defined separately for different ways of masking data for each trait, by a set of BayesB calculations with different numbers of effective genes. For example, for the random masking data for milk yield, we applied BayesB to the data with the number of effective genes 800, 1600, 2400, 3200, 6400, 9600, and 12,800, respectively.
The result in Figure 1 shows that the accuracy for GW-EBV prediction was affected little by the number of markers with an effect. Accuracies varied from 0.56 to 0.58 with respect to numbers of effective genes used. The maximum accuracy achieved for random masking in Figure 1 suggests that 3200 was approximately optimal, and thus γB = 0.169 (3200/18,991) was used for milk yield. The γB value for milk yield for cohort masking of data was determined similarly, and the lower value of γB = 0.126 was found to be slightly better than other values of γB. For other production traits and health traits, the same approach was applied to set γB, and the values are shown in Table 2. Table 2 also lists γM values for MIXTURE, together with 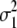 and
and 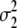 as the variances of the two distributions underlying the model. In contrast to BayesB, optimal numbers of effective genes can be determined during the MIXTURE calculations, and Table 2 shows that the γM value for MIXTURE is mostly lower than the γB for BayesB.
as the variances of the two distributions underlying the model. In contrast to BayesB, optimal numbers of effective genes can be determined during the MIXTURE calculations, and Table 2 shows that the γM value for MIXTURE is mostly lower than the γB for BayesB.
Figure 1.—

Accuracy of GW-EBV prediction with the BayesB method for the milk yield trait with respect to the number of markers with effect.
TABLE 2.
Estimates of γB for BayesB and γM, σ12, and σ22 for MIXTURE for random masking and cohort masking
| Random masking: |
Cohort masking: |
|||||||
|---|---|---|---|---|---|---|---|---|
| BayesB |
MIXTURE |
BayesB |
MIXTURE |
|||||
| Trait | γB | γM | 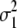 |
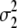 |
γB | γM | 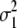 |
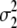 |
| Milk yield | 0.169 | 0.038 | 1.8 × 10−6 | 4.0 × 10−4 | 0.126 | 0.028 | 2.2 × 10−6 | 9.1 × 10−4 |
| Fat yield | 0.169 | 0.041 | 3.0 × 10−3 | 1.6 × 10−1 | 0.506 | 0.031 | 3.0 × 10−3 | 2.2 × 10−1 |
| Protein yield | 0.042 | 0.042 | 1.0 × 10−3 | 7.9 × 10−2 | 0.042 | 0.061 | 1.0 × 10−3 | 6.0 × 10−2 |
| cm1 | 0.042 | 0.062 | 1.0 × 10−8 | 4.5 × 10−5 | 0.084 | 0.017 | 9.8 × 10−9 | 5.0 × 10−5 |
| cm2 | 0.758 | 0.081 | 1.4 × 10−9 | 3.0 × 10−5 | 0.758 | 0.061 | 1.4 × 10−9 | 2.0 × 10−5 |
| cm3 | 0.042 | 0.022 | 5.0 × 10−9 | 1.3 × 10−4 | 0.042 | 0.049 | 4.5 × 10−9 | 1.1 × 10−4 |
| Calving ease |
0.084 |
0.057 |
4.0 × 10−8 |
1.2 × 10−4 |
0.126 |
0.039 |
3.7 × 10−8 |
1.3 × 10−4 |
Accuracy of GW-EBV prediction:
Table 3 shows the accuracy of the GW-EBV prediction by the BayesB, MIXTURE, and G-BLUP methods. For 20% random masking, Table 3 shows the mean predictive accuracy obtained for predicting the 100 individuals in the validation set for analysis of the 400 in the training set. The mean is therefore an average of 30 values, 5 from each random division of the bulls into 5 sets of 100, and then replicated six times. The standard error is based on the variance between the replicate means. However, for cohort masking, the accuracy is combined using the single GW-EBV obtained for each of the 500 bulls. It is shown in Table 4 that in cohort masking, accuracies vary within a considerable range for offspring subsets with different population size and year of progeny test. For most traits, the prediction of the masked sire cohort using phenotypes of their offspring cohorts achieved higher accuracy than those of masked cohort offspring.
