

Abstract
Background:
Frontotemporal lobar degeneration (FTLD) is a genetically and pathologically heterogeneous neurodegenerative disorder.
Methods:
We collected blood samples from a cohort of 225 patients with a diagnosis within the FTLD spectrum and examined the heritability of FTLD by giving each patient a family history score, from 1 (a clear autosomal dominant history of FTLD) through to 4 (no family history of dementia). We also looked for mutations in each of the 5 disease-causing genes (MAPT, GRN, VCP, CHMP2B, and TARDP) and the FUS gene, known to cause motor neuron disease.
Results:
A total of 41.8% of patients had some family history (score of 1, 2, 3, or 3.5), although only 10.2% had a clear autosomal dominant history (score of 1). Heritability varied across the different clinical subtypes of FTLD with the behavioral variant being the most heritable and frontotemporal dementia–motor neuron disease and the language syndromes (particularly semantic dementia) the least heritable. Mutations were found in MAPT (8.9% of the cohort) and GRN (8.4%) but not in any of the other genes. Of the remaining patients without mutations but with a strong family history, 7 had pathologic confirmation, falling into 2 groups: type 3 FTLD-TDP without GRN mutations (6) and FTLD-UPS (1).
Conclusion:
These findings show that frontotemporal lobar degeneration (FTLD) is a highly heritable disorder but heritability varies between the different syndromes. Furthermore, while MAPT and GRN mutations account for a substantial proportion of familial cases, there are other genes yet to be discovered, particularly in patients with type 3 FTLD-TDP without a GRN mutation.
GLOSSARY
- bvFTD
= behavioral variant frontotemporal dementia;
- CBS
= corticobasal syndrome;
- FTLD
= frontotemporal lobar degeneration;
- LPA
= logopenic/phonologic variant of primary progressive aphasia;
- MND
= motor neuron disease;
- PNFA
= progressive nonfluent aphasia;
- PPA
= primary progressive aphasia;
- PSP
= progressive supranuclear palsy;
- SemD
= semantic dementia.
Frontotemporal lobar degeneration (FTLD) is a genetically and pathologically heterogeneous degenerative disorder.1,2 A number of different clinical syndromes fall into the FTLD spectrum: the most common subtype consists of patients who present with behavioral or personality changes (behavioral variant frontotemporal dementia or bvFTD). Less commonly, patients present with language impairment and 3 different disorders are described (often under the collective term primary progressive aphasia or PPA): semantic dementia (SemD), progressive nonfluent aphasia (PNFA), and the logopenic/phonologic variant of PPA (LPA).3,4 There is also an overlap of FTLD with motor disorders: the parkinsonian disorders, corticobasal syndrome (CBS) and progressive supranuclear palsy (PSP) and motor neuron disease (known as FTD-MND when there is an overlap).5,6 FTLD is commonly familial7 and 5 genes are currently known to cause FTLD, of which 2 are relatively common (microtubule-associated protein tau, MAPT, and progranulin, GRN) and 3 are rare causes (valosin-containing protein, VCP, chromatin modifying protein 2B, CHMP2B, and the gene encoding TDP-43, TARDP).2,8 MAPT mutations are associated with tau-positive inclusions pathologically as are the generally sporadic pathologies of corticobasal degeneration, PSP, and Pick disease. GRN, VCP, TARDP, and CHMP2B mutations are associated with the other major pathologic FTLD type where there are tau-negative inclusions.1,9 This second subtype can be further subdivided into those with TDP-43-positive pathology, known as FTLD-TDP (of which there are 4 subtypes; this group includes GRN, VCP, and TARDP mutations) and those without, known as FTLD-UPS (including CHMP2B mutations). The 5 known genes do not account for all familial cases of FTLD, however, suggesting that there are other disease-causing genes yet to be discovered. This study was designed to look at the heritability of FTLD in each of the different clinical syndromes and to investigate to what extent the known genes account for this heritability. We also looked at the patients with strong family histories of FTLD but without known mutations and the underlying pathologic causes in this group.
METHODS
Sample collection.
Blood was collected for DNA extraction from patients attending the Specialist Cognitive Disorders Clinic at the National Hospital for Neurology and Neurosurgery, Queen Square, London, and also from patients involved in studies of FTLD at the Dementia Research Centre, Institute of Neurology, Queen Square, London. Samples were taken from 225 patients who had been diagnosed by clinical, behavioral, and neuropsychologic assessments with a diagnosis within the FTLD spectrum according to consensus criteria.3,10–12 Although samples were collected from patients with and without a family history, there is likely to be some ascertainment bias toward familial cases.
