

Abstract
Although our knowledge on the various aspects of diabetes development in the NOD mouse model is substantial and keeps expanding at a dramatic pace, the dataset on histopathologic features of type 1 diabetes (T1D) in patients remains largely stagnant. Early work has established an array of common aspects that have become epitomic in the absence of new patient material. There is a growing consensus that an updated and more detailed view is required that challenges and expands our understanding. Comprehensive initiatives are currently ongoing to address these issues in pre-diabetic, recent onset and longstanding type 1 diabetic individuals, and some of the old data have been recently revisited. In this review article, we wish to provide an overview of where we stand today and how we can correlate the various cross-sectional studies from the past with contemporary models of the disease. We believe an enhanced understanding of the many histopathological particularities in patients as compared to animal models will ultimately lead, not only to more fundamental insights, but also to an improved ability to translate pre-clinical data from bench to bedside.
Keywords: type 1 diabetes, beta-cell, cytotoxic T lymphocyte, autoantibody, insulitis, virus, pancreas, inflammation, apoptosis, replication
Introduction
Until the mid 1960's, diabetes was recognized by physicians as a single disorder, only roughly characterized by either juvenile or adult onset. Willy Gepts, a Belgian clinician and investigator, was the first to describe infiltration of lymphocytes in pancreatic islets of children who had died within a few days of developing diabetes [1]. This landmark finding, in conjunction with the identification of autoantibodies and HLA association, initiated the immunological study of T1D as a distinct autoimmune entity, characterized by its hallmark association with insulitis [2-5].
Almost half a century later, most of our knowledge on the immunopathology of the condition has been obtained from studies in animal models, while only a limited number of reports have documented the histopathology in patients. Detailed histological study of the disease is of course hampered by the inherent inaccessibility of the target organ, and as a consequence most of what we know is derived from retrospective collections of formalin-fixed, paraffin embedded autopsy pancreases [6]. In recent years, (immuno-) histological techniques have improved dramatically. Taking into account our enhanced understanding of the possible etiological factors and the growing appreciation of the diversity in terms of its course, several leading investigators have expressed a renewed interest in targeted histological research on human pancreas tissue [7]. One of the most promising initiatives is the recent establishment of the Network for Pancreatic Organ Donors with Diabetes (nPOD), a collaborative effort sponsored by the Juvenile Diabetes Research Foundation (JDRF). It aims to support the scientific community by providing the optimal procurement of pancreas specimens and distribution to nearly 25 T1D-focused scientific institutions around the world [8]. In this review, we aim to provide a contemporary context for the data accumulated over the past decades and include new reports into the equation in order to define questions to be addressed by projects such as nPOD.
Insulitis is a hallmark. Or isn’t it?
The original study by Gepts reported an insulitis frequency of 68% in recent-onset cases, of which only a limited number showed marked intra-islet inflammation, while no such events were observed in patients with a longer clinical duration [1]. It took about 2 decades until Alan Foulis and coworkers confirmed these findings and demonstrated the remarkable variability of the inflammatory process on a per-islet basis [9]. Whereas pancreatic autopsy in recent-onset diabetic children showed evidence of insulitis in most of the children, only 18% of insulin-containing islets were inflamed, as compared to 1% of insulin-deficient islets. In view of this, β-cells were likely to be subject to a highly localized and specific autoimmune response, further fueling the differentiation of T1D as an autoimmune condition. Bottazzo and coworkers, in their 1985 case report, detailed the features of the inflammatory infiltrate in the pancreas of a recently diagnosed 12-year old infant [10]. The majority of infiltrating cells were cytotoxic lymphocytes, an observation that would prove to be a characteristic feature in most recent-onset cases.
The collection of samples first described by Foulis et al. in 1986 is still by far the largest collection of samples available worldwide and contains autopsy specimens obtained from various hospitals within the U.K. over a 25-year period between 1960 and 1985 [11]. Their initial paper documented a 78% insulitis occurrence within 60 recent-onset cases, affecting 23% of insulin-positive islets versus 1% of insulin-deficient ones. These authors later estimated the average number of lymphocytes associated with an inflamed islet in recent-onset patients to be about 85 [12]. Surprisingly, when Hanafusa et al. looked at pancreas biopsies 2-4 months after diagnosis in 7 patients, no signs of insulitis were detected [13]. In a subsequent study by the same group, pancreas biopsy specimens from 18 newly diagnosed patients were analyzed and insulitis was found in 8 of them [14]. The degree of infiltration, however, was very limited ranging from 2 to 62 mononuclear cells in 3-33% of the examined islets in these patients. Consistent with most other studies the infiltrate was found to be predominantly composed of CD8 T lymphocytes [15, 16].