TABLE 3.
Accuracy of GW-EBVs obtained by G-BLUP, MIXTURE, and BayesB
| Trait | G-BLUP | MIXTURE | BayesB |
|---|---|---|---|
| Cohort maskinga | |||
| Milk yield | 0.591 | 0.575 | 0.577 |
| Fat yield | 0.617 | 0.591 | 0.590 |
| Protein yield | 0.615 | 0.601 | 0.607 |
| cm1 | 0.282 | 0.278 | 0.272 |
| cm2 | 0.153 | 0.128 | 0.130 |
| cm3 | 0.250 | 0.253 | 0.241 |
| Calving ease | 0.406 | 0.411 | 0.429 |
| Random maskinga | |||
| Milk yield | 0.583 | 0.584 | 0.580 |
| Fat yield | 0.609 | 0.610 | 0.588 |
| Protein yield | 0.603 | 0.612 | 0.601 |
| cm1 | 0.238 | 0.247 | 0.241 |
| cm2 | 0.195 | 0.192 | 0.189 |
| cm3 | 0.265 | 0.276 | 0.263 |
| Calving ease | 0.382 | 0.392 | 0.401 |
| Approximate SE |
0.028 |
0.030 |
0.030 |
The accuracy for cohort masking is shown as a combined accuracy estimated for 500 selected individuals, while the accuracy for random masking is shown as the mean of the accuracies for five training data sets and the pooled approximate standard error.
TABLE 4.
Mean, minimum, and maximum accuracies for six offspring training subsets and accuracy for sire data set in GW-EBV prediction for cohort masking with BayesB
| Offspring |
||||
|---|---|---|---|---|
| Trait | Mean | Min | Max | Sire |
| Milk yield | 0.501 | 0.295 | 0.659 | 0.735 |
| Fat yield | 0.574 | 0.272 | 0.688 | 0.778 |
| Protein yield | 0.482 | 0.252 | 0.654 | 0.718 |
| cm1 | 0.271 | 0.163 | 0.349 | 0.426 |
| cm2 | 0.109 | 0.033 | 0.307 | 0.012 |
| cm3 | 0.251 | 0.140 | 0.311 | 0.525 |
| Calving ease |
0.447 |
0.312 |
0.472 |
0.617 |
The general conclusion from Table 3 is that for the traits studied the differences between the methods are small, and are small compared to their standard errors. For random masking, all three methods used give similar mean accuracy and standard error. There is a trend among the methods for G-BLUP to have a higher accuracy than other methods for cohort masking. Among the three production traits the accuracy for milk yield is in general lower than that for fat yield and for protein yield, but again the difference between the accuracies is within the range of the standard error.
For health traits, Table 3 shows that the accuracies of GW-EBV are considerably lower than the accuracies for production traits. In addition, compared to the production traits, the health traits show bigger differences between mean accuracy and combined accuracy. This can be seen in Table 3 by the difference between mean accuracy and combined accuracy for cm1 and cm2, which is beyond the range of standard error of the accuracy.
Table 5 presents overall average accuracy and bias based on mean accuracies and biases for six replicates of 50% and 20% masking. Results in Table 5 show that accuracy for 20% masking with 400 phenotypes in the training data set is significantly higher compared to 50% masking with 250 phenotypes in the training data set.
TABLE 5.
