

Abstract
Background:
Right temporal frontotemporal dementia (FTD) is an anatomic variant of FTD associated with relatively distinct behavioral and cognitive symptoms. We aimed to determine whether right temporal FTD is a homogeneous clinical, imaging, and pathologic/genetic entity.
Methods:
In this case-control study, 101 subjects with FTD were identified. Atlas-based parcellation generated temporal, frontal, and parietal grey matter volumes which were used to identify subjects with a right temporal dominant atrophy pattern. Clinical, neuropsychological, genetic, and neuropathologic features were reviewed. The subjects with right temporal FTD were grouped by initial clinical diagnosis and voxel-based morphometry was used to assess grey matter loss in the different groups, compared to controls, and each other.
Results:
We identified 20 subjects with right temporal FTD. Twelve had been initially diagnosed with behavioral variant FTD (bvFTD), and the other 8 with semantic dementia (SMD). Personality change and inappropriate behaviors were more frequent in the bvFTD group, while prosopagnosia, word-finding difficulties, comprehension problems, and topographagnosia were more frequent in the SMD group. The bvFTD group showed greater loss in frontal lobes than the SMD group. The SMD group showed greater fusiform loss than the bvFTD group. All 8 bvFTD subjects with pathologic/genetic diagnosis showed abnormalities in tau protein (7 with tau mutations), while all three SMD subjects with pathology showed abnormalities in TDP-43 (p = 0.006).
Conclusions:
We have identified 2 subtypes of right temporal variant frontotemporal dementia (FTD) allowing further differentiation of FTD subjects with underlying tau pathology from those with TDP-43 pathology.
GLOSSARY
- ADPR
= Alzheimer Disease Patient Registry;
- ADRC
= Alzheimer Disease Research Center;
- bvFTD
= behavioral variant frontotemporal dementia;
- CDR-SB
= Clinical Dementia Rating Scale sum of boxes;
- FDR
= False Discovery Rate;
- FTD
= frontotemporal dementia;
- MMSE
= Mini-Mental State Examination;
- NPI
= Neuropsychiatric Inventory;
- SMD
= semantic dementia;
- TPM
= tissue probability map;
- VBM
= voxel-based morphometry.
The frontotemporal dementias (FTD) are a group of clinical syndromes defined by varying degrees of personality change, executive dysfunction, and language impairment.1 Two of the syndromes subsumed under the rubric of FTD are behavioral variant FTD (bvFTD), characterized by changes in personality, behavior, and executive function, and semantic dementia (SMD), characterized by loss of word, visual object, and facial knowledge (prosopagnosia).2 Anatomically, FTD is characterized by varying degrees of left and right frontal and temporal lobe atrophy. A right temporal variant of FTD in which the right temporal lobe is the most atrophic region has been described.3 This variant has been associated with clinical features such as behavioral dyscontrol, personality change, aphasia, and prosopagnosia.3–7 However, it remains unclear whether right temporal variant FTD is a homogeneous entity. Therefore, the aim of this study was to investigate whether right temporal FTD is a homogeneous clinicopathologic/genetic entity.
METHODS
Subject selection.
We identified all subjects from the Mayo Clinic Alzheimer Disease Research Center (ADRC) or Alzheimer Disease Patient Registry (ADPR) recruited between January 1992 and December 2008 who fulfilled clinical criteria for a diagnosis of bvFTD or SMD2 and had a volumetric MRI scan (n = 101). From these 101 subjects, we only selected those who met imaging criteria for right temporal variant FTD (see Atlas-Based Parcellation). A control group of 30 healthy subjects who were age- and gender-matched to the right temporal FTD cohort was also identified from the ADRC/ADPR database.
Standard protocol approvals and patient consents.
Informed consent was obtained from all subjects for participation in the studies, which were approved by the Mayo Institutional Review Board.
Pathologic analysis.