The practice of collecting pancreatic biopsies is still widely considered as too hazardous a procedure for mere research purposes, which is the reason why this approach has never gained wider application. From a researcher's perspective, matters have gradually become more complicated by the near nonexistence of new sample sources, a fact that can be contributed to major advances in disease management [17]. Consequently, researchers have had to rely on reanalysis of old specimens using modern immunohistochemical techniques, such as in a recent study by Willcox et al., which employs the collection first described by Foulis [18]. The authors provided a more refined image of the insulitic islet’s cellular constitution, pinpointing CD8 T cells once again as the predominant lymphocyte subset. Most interestingly, the authors painstakingly stratified the islets according to their percentage insulin-positivity, and correlated the presence of various immune subsets over the spectrum of β-cell destruction. It was revealed that CD8 T cell infiltrates peak according to the degree of β-cell decay, and rapidly disappear when all functional β-cells are destroyed. B cells, which have been until recently grossly ignored in most histopathological datasets, largely follow the same pattern. In contrast, macrophage numbers remain constantly moderate from the moment of infiltration. Likewise, Uno et al. found that macrophages and dendritic cells infiltrate the pancreatic islets at constant rates and produce inflammatory cytokines (TNF-α and IL-1β), irrespective of insulin content or T cell infiltration [19]. Finally, regulatory T (Treg) cells were detected in the islets of only a single patient, suggesting that their default 'territory' may be confined to the pancreatic lymph nodes or spleen or, alternatively, that their aberrant local absence gives free way to insulitis [18].
It is generally accepted that cross-sectional studies around the time of diagnosis represent a picture of the final stages of disease. However, we are in great need of information on the events preceding the clinical phase. This would entail the systematic screening of healthy human organ donors for serum markers or genetic predisposition, followed by sampling of pancreatic tissue from those at risk, an approach that is currently adopted by the nPOD project. Pertaining to this strategy, a Japanese study, examining patients shortly after diagnosis, revealed a close correlation between insulitis and the presence of either anti-GAD or anti-IA-2 antibodies in the serum [20]. Pipeleers and coworkers aimed to exploit the predictive value of serum autoantibodies in an effort to catch diabetes development in its early stages [21]. They screened serum from 1,507 organ donors for ICA, anti-GAD, anti-IA-2, and anti-insulin autoantibodies and collected pancreas tissue from 62 donors that tested positive for at least one antibody. The anticipated outcome was that these pre-diabetic individuals display all the prototypic pathological components acting at full force, as the presence of insulitis around onset is widely considered only a remnant of the entire process. Surprisingly, insulitis was detected in only 2 cases at remarkably low levels in less than 10% of the islets, with the typical predominance of CD8 T cells and presence of macrophages. Although the data on some of the ‘at-risk’ individuals included in the study may be affected by a 'not-so-at-risk' combination of age and HLA-DQ genotype, the results illustrate that insulitis may be a rare phenomenon in at least a subset of autoantibody-positive, non-diabetic individuals.
Collectively, the histological data describing insulitis portray the autoimmune aspect of the disease as a chronic process with a substantially variable course. In this regard, the notoriously lobule-dependent distribution of pathological events throughout the organ makes multiregional sampling mandatory as this may account for a certain portion of the reported variability [22]. Indeed, we know by now that the amount of insulitis not only differs between patients and their respective disease stages, but also on a per-islet basis, supporting the need for a thoughtful sampling design. Certain recurrent parameters such as the predominance of CD8 T cells may serve as a benchmark for future studies. The peri-islet frequency of other potentially important players such as B cells and macrophages should be studied in more detail and correlated with insulin deficiency. Finally, our knowledge on the precise reactivity of the insulitic T cells is still minimal. Velthuis and colleagues recently characterized infiltrating lymphocytes from a pancreatic graft in a recipient with T1D and found that the graft tissue was populated with islet-reactive T cells as assayed by flow cytometry using tetramer staining [23]. These kind of anecdotal reports are obviously far removed from the pathophysiological situation—e.g. the CD8 T cells were not found to associate with islets on a histological level—but nevertheless highlight the chronic, β-cell-specific nature of the disease and strongly indicate that immunotherapy will most likely be part of any future therapeutic strategy.