Accuracy and bias of GW-EBV prediction for random masking with 250 DYDs and 400 DYDs in the training data set
| Overall mean accuracya |
Overall mean biasa |
|||
|---|---|---|---|---|
| Trait | 250 DYDs | 400 DYDs | 250 DYDs | 400 DYDs |
| G-BLUP | ||||
| Milk yield | 0.547 | 0.599 | 1.010 | 1.022 |
| Fat yield | 0.569 | 0.611 | 0.999 | 1.033 |
| Protein yield | 0.550 | 0.594 | 1.006 | 1.035 |
| MIXTURE | ||||
| Milk yield | 0.542 | 0.595 | 1.018 | 1.026 |
| Fat yield | 0.564 | 0.610 | 1.001 | 1.051 |
| Protein yield | 0.545 | 0.594 | 1.004 | 1.037 |
| BayesB | ||||
| Milk yield | 0.538 | 0.585 | 0.957 | 0.967 |
| Fat yield | 0.552 | 0.590 | 0.927 | 0.970 |
| Protein yield | 0.546 | 0.589 | 0.954 | 0.973 |
| Approximate SE |
0.011 |
0.005 |
0.020 |
0.010 |
The accuracy and bias are shown as the overall mean accuracies and biases for five training data sets and six replicates and the pooled approximate standard error.
Bias of GW-EBV prediction:
The degree of bias from the methods is judged by comparing the regression coefficients of phenotypes on predicted phenotypes with the value 1. Table 6 presents the bias of the GW-EBV prediction for the traits studied. As for the presentation of accuracy, for random masking, Table 6 shows the overall mean and the standard error of the bias of the predictions for six replicates of five training data sets, while for cohort masking, it is the combined bias estimated for all 500 selected individuals. The results show that for production traits the GW-EBV predictions for random masking had a lower bias compared to those for cohort masking, but these differences were within the standard errors. It is observed for the three mastitis traits in Table 6 that there is less bias if the prediction is more accurate. For example, for cm1, Table 3 shows the combined accuracy of the prediction for cohort masking is higher than the mean accuracy for random masking, and Table 6 shows the combined bias of cohort masking is lower than the mean bias for random masking. Among the four health traits, GW-EBV prediction for calving ease has the highest accuracy (Table 3), and the prediction is in general least biased (Table 6).
TABLE 6.
Bias of GW-EBVs obtained by G-BLUP, MIXTURE, and BayesB
| G-BLUP | MIXTURE | BayesB | |
|---|---|---|---|
| Cohort maskinga | |||
| Milk yield | 1.037 | 1.047 | 1.008 |
| Fat yield | 1.075 | 1.119 | 1.020 |
| Protein yield | 1.080 | 1.106 | 1.044 |
| cm1 | 0.693 | 0.849 | 0.502 |
| cm2 | 0.550 | 0.546 | 0.323 |
| cm3 | 0.718 | 0.831 | 0.475 |
| Calving ease | 1.192 | 0.932 | 0.998 |
| Random maskinga | |||
| Milk yield | 0.984 | 0.995 | 0.951 |
| Fat yield | 1.034 | 1.056 | 0.967 |
| Protein yield | 1.046 | 1.075 | 0.992 |
| cm1 | 0.628 | 0.842 | 0.467 |
| cm2 | 0.693 | 0.814 | 0.466 |
| cm3 | 0.771 | 0.921 | 0.525 |
| Calving ease | 1.104 | 0.953 | 0.862 |
| Approximate SE |
0.089 |
0.119 |
0.070 |
The bias for cohort masking is shown as a combined bias estimated for 500 selected individuals, while the bias for random masking is shown as the mean of the biases for five training data sets and the pooled approximate standard error.
Accuracy and bias for a subset of markers:
To investigate the effect of the number of markers fitted on the accuracy of the GW-EBV, for production traits, we randomly removed markers in the complete data sets and repeated the analyses described in materials and methods. In this work, 25, 50, and 75% of 18,991 markers were randomly selected and removed. This process was replicated six times to obtain standard errors and results for cohort masking are shown in Tables 7 and 8. In general the results for the reduced marker data sets show similar features to those for the complete data with respect to the different traits, methods of analysis, and masking of the phenotypes. As expected, the accuracy of the prediction reduces as the number of markers becomes smaller. However, the decrease of the accuracy was small. For example, for G-BLUP applied to the three production traits, the combined accuracies of the GW-EBV decrease <9% of their original value for the subset with 75% of the markers removed.