Subjects enrolled in the ADRC and ADPR are consented for future pathologic and DNA analysis. Pathologic examination is conducted according to the recommendations of the Consortium to Establish a Registry for AD8 by 1 of 2 expert neuropathologists (D.W.D. or J.E.P.) as previously described.9 Final pathologic diagnoses rendered were based on the most recent published criteria.10 Pathologic nomenclature throughout the article is based on recently published consensus.11 Given that a previous study found an association between typical left-sided SMD and frontotemporal lobar degeneration with TAR DNA binding protein 43 (FTLD-TDP), type 212 (Sampathu type 113), we also performed TDP-43 typing on all subjects who were pathologically confirmed to have FTLD-TDP to determine if there was any association between FTLD-TDP type and right temporal variant FTD.
Genetic analysis.
All subjects with a positive family history of a neurodegenerative disease were screened for mutations in the microtubule associated protein tau (MAPT)14 gene and for mutations in the progranulin gene, as previously described.14 In addition, all subjects with a pathologically confirmed diagnosis of FTLD-TDP were screened for progranulin mutations.
MRI acquisition.
All subjects had a T1-weighted volumetric MRI performed with a standardized imaging protocol.14 All scanners undergo a standardized quality control calibration daily. The first MRI after presentation was used in all cases.
Atlas-based parcellation.
All images underwent preprocessing correction for gradient nonlinearity and intensity nonuniformity.15 An atlas-based parcellation technique was employed using SPM5 and the automated anatomic labeling atlas16 in order to generate grey matter volumes for the frontal, temporal, and parietal lobes for each subject. Left and right hemispheres were assessed separately for each region. Total intracranial volume was measured and used to correct regional grey matter volumes for differences in head size. Regional volumes for each of the 101 FTD subjects were compared to the 30 controls, and Z scores were calculated for each region (left and right frontal, temporal, and parietal lobes). Z scores denote how many standard deviations each subjects’ region of interest volumes were below the mean of the control group.
In order to identify right temporal variant FTD, we selected subjects who fulfilled the following criteria: 1) Z score for the right temporal lobe >0.5 standard deviations from the Z score of the left frontal lobe, right frontal lobe, left parietal lobe, and right parietal lobe; 2) Z score in the right temporal lobe > left temporal lobe. These steps identified a total of 20 subjects classified as right temporal variant FTD in which the mean right temporal lobe had a Z score of −3.8 (SD: 1.2) compared to −2.6 (1.1) for the left temporal lobe; −0.8 (1.5) for left frontal lobe; −1.2 (1.4) for right frontal lobe; −0.7 (1.5) for left parietal lobe; and −0.9 (1.1) for right parietal lobe.
Data collection.
All 20 subjects had been followed throughout their disease course by an experienced behavioral neurologist. The neurologists’ initial and annual reports were reviewed by a neurodegenerative specialist (K.A.J.) blinded to pathologic and genetic information. The following data were abstracted on all subjects at the time of MRI: demographic features (sex, age at onset, age at MRI, time from disease onset to MRI, education), clinical diagnosis at the time of presentation, measures of cognitive (Mini-Mental State Examination [MMSE]17) and functional severity (Clinical Dementia Rating Scale sum of boxes [CDR-SB]),18 and measures of executive function (Trail Making Test B and Control Oral Word Association Test),19 language function (Boston Naming Tests),19 and behavioral features (questionnaire version of the Neuropsychiatric Inventory [NPI-Q]).20 In addition, data were abstracted on 16 specific clinical features (table 1), recorded as present or absent, within the first 2 years from disease onset, and at any time during the disease course. Family history of any neurodegenerative disease was recorded. Motor neuron disease was assessed but found to be absent in all subjects.
Table 1 Frequency and definitions of symptoms in all 20 subjects with right temporal FTD

Statistical analysis.