Standard protocol approvals, registrations, and patient consents.
Ethical approval for the study was obtained from the National Hospital for Neurology and Neurosurgery Local Research Ethics Committee. Written research consent was obtained from all patients participating in the study.
Analysis of family history.
All patients were given a modified Goldman score between 1 and 4 as per Goldman et al.,13,14 where 1 is an autosomal dominant family history of FTLD, MND, CBS, or PSP, defined as the presence of at least 3 affected people in 2 generations with 1 person being a first-degree relative of the other 2, 2 is familial aggregation of 3 of more family members with dementia but not meeting criteria for 1, 3 is 1 other affected family member with dementia (modified to give a score of 3 only if there is a history of young-onset dementia within the family, i.e., less than 65, and 3.5 if onset above 65), and 4 is no or unknown family history. All patients had had a structured clinical interview which had included a detailed family tree. This had been discussed with the patient and family members (a minimum of 1 other person). The data for this study were ascertained from a review of all of the clinical notes: data were available on 222 of the cases with only 3 patients scoring 4 because of an unknown family history (2 with bvFTD and 1 with FTD-MND).
Genetic analysis.
All 225 patients were screened for mutations in MAPT and GRN, detecting 39 pathogenic mutations. Of the remaining 186 mutation negative patients, sequencing was obtained for VCP exons 3, 5, 6, and 10 in 160 patients, TARDBP exons 4 and 6 in 179 patients, and CHMP2B in 92 patients. We also sequenced exon 15 of the FUS gene in 183 patients, which has previously been shown to be causative of motor neuron disease, although currently no mutations have been found in FTLD.15,16 PCR amplicons were generated using primers at 500 μM in MegaMix Blue PCR cocktail (Microzone). Details of primers and PCR conditions are available on request. Amplicons cleaned with Microclean (Microzone) were sequenced using Applied Biosystems BigDye v1.1 cycle sequence chemistry with 1 μL BigDye, 5 μL BetterBuffer (Microzone), 0.75 μL sequencing primer at 5 pmol/μL, 2.5 ng of cleaned amplicon, and ddH2O to a final volume of 15 μL. Following 25 thermal cycles, sequencing products were precipitated and cleaned used standard EtOH/EDTA methodology and electrophoresed on an ABI 3130xl automated sequencer. Data were analyzed using ABI Seqscape software v2.5. For details of GRN and MAPT sequencing, see Beck et al., 2008,14 and Janssen et al., 2002.17
RESULTS
Demographic and family history data.
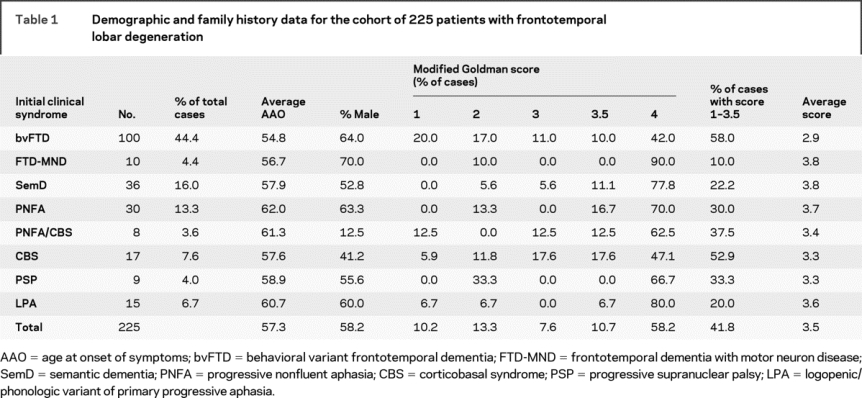
Almost half of the 225 patients had bvFTD as their initial clinical syndrome (44.4%) with the next most common disorders being SemD (16.0%) and PNFA (13.3%). Smaller numbers had CBS or a CBS/PNFA overlap syndrome, PSP or LPA. Average age at onset for the different groups was between 54.8 years (bvFTD) and 62.0 years (PNFA) with a total mean of 57.3 years. In total, 58.2% of the patients were male, with more male patients in each of the groups apart from the CBS and CBS/PNFA overlap groups. A total of 10.2% of patients had an autosomal dominant inheritance (as defined by a modified Goldman score of 1) but the heritability of FTLD was substantially higher (41.8%) when a family history was defined by a modified Goldman score of 1, 2, 3, or 3.5. The bvFTD group had the largest proportion of cases with a family history (58.0% with modified Goldman score 1 to 3.5 and an average modified Goldman score of 2.9) and the least familial of the disorders were FTD-MND (10.0%, 3.8), LPA (20.0%, 3.6), and SemD (22.2%, 3.8) (table 1).