The fate of β-cells
At the time of diagnosis, 60-90% of the β-cells are already destroyed or dysfunctional. At present, there are no adequate non-invasive technologies available to assess how pancreatic β-cell mass fluctuates during the course of diabetes development, and hence what we know is based largely on indirect evidence [24]. The discussion on how β-cell mass may be monitored falls beyond the scope of this article, instead we will focus on histological evidence of β-cell survival pathways, their molecular phenotype when ‘under attack’ and evidence for apoptosis.
Molecular phenotype
Assuming that CD8 T cell mediated autoimmunity occurs in all T1D patients, at least at some point during disease development, it is crucial to know what specifically attracts these cells to the islets and why β-cells are selectively targeted. In their 1987 article, the Foulis group first documented the dramatic hyperexpression of MHC Class I molecules on cells of insulin-positive islets [25]. Although Bottazzo et al. previously suggested this potentially pivotal alteration in their case report, the Foulis study established that this was in fact a common property of diabetic islets around the time of disease onset. This has been consistently confirmed in later publications by other groups [10, 14, 26]. Curiously, MHC Class I hyperexpression was not confined to β-cells, as it was present on all endocrine cells in insulin-containing islets. This was in contrast with the observed pattern of MHC Class II hyperexpression, which was found exclusively on β-cells [25, 27]. A potential explanation for this differential expression was given by the subsequent finding that the inflammatory cytokine interferon-α is produced by MHC Class I overexpressing cells, suggesting a paracrine effect on adjacent A and D cells [28-30]. In combination, these findings led researchers to adopt a serial model for the pathology of T1D, including a sequence of events rather than a single-step mechanism (Figure 1).
Figure 1. Local early events in islets leading to β-cell death.
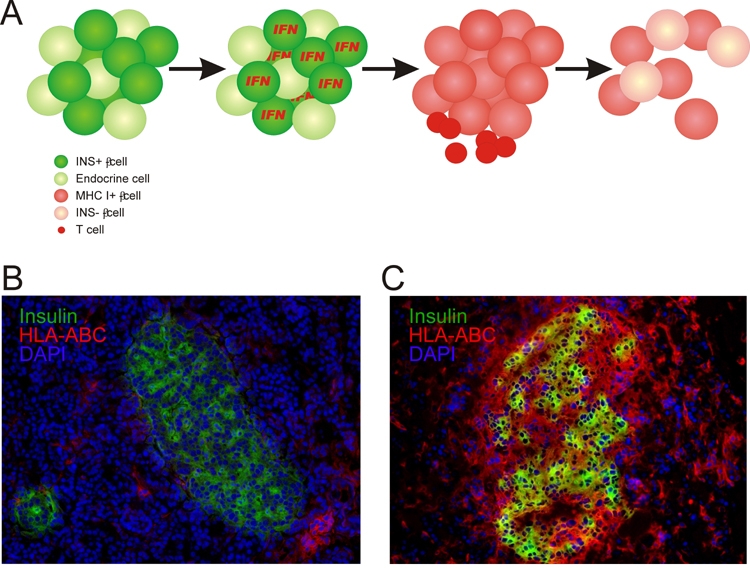
A: Model for a possible sequence causing the β-cells to undergo apoptosis. From left to right, normal islets are challenged by an environmental factor, presumably viral infection, and upregulate IFN-α. This cytokine exerts its effects in a paracrine fashion, inducing upregulation of MHC Class I in all islet cells and the subsequent influx of β-cell-specific CD8 T cells in genetically at-risk individuals. Finally, β-cells undergo apoptosis and most islets become devoid of β-cells, with potentially only a remnant pool of insulin-deficient β-cells that briefly restores insulin sufficiency during the honeymoon phase. B: Section from the pancreatic head region of a normal 72 year old female individual. Note the abundant quantities of functional β-cells and low levels of MHC Class I+ (HLA-ABC) cells, which are scattered in the exocrine region. C: Section from the pancreatic tail region of a 12 year old male with 1 year of clinical diabetes, who died of ketoacidosis. The strong hyperexpression of MHC Class I (HLA-ABC) on all islet cells is obvious, while many β-cells continue to produce insulin. This islet was found to contain limited numbers of infiltrating CD8 T cells.