Differences in categorical variables between groups were assessed with χ2 tests (Fisher exact tests if cells consisted of small numbers). Differences in continuous variables between groups were assessed using logistic regression analysis with group as the outcome variable adjusting for age, given that the age difference between the 2 groups approached significance. Wilcoxon signed-rank test was used to compare all paired data. Statistical analyses were performed utilizing JMP computer software (JMP Software, version 6.0.0; SAS Institute Inc., Cary, NC) with α set at 0.05.
Voxel-based morphometry.
Voxel-based morphometry (VBM),21 using SPM5, was used to assess patterns of grey matter loss as previously described.22 Customized templates and tissue probability maps (TPMs) were created by normalizing and segmenting all scans using the unified segmentation model in SPM523 with the standard MNI template and TPMs, and the normalized patient TPMs were averaged. All images were normalized to the customized template and segmented using the customized TPMs into grey matter, white matter, and CSF. All grey matter images were then modulated and smoothed with a Gaussian kernel of 8 mm full-width at half maximum.
Grey matter differences were assessed between right temporal FTD groups and controls using one-sided t-tests corrected for multiple comparisons using the False Discovery Rate (FDR) correction at p < 0.0001. Direct comparisons were performed between the right temporal FTD groups at a threshold of p < 0.005 (uncorrected) and were inclusively masked by the relevant comparisons to controls. All analyses were adjusted for the potential confounding effects of age and gender.
RESULTS
Right temporal FTD subjects.
We identified 20 subjects who met our criteria for right temporal variant FTD; 11 (55%) were female. The median age at onset was 54 years (range: 31–82). Subjects had been followed by a behavioral neurologist for median 5 years (range: 1–9). Nine (45%) of the 20 right temporal variant FTD subjects had a positive family history. The 2 most common symptoms recorded within the first 2 years of onset were personality change and inappropriate behaviors. Over the disease course, 100% of the subjects developed inappropriate behaviors (table 1). Word-finding difficulties, comprehension problems, and episodic memory loss were frequent, occurring in 60% or more of all right temporal FTD subjects over the disease course. Of the 20 subjects, 12 had been diagnosed with bvFTD, the other 8 with SMD. We therefore divided the 20 right temporal variant FTD subjects into 2 groups based on clinical diagnosis; a bvFTD group (n = 12) and a SMD group (n = 8).
Pathologic and genetic findings.
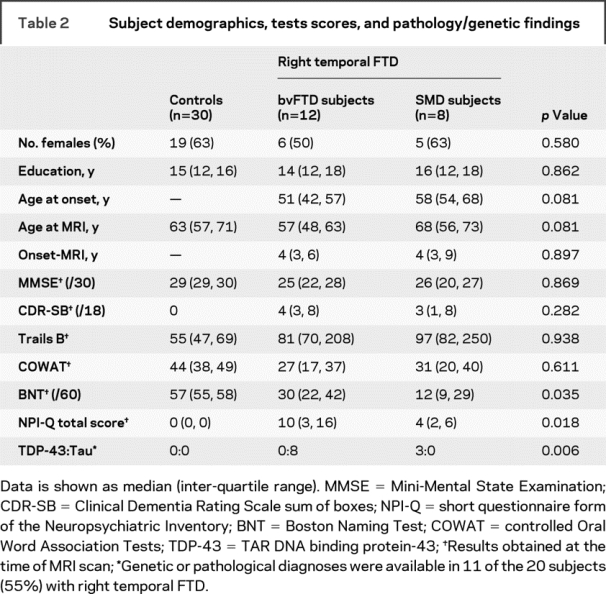
Eleven of the 20 subjects had been given a pathologic or genetic diagnosis (8 from the bvFTD group and 3 from the SMD group). An association was observed between pathology/genetic diagnosis and group (p = 0.006). All 8 of the 12 bvFTD subjects showed abnormalities in tau (table 2). One had autopsy examination showing sliver, and tau-positive rounded inclusions consistent with a diagnosis of Pick disease, while the other 7 had genetic confirmation of a MAPT mutation, from 5 different families. Conversely, all 3 SMD subjects who had brain autopsy were found to have abnormalities in TDP-43 and hence were diagnosed with FTLD-TDP. In all 3 SMD subjects, there were ubiquitin/TDP-43 immunoreactive neuronal cytoplasmic inclusions and dystrophic neurites with a distribution consistent with FTLD-TDP type 212 (Sampathu type 1).13 None had tau pathology or a mutation in progranulin.