Table 1 Demographic and family history data for the cohort of 225 patients with frontotemporal lobar degeneration
Genetic analysis.
Mutations were found in the MAPT and GRN genes but no mutations were found in the CHMP2B, VCP, TARDBP, or FUS genes. In total, 20 patients (8.9%) had mutations in MAPT (15 probands) and 19 patients (8.4%) had mutations in GRN (13 probands). Of the MAPT mutations, 13 (from 8 families) had an intronic 10 + 16 mutation of which 7 families have previously been described.17 The other previously described mutations were an intronic 10 + 19 mutation as well as deltaK280, L284R, N296N,18 S320F, and G389R mutations. A novel MAPT variant was also found, N286N, a synonymous change similar to the N296N which is thought to be pathogenic via its effect on the splicing of exon 10.18 Most patients presented with bvFTD (although many developed semantic impairment as the disease progressed) apart from the N296N (CBS) and the L284R (PSP) mutations. The GRN mutations were 10 C31fs mutations (from 4 families) and 2 385_388delAGTC mutations (from 2 families).14 Other mutations were 603_603insC, 1494_ 1498delAGTGG, Q300X, L469F, A199V mutations (all previously described in Beck et al., 200814) as well as 1048_1049insG19 and R493X mutations. Patients were diagnosed with bvFTD (C31fs, 385_388delAGTC, Q300X), PPA (PNFA or LPA: C31fs, L469F, R493X, 603_603insC), or CBS (1048_ 1049insG, A199V, C31fs, 1494_1498delAGTGG) with 2 patients having a PNFA/CBS overlap (C31fs, 1494_1498delAGTGG).
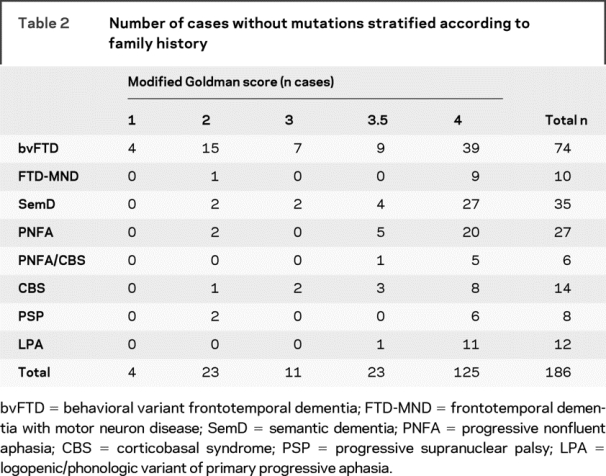
Of the 186 patients without a known mutation, we re-examined the number of cases with a family history and in particular we looked at whether any of the “familial” cases had postmortem confirmation of FTLD pathology (table 2). The majority of these cases (125) had a modified Goldman score of 4 but 4 cases with an autosomal dominant family history (modified Goldman score of 1) were still without a known mutation (all with bvFTD) and in total 61 cases with a modified Goldman score of 1, 2, 3, or 3.5 were not known to have a mutation. We looked particularly at those cases with a score of 1, 2, or 3 (38 in total) as a score of 3.5 may well represent another family member with old-age onset dementia and therefore less likely to be a true familial history of FTLD. Of these 38 cases, 7 had pathologic confirmation of disease (6 bvFTD and 1 FTD-MND). All of these cases had tau-negative FTLD pathology: 1 case with bvFTD was known to have ubiquitin-positive, TDP-43 negative pathology (FTLD-UPS) without intranuclear inclusions similar to the pathology found in the Danish family with a mutation in CHMP2B (but in this case without a CHMP2B mutation) but the other 6 all had FTLD-TDP (i.e., TDP-43 pathology) with all having type 3 pathology according to consensus criteria.1,9
Table 2 Number of cases without mutations stratified according to family history
DISCUSSION
Our results confirm previous findings that FTLD is a highly heritable degenerative disorder. Heritability varies between the different clinical syndromes, with SemD and LPA having a much lower percentage of cases with a family history compared with bvFTD. SemD is typically a sporadic disease and is associated with type 1 FTLD-TDP20 although patients with MAPT mutations will often develop semantic impairment later in the disease (with behavioral symptoms initially). There are few reports of the associations of LPA with family history although some studies suggest that the majority of cases of LPA actually have Alzheimer-type pathology rather than an FTLD pathology, which would account for the lower rate of reported family history in this group.4,21 A previous study suggested that FTD-MND was the most heritable of the FTLD syndromes13 but in our series it was the least heritable, albeit with low numbers in our cohort. Inconsistent results with other series may reflect ethnogeographic clustering of particular causal mutations. Numbers were also low in the PSP group, limiting the ability to interpret these data.