This early hypothesis still serves as a framework for mechanistic studies to some extent, as it provides a rationale for the involvement of viruses in the etiology of the disease (see section "Viral footprints" below). Nevertheless, caution is necessary to avoid over-interpretation of this postulated scheme. The concept that temporally distinct phases of interferon-α and MHC Class I upregulation followed by insulitis lead to β-cell destruction originated from the studies by Foulis et al. looking at the molecular phenotype of individual islets. In support of this sequential model, some islets were found to express MHC Class I in the absence of insulitis, whereas MHC Class II hyperexpression was always accompanied by MHC Class I expression. However, it is easy to conceive that the upregulation of MHC molecules could be a mere consequence of (transient) insulitis rather than vice versa, at least in a fraction of the islet population, so more concrete evidence is required to back up this hypothesis. Moreover, with regard to MHC Class II hyperexpression, β-cells were found to be devoid of the co-stimulatory molecules CD80 and CD86, which are required for efficient antigen presentation, virtually excluding their role in direct activation of CD4 T cells [31]. Due to the aforementioned difficulties associated with obtaining new samples, the sequential model has never been thoroughly challenged and thus should be regarded as a valuable indication rather than as universal evidence of this cascade of events.
Apoptosis
Whatever the underlying cause, apoptosis signifies the end for β-cells in T1D. Isolated human islets seem to be particularly prone to this pathway. Thus, one would expect high levels of β-cell apoptosis under conditions of insulitis, especially in most of the available histological specimens isolated from deceased patients suffering marked hyperglycemia and diabetic ketoacidosis [32-34]. The Osaka group found no evidence of β-cell apoptosis in a set of 13 biopsies from recent-onset patients [35]. Subsequent studies did find detectable levels of β-cell apoptosis, which were estimated at around 6% of all β-cells by Meier et al., approximately double those found in control patients [36, 37]. The latter study deserves merit for its inclusion of cleaved caspase-3 staining for apoptosis detection in addition to TUNEL, which may aberrantly pick up post-mortem artefacts and lead to overestimation. The most recent paper from the same laboratory, however, showed lower levels of β-cell apoptosis, averaging 0.2 TUNEL-positive β-cells per islet as compared to virtually none in healthy controls [22]. Given the significantly increased blood glucose values and acidosis in this cohort, these results emphasize that β-cells may be more robust cells than generally thought, and probably represent yet another indication as to why T1D may take several years to become clinically apparent. In view of the data on β-cell proliferation rates (see subsection "β-cell survival" below), the results from the last study may seem more plausible, as higher rates of apoptosis would not correlate with the disease’s slow acting course, at least not in the absence of substantial β-cell replenishment.
A limited number of articles detailed the path to apoptosis, chiefly highlighting the pivotal role of Fas-Fas ligand (CD95-CD95L) interactions. In support of this simple death mechanism, β-cells were shown to express Fas, most likely in response to cytokines produced by infiltrating immune cells. This would render them susceptible to apoptosis by way of interacting with Fas ligand (Fas-L) expressing lymphocytes [35, 36]. The expression of Fas-L on peri-islet lymphocytes is in notable contrast with its downmodulation on peripheral blood lymphocytes, where lower levels may account for decreased apoptosis of auto-aggressive T cells [38, 39]. One explanation for this difference may be that highly activated diabetogenic T cells locally acquire Fas-L expression upon antigen recognition or, as described by DeFranco et al., peripheral downregulation may only occur in a subset of patients [38].
The relative contribution of the Fas pathway to the total ratio of β-cell apoptosis in humans is unknown, but most data in the NOD mouse indicate it serves a pivotal role [40-42]. However, as evidenced by the discovery of TNF-related apoptosis-inducing ligand (TRAIL) CD253 on β-cells, it is very possible that other apoptotic pathways act in concert during β-cell decay, likely at variable extents in different patients [43].
In conclusion, the limited frequency of ongoing apoptosis as revealed in recent-onset patients correlates with the slow progression of β-cell degeneration. Variability between patients may be ascribed to the chronic nature of the disease and its relapsing-remitting course (see section "Emerging diversity and the relapsing-remitting hypothesis" below). The relative contribution of e.g. direct cytokine killing or cytotoxic T lymphocyte (CTL) lysis as initiating events in the apoptosis cascade are difficult to address in humans and will likely need to be studied in the NOD model.