Table 2 Subject demographics, tests scores, and pathology/genetic findings
Clinical findings.
Clinical features abstracted from the medical records differed across the 20 right temporal FTD subjects when divided into the 2 groups based on clinical diagnosis (figure 1). During the first 2 years from onset, personality change and inappropriate behaviors were significantly more common in the bvFTD group, while prosopagnosia, word-finding difficulties, comprehension problems, and topographagnosia (landmark agnosia resulting in geographic orientation) were significantly more frequent in the SMD group. Over the entire disease course, sweet tooth and parkinsonism were significantly more common in the bvFTD group, while prosopagnosia and topographagnosia remained significantly more frequent in the SMD group. There was a difference in positive family history (bvFTD, n = 9; SMD, n = 0; p = 0.001).

Figure 1 Clinical features abstracted from the medical records
Bar graphs showing the proportion of subjects in the behavioral variant frontotemporal dementia (bvFTD) and semantic dementia (SMD) groups with clinical symptoms within the first 2 years of onset (A) and throughout the entire disease (B). *Difference between the bvFTD and SMD groups at p < 0.05. **Difference between the bvFTD and SMD groups at p < 0.001.
There were significant differences in neuropsychological and behavioral features when the SMD group was directly compared to the bvFTD group (table 2). The bvFTD group performed significantly worse on the NPI-Q, a measure of behavioral dyscontrol, while the SMD group performed significantly worse on the Boston Naming Test.
There was a trend for younger age at onset, and time of MRI scan, in the bvFTD group compared to the SMD group (table 2).
Atlas-based parcellation.
Actual Z scores for all regions of interest can be found in table e-1 on the Neurology® Web site at www.neurology.org. There was a difference between mean right and left temporal lobe Z scores for the bvFTD (p = 0.0006) and the SMD groups (p < 0.0001). The mean difference between right and left temporal lobe Z scores was greater in the SMD group (Z score = 1.7) than the mean difference between right and left temporal lobe for the bvFTD group (Z score = 0.9; p = 0.004).
Voxel-based morphometry results.
The VBM analysis showed both similarities and differences across the right temporal FTD subjects when divided into the 2 groups. As expected from the manner in which we defined the subjects, both groups showed the greatest grey matter loss in the right temporal lobe when compared to controls (figure 2). The left temporal lobe was also involved in both groups, but to a lesser degree than the right (figure 2). Some grey matter loss was observed outside the temporal lobe in the bvFTD group, involving medial and dorsolateral frontal lobes, orbitofrontal cortex, anterior cingulate, insula, caudate nucleus, and parietal lobes (figure 2). Grey matter loss in the SMD group was almost exclusively located in temporal lobes, with only minor loss observed in medial orbitofrontal cortex and insula. The bvFTD group showed greater loss in frontal lobes and left anteromedial temporal lobe (amygdala/hippocampus) than the SMD group on direct comparison (figure 3). The SMD group showed greater loss in right lateral temporal and fusiform gyrus than the bvFTD group (figure 3).

Figure 2 Patterns of atrophy in the right temporal groups compared to controls
Regions of grey matter loss identified in the behavioral variant frontotemporal dementia (A) and semantic dementia (B) groups of right temporal frontotemporal dementia compared to controls. Results are shown on 3-dimensional renders of the brain and representative coronal slices at p < 0.0001 (corrected for multiple comparisons using False Discovery Rate). Age and gender were included in the model as nuisance variables.