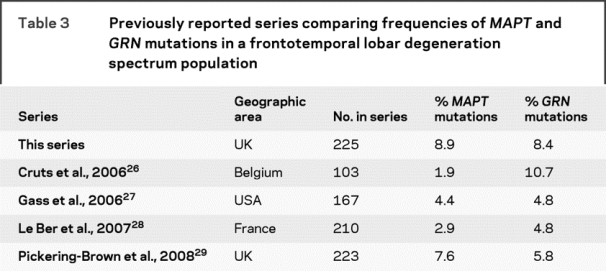
Mutations in MAPT and GRN are relatively common and have a similar prevalence in our series. However, there is variability in the prevalence geographically across different reported series with MAPT mutation frequency between 3% and 14%22–25 and GRN mutation frequency between 1% and 16%,26–31 with some series reporting vastly different frequencies within the same country, e.g., 2 studies of FTLD patients in Italy found GRN mutation frequencies of 1.6% and 15.2%.30,31 A number of recent series have compared the frequency of MAPT and GRN mutations in FTLD populations26–29 (table 3): some countries have families with a founder effect causing higher GRN mutation prevalence than MAPT, e.g., in Belgium.26 In the United Kingdom, the 10 + 16 MAPT mutation is common with a known founder effect32 which is likely to account for the higher frequency of MAPT mutations in some series,29 although we have also previously shown that patients in our series with the GRN C31fs mutation are part of the same family,14 which is likely to at least partly account for the similar prevalence of GRN and MAPT mutations in our series.
Table 3 Previously reported series comparing frequencies of MAPT and GRN mutations in a frontotemporal lobar degeneration spectrum population
We found no mutations in VCP, CHMP2B, or TARDP consistent with previous series suggesting that these are rare causes of FTLD.2 It remains possible that causal mutations in these genes are present in unscreened exons of these 3 genes, although our sequencing strategy covered all known mutations in these genes to date. We also found no mutations in FUS, suggesting that mutations in this gene are either not causative or are a very rare cause of FTLD.
Taking into account the known mutations, many patients were still found to have a strong family history, suggesting that there are still unknown genes that cause FTLD.33 One locus (on chromosome 9) is known for patients with a clinical phenotype of FTD-MND34; however, this is associated with type 2 FTLD-TDP. Analysis of the pathologic cases within this subgroup suggests that there are at least 2 other groups of patients with a family history without a known mutation: those with FTLD-UPS and those with type 3 FTLD-TDP without a GRN mutation (some of whom have a clinical phenotype of FTD-MND). A number of series have now been reported with FTLD-UPS (ubiquitin-positive, TDP-43 negative) pathology although these have all been reported as sporadic cases.35,36 The family reported here is pathologically distinct from these patients, lacking the intranuclear inclusions seen in these cases. Although similar pathologically to cases with CHMP2B mutations, the single case in our series was negative for mutations in this gene. Studies of type 3 FTLD-TDP suggest that between 30% and 60% of such patients have GRN mutations but some patients negative for GRN mutations still have a family history.37,38,39 There are no known loci associated with such patients, suggesting further work needs to be done to clarify the genetic cause in this group as well as patients with familial FTLD-UPS.
ACKNOWLEDGMENT
This work was undertaken at UCLH/UCL, which received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme. The Dementia Research Centre is an Alzheimer's Research Trust Co-ordinating Centre.