TABLE 7.
Accuracy of GW-EBV prediction for cohort masking with 25, 50, and 75% of all markers masked
| % of all markers masked |
|||
|---|---|---|---|
| 25 | 50 | 75 | |
| G-BLUP | |||
| Milk yield | 0.587 | 0.581 | 0.554 |
| Fat yield | 0.611 | 0.608 | 0.589 |
| Protein yield | 0.610 | 0.603 | 0.596 |
| MIXTURE | |||
| Milk yield | 0.571 | 0.563 | 0.538 |
| Fat yield | 0.588 | 0.584 | 0.567 |
| Protein yield | 0.596 | 0.586 | 0.576 |
| BayesB | |||
| Milk yield | 0.562 | 0.553 | 0.518 |
| Fat yield | 0.563 | 0.569 | 0.539 |
| Protein yield | 0.595 | 0.577 | 0.544 |
| Approximate SE |
0.003 |
0.004 |
0.006 |
The accuracy is shown as the average combined accuracies across six different replicated marker maskings (see results) and the pooled approximate standard error.
TABLE 8.
Bias of GW-EBV prediction for cohort masking with 25, 50, and 75% of all markers masked
| % of all markers masked |
|||
|---|---|---|---|
| 25 | 50 | 75 | |
| G-BLUP | |||
| Milk yield | 1.028 | 1.013 | 0.981 |
| Fat yield | 1.064 | 1.049 | 1.011 |
| Protein yield | 1.082 | 1.061 | 1.060 |
| MIXTURE | |||
| Milk yield | 1.035 | 1.016 | 0.976 |
| Fat yield | 1.109 | 1.095 | 1.061 |
| Protein yield | 1.088 | 1.070 | 1.038 |
| BayesB | |||
| Milk yield | 0.962 | 0.926 | 0.826 |
| Fat yield | 0.976 | 0.969 | 0.878 |
| Protein yield | 1.003 | 0.956 | 0.873 |
| Approximate SE |
0.006 |
0.007 |
0.010 |
The bias is shown as the average combined biases across six different replicated marker maskings and the pooled approximate standard error.
DISCUSSION
This study applied genomic selection to real rather than simulated phenotypes in a setting in which it would be used in practice and where the predictive accuracy can be assessed by comparison with relatively precise estimates of breeding values obtained from phenotypic measurements and genetic evaluation using pedigrees. The existence of a relatively precise comparison allowed this study to compare the effectiveness of the different methods that might be employed. A total of ∼519,000 measurements were made on individual dairy cows, to obtain the 500 DYD phenotypes on individual bulls used in the analysis. The GW-EBVs for individual bulls were calculated from the estimates of effects of 19,000 SNP markers alone and the accuracy of the estimates was determined by the correlation between predicted and realized DYDs, rDYD,GW-EBV. The estimates of the marker effects came from training data sets and rDYD,GW-EBV derived from using cross-validation.
Generally, the estimated accuracies will underpredict the observed accuracy of selection, i.e., the correlation between GW-EBV and true breeding values, rGW-EBV,TBV, because the realized DYDs are not perfectly predicting the true breeding values. The expected correlations between the DYDs and genetic value, rDYD,TBV, are given in Table 9. A better estimate of the accuracy of the GW-EBV (rGW-EBV,TBV) may be obtained by calculating rDYD,GW-EBV/rDYD,TBV, where rDYD,TBV equals the accuracy of DYD (Table 1). In Table 1, there is a strong relationship between heritability and the accuracy of DYD, i.e., rGW-EBV,TBV. This suggests that data sets of >500 bulls are needed to achieve comparable accuracies for the less heritable traits, i.e., the health traits as shown by Daetwyler et al. (2008).
TABLE 9.