Figure 3 Patterns of atrophy in the right temporal groups compared to each other
Regions of grey matter loss in each group contrasted to the other were identified by performing direct comparisons between the 2 groups across the whole brain and then masking these comparisons by the relevant comparisons to controls. Therefore, in order to identify regions of greater loss in the behavioral variant frontotemporal dementia (bvFTD) group compared to the semantic dementia (SMD) group, we performed a contrast of bvFTD vs SMD and inclusively masked it with the bvFTD vs controls contrast (A). Conversely, in order to identify regions of greater loss in the SMD group compared to the bvFTD group, we performed a contrast of SMD vs bvFTD and inclusively masked it with the SMD vs controls contrast (B). The direct comparison analyses were performed across the whole brain at p < 0.005 uncorrected for multiple comparisons. A p value of p < 0.001 uncorrected was applied to the masks. Age and gender were included in the model as nuisance variables. The results remained the same if time from onset to scan was also included as a nuisance variable. Results are shown on representative coronal slices.
DISCUSSION
In this study we have identified 2 distinct clinicopathologic subtypes of right temporal variant FTD, one associated with tau pathology and the other with TDP-43 pathology. These results may advance our knowledge of right temporal variant FTD4,6,7 and FTD in general and could have significant implications for differentiating tau from TDP-43 as the underlying substrate of FTD.
Using atlas-based parcellation, we identified 20 FTD subjects in which the right temporal lobe was the most atrophic region, hence our designation of the cohort as right temporal variant FTD. Similar to other studies, our cohort was characterized by personality change, inappropriate behaviors, episodic memory loss, prosopagnosia, and topographagnosia.3–7 We found personality change and inappropriate behaviors to be the most frequent features of the cohort.4,6 Other features present at any time during the disease course were parkinsonism, compulsive behaviors, craving for sweet foods (sweet tooth), and obsession with puzzles/jigsaws. A few subjects had indiscriminate eating behaviors such as eating soap and dirt, 2 of whom were noted to have persistent hunger. We hypothesize that indiscriminate eating may be occurring secondary to loss of food knowledge and possibly a feeling of persistent hunger. Stereotypical behaviors, previously demonstrated to be associated with striatal atrophy,24 are rare in right temporal variant FTD.
Our 20 right temporal FTD subjects were divided into 2 groups based on clinical diagnosis. Taking into account the significant differences identified in clinical, neuropsychological, and neurobehavioral features between both groups, it is reasonable to suggest two distinct clinico-psychological profiles of right temporal variant FTD. Therefore, at onset, the bvFTD phenotype is best defined by personality change, inappropriate behaviors, positive family history, and relatively poor performance on neurobehavioral testing with better performance on confrontational naming tests. The SMD phenotype is defined by word-finding and comprehension difficulties, prosopagnosia, and topographagnosia,2 absent family history, relatively better performance on neurobehavioral testing, and poor performance on naming tests. As disease progresses, the development of sweet tooth and parkinsonism suggest the bvFTD phenotype, although personality change and inappropriate behaviors will be less helpful to differentiate the 2 phenotypes, as these 2 features will occur later in the SMD subjects.
The results of the VBM analysis shed light on some of these findings. We identified significantly more frontal lobe atrophy, on direct comparison, in the bvFTD group than the SMD group, which concurs with the fact that we found more behavioral dyscontrol in the bvFTD group. Similarly, the presence of sweet tooth, which was common in our bvFTD subjects, has been associated with atrophy of the orbitofrontal cortex,25 and indeed the bvFTD group showed a greater degree of orbitofrontal loss. Conversely, the SMD group showed greater loss in the right fusiform and lateral temporal lobes on direct comparison. We have previously shown that prosopagnosia may be associated with atrophy of the right fusiform and parahippocampal gyri, as well as mesial temporal structures.26 Of these structures, only the fusiform gyrus was involved to a greater degree in the SMD group with prosopagnosia compared to the bvFTD group, suggesting that the fusiform gyrus is the most likely region accounting for prosopagnosia in SMD. Another imaging characteristic differentiating the SMD subjects from the bvFTD subjects, in right temporal FTD, is greater temporal lobe asymmetry in those with SMD compared to lesser temporal lobe asymmetry in those with bvFTD. The bvFTD subjects in this study overlap with those reported in a recent cluster-based study in which we demonstrated that bvFTD subjects can show four distinct patterns of atrophy.27 The majority of the right temporal FTD subjects were indeed classified as showing a predominantly temporal pattern of atrophy, although some were classified as showing a frontotemporal pattern reflecting the additional involvement of the frontal lobes.