DISCLOSURE
Dr. Rohrer has received research support from the Wellcome Trust (Clinical Research Fellowship) and Brain (Exit Scholarship). R. Guerreiro, Dr. Vandrovcova, J. Uphill, D. Reiman, and J. Beck report no disclosures. Dr. Isaacs receives research support from the Medical Research Council Prion Unit, Alzheimer's Research Trust, and UCL Hospitals Clinical Research and Development Committee. A. Authier and R. Ferrari report no disclosures. Dr. Fox has served on scientific advisory boards for the Alzheimer's Research Forum and GE Healthcare; may accrue revenue on Patent PCT/GB2008/001537 (issued: 04/05/2007): QA Box; has received honoraria from GE Healthcare and Lancet Neurology (reviewer fee); has served as a consultant to Eli Lilly and Company, Abbott, and Lundbeck Inc.; has received research support (to the Dementia Research Centre) from Elan Corporation, Wyeth, Lundbeck Inc., Sanofi-Aventis, IXICO Ltd., Pfizer Inc., and Neurochem Inc.; and receives research support from the MRC [G0801306 (PI) and G0601846 (PI)], the NIH [U01 AG024904 (Co-I)], and the Alzheimer Research Trust [ART/RF/2007/1 (PI)]. Dr. Mackenzie serves on the advisory board of the 2009 International Conference on Alzheimer's Disease; has received funding for travel and speaker honoraria for non-industry-sponsored activities; serves on editorial boards for the Journal of Neuropathology & Experimental Neurology, the Journal of Neuroinflammation, and Clinical Neuropathology; and receives research support from Canadian Institutes of Health Research, Pacific Alzheimer Research Foundation, and Michael Smith Foundation for Health Research. Dr. Warren and Dr. De Silva report no disclosures. Dr. Holton serves on the editorial board of Neuropathology & Applied Neurobiology and has received research support from the Alzheimer's Research Trust, The Sarah Matheson Trust, Action Medical Research, BrainNet Europe, and the Myositis Support Group. Dr. Revesz serves on editorial boards for Acta Neuropathological, Clinical Neuropathology, and Neuropathology & Applied Neurobiology; has received speaker honoraria from Boehringer Ingelheim; and receives research support from Orion Pharma, the Alzheimer's Research Trust, the European Commission, BrainNet Europe II, Network of Excellence, the Sarah Matheson Trust, and the Parkinson's Disease Society, UK. Dr. Hardy serves as a consultant to Eisai Inc. Dr. Mead receives research support from the Medical Research Council Prion Unit and Wellcome Trust. Dr. Rossor serves on a scientific advisory board for Elan Corporation and Wyeth; serves as Editor-in-Chief of the Journal of Neurology, Neurosurgery and Psychiatry, and on the editorial boards of Practical Neurology, Dementia and Geriatric Cognitive Disorders, Neurodegenerative Diseases, and the British Medical Journal; receives royalties from publishing Brain's Diseases of the Nervous System (11th edition), Oxford University Press, 2001, and Brain's Diseases of the Nervous System (12th edition), Oxford University Press, 2009; and receives research support from the Department of Health and the Alzheimer's Research Trust.
Address correspondence and reprint requests to Professor Martin N. Rossor, Dementia Research Centre, Institute of Neurology, Queen Square, London WC1N 3BG, UK mrossor@dementia.ion.ucl.ac.uk
Disclosure: Author disclosures are provided at the end of the article.
Received May 1, 2009. Accepted in final form August 3, 2009.
REFERENCES
- 1.Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 2007;114:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mackenzie IR, Rademakers R. The molecular genetics and neuropathology of frontotemporal lobar degeneration: recent developments. Neurogenetics 2007;8:237–248. [DOI] [PubMed] [Google Scholar]
- 3.Gorno-Tempini ML, Dronkers NF, Rankin KP, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol 2004;55:335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gorno-Tempini ML, Brambati SM, Ginex V, et al. The logopenic/phonological variant of primary progressive aphasia. Neurology 2008;71:1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kertesz A, Davidson W, Munoz DG. Clinical and pathological overlap between frontotemporal dementia, primary progressive aphasia and corticobasal degeneration: The Pick complex. Dement Geriatr Cogn Disord 1999;10:46–49. [DOI] [PubMed] [Google Scholar]
- 6.Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002;59:1077–1079. [DOI] [PubMed] [Google Scholar]
- 7.Chow TW, Miller BL, Hayashi VN, Geschwind DH. Inheritance of frontotemporal dementia. Arch Neurol 1999;56:817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benajiba L, Le Ber I, Camuzat A, et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol 2009;65:470–473. [DOI] [PubMed] [Google Scholar]