Accuracy of the GW-EBV prediction (rDYD,GW-EBV), the expected correlation between the DYDs and genetic value (rDYD,TBV), and rGW-EBV,TBV for studied traits
| Trait | rDYD,TBV | rDYD,GW-EBVa | rGW-EBV,TBV |
|---|---|---|---|
| Milk yield | 0.972 | 0.591 | 0.608 |
| Fat yield | 0.968 | 0.617 | 0.637 |
| Protein yield | 0.965 | 0.615 | 0.637 |
| cm1 | 0.803 | 0.282 | 0.351 |
| cm2 | 0.583 | 0.153 | 0.262 |
| cm3 | 0.668 | 0.250 | 0.374 |
| Calving ease |
0.812 |
0.406 |
0.500 |
rDYD.GW-EBV is represented by the combined accuracy of the G-BLUP method applied to the cohort masking data set.
The G-BLUP method gave overall the highest rDYD,GW-EBV and little bias of the GW-EBV. The G-BLUP method makes no assumptions about the distribution of the sizes of the SNP effects. BayesB and MIXTURE assumed that some SNPs explain more variance than others. This outcome suggests that the distribution of true effects is sufficiently spread among loci, that there is insufficient benefit from fitting the more complex models, that at least the majority of the SNPs explain a small amount of genetic variance, and that accounting for some outlier SNPs that explain substantially more variance, as in the BayesB model, does not improve GW-EBVs. The latter is probably because there are too few such outlier SNPs and the genetic variance they explain is too small relative to that explained by all the SNPs with small effects. A contributing factor to this outcome may be that the SNP density is also too low for the benefits of BayesB or MIXTURE to be fully apparent. In the absence of sequence data, the causative SNPs are unlikely to be in the data and at low density more SNPs will be required to capture a QTL. This result that most genetic effects are small agrees with a recent large-scale genomewide association study conducted for height in humans, where 20 variants were detected that explained together only 3% of the variation (Weedon et al. 2008).
For a trait whose genetic variance can be explained by a small number of genes, BayesB might be expected to do better than G-BLUP. For example, a previous study with the Holstein–Friesian cattle breed (Riquet et al. 1999) identified a QTL for milk production traits, especially milk fat. The positional candidate cloning of the QTL identified the candidate gene coding acylcoA: diacylglycerol acyltransferase 1 (DGAT1) (Grisart et al. 2002). One may expect that BayesB might achieve higher accuracy than G-BLUP for the prediction of breeding values for milk production traits of Holstein–Friesians. However, so far there is no published result available showing the segregation of the DGAT1 gene in the sample of Norwegian Red cattle used for the present work. It may be that the DGAT1 gene is not segregating in Norwegian Red cattle, the genetic variance in all the milk production traits of Norwegian Red cattle might be explained by many small genes, and hence G-BLUP might be in favor of achieving higher accuracy for the GW-EBV prediction than BayesB as observed here.
When reducing the number of markers by a factor of 2 or 4, rDYD,GW-EBV was not much reduced (Table 7). This may be because the G-BLUP, which was found to give the highest rDYD,GW-EBV, merely uses the markers to estimate the relationship between the bulls, i.e., to estimate the fraction of alleles the animals have in common (Habier et al. 2007). Thus the use of fewer markers did not reduce the accuracy of the estimate of the relationship matrix much (Hayes et al. 2003), which is central to G-BLUP. The SNP detection method may also have affected this result, in that the sequencing of small chromosomal segments results in some of the detected SNPs being very close to each other; i.e., the SNPs are unevenly distributed across the chromosome and clusters of very closely linked SNPs occur. If one or a few of the SNPs within such a cluster are omitted, this would not reduce the marker information content very much, since often several SNPs remain within the cluster in close LD with the one omitted; i.e., the cluster is still informative.
Daetwyler et al. (2008) studied factors that affect the accuracy of a prediction, using a genomewide approach. With the formula derived by the authors we can calculate an estimate of the number of independent loci that contribute to the genetic variance for a trait (nG) by the accuracy of the breeding value prediction (r), the number of phenotypes used for the prediction (nP), and the heritability of the trait (h2, obtained here as square of the accuracy of DYD in Table 1) as nG = nP[h2/r2 − h2]. We applied the formula to the result of G-BLUP for random masking of data for milk yield and got nG = 734. We also had available a smaller data set of Holstein cattle and we used the estimate of nG to predict what accuracy might be obtained for milk yield using the methods described here, from a training set of 255 records. When this was done, we obtained a predicted value of 0.46, which compares to the observed value of 0.39. This gives us some optimism that a degree of predictability of these accuracies might be obtained.