The most important finding of this study was the very significant separation of pathologies. All subjects in the bvFTD group with pathologic or genetic confirmation had tau abnormalities, the majority of which were related to mutations in the MAPT gene. Conversely, all 3 SMD subjects who had pathology were found to have pathology related to TDP-43. More impressive, all 3 subjects in the SMD group with FTLD-TDP shared identical TDP-43 typing: FTLD-TDP type 212 (Sampathu type 113). These pathologic/genetic differences also shed light on some of the clinical findings. MAPT mutations were common in the bvFTD group and likely account for the high frequency of parkinsonism observed in the bvFTD group,28 the high frequency of a positive family history,29 and the trend for the bvFTD subjects to have a slightly young age at onset.30 We have also previously found that MAPT mutations are associated with anteromedial temporal atrophy which concurs with the finding of greater left anteromedial temporal loss in the bvFTD group.14 It should be noted, however, that although MAPT mutations are associated with right temporal atrophy in this study, we cannot conclude that all subjects with a MAPT mutation show dominant right temporal lobe atrophy.14 Given the relatively high frequency of MAPT mutation in our bvFTD group, it is unclear if these results generalize to pure sporadic FTD cohorts. Specifically, it is unclear whether there would be any association between bvFTD and tau in a right temporal variant FTD cohort without MAPT mutations, although one subject did show sporadic Pick disease. It is actually more likely that SMD with TDP-43 will underlie right temporal variant sporadic FTD. Given that we had such a high proportion of MAPT mutations in our cohort, and that we only had 3 pathologically confirmed subjects in the SMD cohort, these results will need to be replicated in a larger cohort, with comprehensive genetic analysis and pathologic confirmation. We are not surprised that we did not find any mutations in the progranulin gene since dominant right temporal lobe atrophy is not a feature of progranulin gene mutations.14
The findings from this study have significant implications and further help to decipher the complexity of the FTD field. Differentiating subjects with underlying tau pathology from those with TDP-43 pathology is critical to the field,1 especially in the current era of developing treatment which appears to be focused at the protein level. The findings from this study suggest that subjects with FTD with right temporal lobe atrophy, prominent behavioral features, a positive family history, some frontal lobe atrophy, and modest temporal lobe asymmetry will more likely have underling tau pathology, whereas subjects with prosopagnosia, word-finding and comprehension difficulties, topographagnosia, absent family history, absent frontal lobe atrophy, and marked temporal lobe asymmetry will more likely have TDP-43 pathology. It is therefore important to understand that although predicting pathology is difficult in bvFTD subjects as a whole, it is possible that prediction may be improved if MRI is used to identify those with a pattern of atrophy consistent with right temporal variant FTD. This approach would be similar to the approach used in FTD subjects with left hemisphere atrophy and an aphasia presentation, where those with a nonfluent aphasia and perisylvian atrophy with motor speech impairment associate with tau pathology, while those with a fluent aphasia and left anterior temporal lobe atrophy associate with TDP-43 pathology.1
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Dr. Keith A. Josephs.
ACKNOWLEDGMENT
The authors thank Rosa Rademakers, PhD, and Matthew Baker, BS, for conducting the genetic analyses.