- 9.Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 2009;117:15–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 11.Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 2003;54 suppl 5:S15–S19. [DOI] [PubMed] [Google Scholar]
- 12.Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 1996;47:1–9. [DOI] [PubMed] [Google Scholar]
- 13.Goldman JS, Farmer JM, Wood EM, et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology 2005;65:1817–1819. [DOI] [PubMed] [Google Scholar]
- 14.Beck J, Rohrer JD, Campbell T, et al. A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain 2008;131(Pt 3):706–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009 27;323:1208–1211. [DOI] [PMC free article] [PubMed]
- 16.Kwiatkowski TJ, Jr., Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323:1205–1208. [DOI] [PubMed] [Google Scholar]
- 17.Janssen JC, Warrington EK, Morris HR, et al. Clinical features of frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology 2002;58:1161–1168. [DOI] [PubMed] [Google Scholar]
- 18.Spillantini MG, Yoshida H, Rizzini C, et al. A novel tau mutation (N296N) in familial dementia with swollen achromatic neurons and corticobasal inclusion bodies. Ann Neurol 2000;48:939–943. [DOI] [PubMed] [Google Scholar]
- 19.Rohrer JD, Beck J, Warren JD, et al. Corticobasal syndrome associated with a novel 1048_1049insG progranulin mutation. JNNP 2009 (in press). [DOI] [PubMed]
- 20.Snowden J, Neary D, Mann D. Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol 2007;114:31–38. [DOI] [PubMed] [Google Scholar]
- 21.Mesulam M, Wicklund A, Johnson N, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol 2008;63:709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houlden H, Baker M, Adamson J, et al. Frequency of tau mutations in three series of non-Alzheimer's degenerative dementia. Ann Neurol 1999;46:243–248. [DOI] [PubMed] [Google Scholar]
- 23.Rosso SM, Donker Kaat L, Baks T, et al. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain 2003;126:2016–2022. [DOI] [PubMed] [Google Scholar]
- 24.Stanford PM, Brooks WS, Teber ET, et al. Frequency of tau mutations in familial and sporadic frontotemporal dementia and other tauopathies. J Neurol 2004;251:1098–1104. [DOI] [PubMed] [Google Scholar]
- 25.Signorini S, Ghidoni R, Barbiero L, et al. Prevalence of pathogenic mutations in an Italian clinical series of patients with familial dementia. Curr Alzheimer Res 2004;1:215–218. [DOI] [PubMed] [Google Scholar]
- 26.Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006;442:920–924. [DOI] [PubMed] [Google Scholar]
- 27.Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 2006;15:2988–3001. [DOI] [PubMed] [Google Scholar]
- 28.Le Ber I, van der Zee J, Hannequin D, et al. Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum Mutat 2007;28:846–855. [DOI] [PubMed] [Google Scholar]
- 29.Pickering-Brown SM, Rollinson S, Du Plessis D, et al. Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 2008;131:721–731. [DOI] [PubMed] [Google Scholar]
- 30.Borroni B, Archetti S, Alberici A, et al. Progranulin genetic variations in frontotemporal lobar degeneration: evidence for low mutation frequency in an Italian clinical series. Neurogenetics 2008;9:197–205. [DOI] [PubMed] [Google Scholar]
- 31.Benussi L, Ghidoni R, Pegoiani E, et al. Progranulin Leu271LeufsX10 is one of the most common FTLD and CBS associated mutations worldwide. Neurobiol Dis 2009;33:379–385. [DOI] [PubMed] [Google Scholar]
- 32.Pickering-Brown S, Baker M, Bird T, et al. Evidence of a founder effect in families with frontotemporal dementia that harbor the tau +16 splice mutation. Am J Med Genet B Neuropsychiatr Genet 2004;125B:79–82. [DOI] [PubMed] [Google Scholar]
- 33.Seelaar H, Kamphorst W, Rosso SM, et al. Distinct genetic forms of frontotemporal dementia. Neurology 2008;71:1220–1226. [DOI] [PubMed] [Google Scholar]
- 34.Le Ber I, Camuzat A, Berger E, et al. Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology 2009;72:1669–1676. [DOI] [PubMed] [Google Scholar]
- 35.Mackenzie IR, Foti D, Woulfe J, Hurwitz TA. Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions Brain 2008;131:1282–1293. [DOI] [PubMed] [Google Scholar]
- 36.Roeber S, Mackenzie IR, Kretzschmar HA, Neumann M. TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol 2008;116:147–157. [DOI] [PubMed] [Google Scholar]
- 37.Geser F, Martinez-Lage M, Robinson J, et al. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol 2009;66:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Josephs KA, Ahmed Z, Katsuse O, et al. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol 2007;66:142–151. [DOI] [PubMed] [Google Scholar]
- 39.Josephs KA, Stroh A, Dugger B, Dickson DW. Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol Epub 2009 May 20. [DOI] [PMC free article] [PubMed]