Cohort masking, grouping the DYDs by year of progeny test of the bulls, in the cross-validation more closely resembles the practical application of genomic selection, where GW-EBV of contemporaries is required. The decreasing accuracy for the mean of the cohorts is first explained by the smaller variance of the EBVs within a cohort, ∼80% of the whole set: assuming a constant regression line, then the first approximation of this impact is to reduce the accuracies, being correlations, by ∼10%. Further reductions of the correlation within each cohort might be expected because of changes in haplotype and allele frequencies arising from genetic change over time in this selected population and incomplete mixing of alleles over the different year groups. Overall our results indicated that cohort masking was not substantially worse than random masking of the data, which suggests that the population structure for predicting GW-EBV of contemporary bulls is approximately as equally suited to genomic selection as the prediction of a random set of Norwegian Red bulls. The latter may be different in different species, breeds, and/or selection programs.
In general, the accuracies of the GW-EBV vary widely between 0.12 and 0.62, where the lower accuracies apply to the mastitis traits that have low heritability, i.e., down to as low as 0.008. An accuracy of ∼0.75 is probably sufficient for the (pre)selection of young bulls at a young age (Schaeffer 2006). To achieve this for the population structure of the 500 bulls, it is predicted that a training set of 1000 bulls would be required for milk production traits, using Daetwyler et al. (2008), and more bulls for health traits. However, in practice larger training sets would be required, first to be confident of achieving comparable accuracies within a cohort and second to achieve accuracies for health and fitness traits that are comparable to those for milk production. Therefore this article demonstrates the feasibility of developing GW-EBV in practice, but at the same time indicates the scale of training data sets required to compete with existing pedigree and phenotype approaches in dairy cattle breeding. Such approaches in dairy cattle are generally considered to be well optimized, and so in other species and for other objectives the size of the training set may not necessarily be as large to be competitive.
Acknowledgments
This article represents the authors' views and does not necessarily represent a position of the European Commission, who are not liable for the use made of such information. Helpful comments of two anonymous reviewers are gratefully acknowledged. This research project has been cofinanced by the European Commission, within the 6th Framework Programme, contract no. FOOD-CT-2006-016250 (“SABRE”—Cutting Edge Genomics for Sustainable Animal Breeding).
References
- Calus, M. P. L., and R. F. Veerkamp, 2007. Accuracy of breeding values when using and ignoring the polygenic effect in genomic breeding value estimation with a marker density of one SNP per cM. J. Anim. Breed. Genet. 124 362–368. [DOI] [PubMed] [Google Scholar]
- Calus, M. P. L., T. H. E. Meuwissen, A. P. W. de Roos and R. F. Veerkamp, 2008. Accuracy of genomic selection using different methods to define haplotypes. Genetics 178 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daetwyler, H. D., B. Villanueva and J. A. Woolliams, 2008. Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLoS ONE 3 e3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George, E. I., and R. E. McCulloch, 1996. Stochastic search variable selection, pp. 441–461 in Markov Chain Monte Carlo in Practice, edited by W. R. Gilks, S. Richardson and D. J. Spiegelhalter. Chapman & Hall/CRC, London/New York.