DISCLOSURE
Dr. Whitwell reports no disclosures. Dr. Knopman serves as an Associate Editor for Neurology®; has served on a data safety monitoring board for Sanofi-Aventis Pharmaceuticals; is an investigator in a clinical trial sponsored by Elan Pharmaceuticals and Forest Pharmaceuticals; and receives research support from the NIH [R01-AG023195 (PI), R01-AG11378 (Co-I), P50 AG16574 (Co-I), U01 AG 06786 (Co-I), and R01 HL70825 (Co-I)]. Dr. Boeve has served as a consultant to GE Healthcare; and receives research support from Myriad Genetics Inc., the Alzheimer's Association (PI), and from the NIH as a Co-I [P50 AG16574, UO1 AG06786, and RO1 AG15866.] Dr. Vemuri and M.L. Senjem report no disclosures. Dr. Parisi serves as a Section Editor for Neurology®; serves on the US Government Defense Health Board and as Chair of the Subcommittee for Laboratory Services and Pathology; receives royalties from publishing Principles & Practice of Neuropathology, 2nd ed. (Oxford University Press, 2003); and receives research support from the NIH [P50 AG16574 (Co-I) and U01 AG03949 (Co-I)]. Dr. Ivnik serves on the editorial boards of The Clinical Neuropsychologist and Aging, Neuropsychology, and Cognition; and receives research support from the NIA as Co-I [U01 AG06786 and P50 AG16574]. Dr. Dickson serves on the editorial boards of the American Journal of Pathology, Journal of Neuropathology and Experimental Neurology, Brain Pathology, Neurobiology of Aging, Journal of Neurology, Neurosurgery, and Psychiatry, Annals of Neurology, and Neuropathology; and receives research support from the NIH [P50-AG25711 (CL), P50-AG16574 (CL), P50-NS40256 (PI), P01-AG17216 (PI), P01- AG03949 (Co-I), and R01-AG15866 (Co-I)]. Dr. Petersen serves on scientific advisory boards for Elan Pharmaceuticals, Wyeth Pharmaceuticals, and GE Healthcare; receives royalties from publishing Mild Cognitive Impairment (Oxford University Press, 2003); and receives research support from the NIH [P50-AG16574 (PI) and U01-AG06786 (PI), R01-AG11378 (Co-I), and U01–24904 (Co-I)]. Dr. Jack serves as a consultant for Elan Corporation; and receives research support from Pfizer, Inc., the NIA [AG11378 (PI), P50-AG16574 (Co-I), and U01 AG024904-01 (Co-I)], the NIH [R01-AG11378], and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation. Dr. Josephs is funded by the NIH Roadmap Multidisciplinary Clinical Research Career Development Award Grant (K12/NICHD)-HD49078 (PI) and the Morris K. Udall PD Research Center of Excellence NIH/NINDS P50 NS40256 (Co-I).
Supplementary Material
Address correspondence and reprint requests to Dr. Keith A. Josephs, Department of Neurology, Mayo Clinic, Rochester, MN 55905 josephs.keith@mayo.edu
Supplemental data at www.neurology.org
Supported by the NIH Roadmap Multidisciplinary Clinical Research Career Development Award Grant (K12/NICHD)-HD49078, by grants P50 AG16574, U01 AG06786, and R01 AG11378 from the National Institute on Aging, Bethesda, MD, and the generous support of the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation, USA.
Disclosure: Author disclosures are provided at the end of the article.
Received April 20, 2009. Accepted in final form July 17, 2009.
REFERENCES
- 1.Josephs KA. Frontotemporal dementia and related disorders: deciphering the enigma. Ann Neurol 2008;64:4–14. [DOI] [PubMed] [Google Scholar]
- 2.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 3.Miller BL, Chang L, Mena I, Boone K, Lesser IM. Progressive right frontotemporal degeneration: clinical, neuropsychological and SPECT characteristics. Dementia 1993;4:204–213. [DOI] [PubMed] [Google Scholar]
- 4.Chan D, Anderson V, Pijnenburg Y, et al. The clinical profile of right temporal lobe atrophy. Brain Epub 2009. [DOI] [PubMed]
- 5.Edwards-Lee T, Miller BL, Benson DF, et al. The temporal variant of frontotemporal dementia. Brain 1997;120:1027–1040. [DOI] [PubMed] [Google Scholar]
- 6.Seeley WW, Bauer AM, Miller BL, et al. The natural history of temporal variant frontotemporal dementia. Neurology 2005;64:1384–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson SA, Patterson K, Hodges JR. Left/right asymmetry of atrophy in semantic dementia: behavioral-cognitive implications. Neurology 2003;61:1196–1203. [DOI] [PubMed] [Google Scholar]
- 8.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD): part II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486. [DOI] [PubMed] [Google Scholar]
- 9.Knopman DS, Parisi JE, Salviati A, et al. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol 2003;62:1087–1095. [DOI] [PubMed] [Google Scholar]
- 10.Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 2007;114:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 2009;117:15–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mackenzie IR, Baborie A, Pickering-Brown S, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol 2006;112:539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sampathu DM, Neumann M, Kwong LK, et al. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol 2006;169:1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitwell JL, Jack CR, Jr., Boeve BF, et al. Voxel-based morphometry patterns of atrophy in FTLD with mutations in MAPT or PGRN. Neurology 2009;72:813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jack CR, Jr., Bernstein MA, Fox NC, et al. The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging 2008;27:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tzourio-Mazoyer N, Landeau B, Papathanassiou D, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage 2002;15:273–289. [DOI] [PubMed] [Google Scholar]
- 17.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. Journal Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 18.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry 1982;140:566–572. [DOI] [PubMed] [Google Scholar]
- 19.Lezak MD, Howieson DB, Loring DW. Neuropsychological Assessment. 4th ed. New York: Oxford University Press, 2004. [Google Scholar]
- 20.Kaufer DI, Cummings JL, Ketchel P, et al. Validation of the NPI-Q, a brief clinical form of the Neuropsychiatric Inventory. J Neuropsychiatry Clin Neurosci 2000;12:233–239. [DOI] [PubMed] [Google Scholar]
- 21.Ashburner J, Friston KJ. Voxel-based morphometry: the methods. NeuroImage 2000;11:805–821. [DOI] [PubMed] [Google Scholar]
- 22.Jack CR Jr., Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain 2008;131:665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashburner J, Friston KJ. Unified segmentation. NeuroImage 2005;26:839–851. [DOI] [PubMed] [Google Scholar]
- 24.Josephs KA, Whitwell JL, Jack CR, Jr. Anatomic correlates of stereotypies in frontotemporal lobar degeneration. Neurobiol Aging 2008;29:1859–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitwell JL, Sampson EL, Loy CT, et al. VBM signatures of abnormal eating behaviours in frontotemporal lobar degeneration. Neuroimage 2007;35:207–213. [DOI] [PubMed] [Google Scholar]
- 26.Josephs KA, Whitwell JL, Vemuri P, et al. The anatomic correlate of prosopagnosia in semantic dementia. Neurology 2008;71:1628–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whitwell JL, Przybelski SA, Weigand SA, et al. Distinct anatomical subtypes of the behavioral variant of frontotemporal dementia: a cluster analysis study. Brain Epub 2009 Sep 17. [DOI] [PMC free article] [PubMed]
- 28.Wszolek ZK, Pfeiffer RF, Bhatt MH, et al. Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann Neurol 1992;32:312–320. [DOI] [PubMed] [Google Scholar]
- 29.Rizzu P, Van Swieten JC, Joosse M, et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet 1999;64:414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pickering-Brown SM, Rollinson S, Du Plessis D, et al. Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 2008;131:721–731. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.