- Goddard, M. E., and B. J. Hayes, 2007. Genomic selection. J. Anim. Breed. Genet. 124 323–330. [DOI] [PubMed] [Google Scholar]
- Goddard, M. E., and B. J. Hayes, 2009. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 10 381–391. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Recio, O., D. Gianola, G. J. M. Rosa, K. A. Weigel and A. Kranis, 2009. Genome-assisted prediction of a quantitative trait measured in parents and progeny: application to food conversion rate in chickens. Genet. Sel. Evol. 41 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisart, B., W. Coppieters, F. Farnir, L. Karim, C. Ford et al., 2002. Positional candidate cloning of a QTL in dairy cattle: identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res. 12 222–231. [DOI] [PubMed] [Google Scholar]
- Habier, D., R. L. Fernando and J. C. M. Dekkers, 2007. The impact of genetic relationship information on genome-assisted breeding values. Genetics 177 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, B. L., D. L. Johnson and R. J. Spelman, 2008. Genomic selection in New Zealand and the implications for national genetic evaluation. Proceedings of the Interbull Meeting, Niagara Falls, NY.
- Hayes, B. J., M. Carrick, P. Bowman and M. E. Goddard, 2003. Genotype x environment interaction for milk production of daughters of Australian dairy sires from test-day records. J. Dairy Sci. 86 3736–3744. [DOI] [PubMed] [Google Scholar]
- Hayes, B. J., P. J. Bowman, A. J. Chamberlain and M. E. Goddard, 2009. Invited review: genomic selection in dairy cattle: progress and challenges. J. Dairy Sci. 92 433–443. [DOI] [PubMed] [Google Scholar]
- Henderson, C. R., 1975. Best linear unbiased estimation and prediction under a selection model. Biometrics 31 423–447. [PubMed] [Google Scholar]
- Kolbehdari, D., L. R. Schaeffer and J. A. B. Robinson, 2007. Estimation of genome-wide haplotype effects in half-sib designs. J. Anim. Breed. Genet. 124 356–361. [DOI] [PubMed] [Google Scholar]
- Lee, S. H., J. H. J. van der Werf, B. J. Hayes, M. E. Goddard and P. M. Visscher, 2008. Predicting unobserved phenotypes for complex traits from whole-genome SNP data. PLoS Genet. 4 e1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legarra, A., C. Robert-Granie, E. Manfredi and J. M. Elsen, 2008. Performance of genomic selection in mice. Genetics 180 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., B. J. Hayes and M. E. Goddard, 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquet, J., W. Coppieters, N. Cambisano, J. J. Arranz, P. Berzi et al., 1999. Fine-mapping of quantitative trait loci by identity by descent in outbred populations: application to milk production in dairy cattle. Proc. Natl. Acad. Sci. USA 96 9252–9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer, L. R., 2006. Strategy for applying genome-wide selection in dairy cattle. J. Anim. Breed. Genet. 123 218–223. [DOI] [PubMed] [Google Scholar]
- Silverman, B. W., 1996. Smoothed functional principal, components analysis by choice of norm. Ann. Stat. 24 1–24. [Google Scholar]
- Solberg, T. R., A. K. Sonesson, J. A. Woolliams and T. H. E. Meuwissen, 2008. Genomic selection using different marker types and densities. J. Anim. Sci. 86 2447–2454. [DOI] [PubMed] [Google Scholar]
- Sorensen, D., and D. Gianola, 2007. Likelihood, Bayesian and MCMC Methods in Quantitative Genetics. Springer-Verlag, New York.
- VanRaden, P. M., C. P. Van Tassell, G. R. Wiggans, T. S. Sonstegard, R. D. Schnabel et al., 2009. Invited review: reliability of genomic predictions for North American Holstein bulls. J. Dairy Sci. 92 16–24. [DOI] [PubMed] [Google Scholar]
- Weedon, M. N., H. Lango, C. M. Lindgren, C. Wallace, D. M. Evans et al., 2008. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 40 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggans, G. R., P. M. VanRaden and R. L. Powell, 1992. A method for combining United-States and Canadian bull evaluations. J. Dairy Sci. 75 2834–2839. [DOI] [PubMed] [Google Scholar]
- Zhong, S., J. C. M. Dekkers, R. L. Fernando and J. L. Jannink, 2009. Factors affecting accuracy from genomic selection in populations derived from multiple inbred lines: a barley case study. Genetics 182 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]