β-cell survival
Just like every other tissue type, pancreatic islets are capable of regeneration after insult. A considerable body of knowledge exists on the powerful regenerative capacity of β-cells in the NOD mouse, mostly favoring β-cell proliferation as the driving force [44]. Considering the low rate of apoptosis as discussed above, the degree of β-cell renewal can be projected to be limited. Indeed, the available data demonstrate varying levels of β-cell proliferation (none [22, 37], limited [21] and occasionally extensive [45]) at various stages of disease, all using the Ki-67 proliferative marker as a readout measure.
What could be the reasons for this discrepancy between rodents and humans, and what can we infer in relation to our hopes to therapeutically replenish the β-cell pool? First, although β-cell proliferation may be the default mechanism of β-cell expansion under normal conditions [46], other mechanisms, such as progenitor differentiation, may be at work that have currently escaped our attention [47]. Second, the abundant influx of inflammatory cells was shown to be a potent driver of β-cell replication in the NOD, and anecdotal evidence exists in humans [48, 49]. Considering the usually limited degree of insulitis in humans, an important stimulus for proliferation may be largely lacking. Further to this consideration, successful immunosuppressive therapy would be accompanied by a deteriorated proliferative β-cell response, as has been reported in the NOD mouse [50]. This suggests the application of combinatorial strategies targeting both the immune system and stimulating β-cell expansion [51]. Third, the inherent capacity of murine β-cells to enter the cell cycle is likely to surpass their human counterparts, as only mouse β-cells are able to recover from serious mechanical insult [52]. Fourth, the modest proliferation rates in humans may actually represent a reason why the disease acts so slowly, as the constant provision of newly formed β-cells would conceivably trigger enhanced release of antigens leading to epitope spreading [53]. Indeed, it has been suggested that this population of newly formed β-cells is most vulnerable to rapid destruction [54]. Finally, one of the major reasons why the NOD mouse displays such an exceptional plasticity in terms of β-cell recovery is the presence of a vast amount of non-functional, insulin-depleted β-cells at the onset of hyperglycemia [49]. Future studies should address whether this population exists in humans using non-functional β-cell markers such as glucose transporter 2 (GLUT-2).
Altogether, we believe our current understanding of how β-cells can rebound from inflammatory damage provides reason for cautious optimism. It is now established that the majority of patients maintains at least a fraction of their β-cell population, pointing towards the existence of powerful β-cell survival pathways [37]. However, animal models may overestimate the magnitude of such mechanisms warranting the design of combinatorial strategies tackling autoimmunity while giving β-cells a gentle push in the right direction.
Viral footprints
In their landmark paper from 1987, Foulis and colleagues formulated the working hypothesis of viral infection as an underlying factor in T1D development [28]. Albeit indirectly, the outcome of their research offered for the first time a mechanistic clue to a viral etiology hypothesis that had been so far supported by epidemiological data, including analysis of anti-viral antibodies in patient sera [55]. A plethora of publications have since elaborated on various aspects of the virus-β cell relationship, most of which were conducted in animal models. We will confine our review to the most direct evidence obtained in humans by way of histological analysis, as only this type of experimental approach specifically unveils events at the β-cell level.
Most studies at present were aimed at the detection of enteroviruses, a viral genus prominently found to induce diabetes in mice upon culturing from the pancreas of a recent-onset diabetic child [56]. The first data from the Foulis cohort used an antiserum raised against a coxsackie virus capsid protein, viral protein 1 (VP1), to examine the pancreas of 88 patients who had died at clinical presentation [57]. However, no evidence of VP1 presence was found. Another attempt employed in situ hybridization to detect enteroviral RNA in the same collection but again no sign of infection was found [58]. This negative outcome was arguably due to the compromised quality of RNA isolated from these formalin-fixed, paraffin-embedded autopsy specimens.
Reflecting on these early methodologies many years later, doubt was cast over the sensitivity of the immunohistochemical technique, leading Richardson et al. to revisit the issue in a recent publication, using the same Foulis sample collection [59]. They found islet VP1 immunopositivity in ~61% of 72 recent-onset type 1 patients with almost no equivalent staining in the pancreases of neonatal and pediatric non-diabetic controls. Furthermore, the detection was confined to β-cells and confirmed by staining with two additional antisera and an antibody against RNA-dependent protein kinase (PKR), a protein that is upregulated in response to enteroviral infection. The finding that 40% of adult type 2 diabetes cases displayed focal staining for VP1 in their islets was unanticipated, but may be interpreted in support of the view that type 1 and type 2 diabetes are poles of a single spectrum and that viral infection is merely the straw that breaks the camel's back around the time of clinical presentation (see section 5). Another group assayed pancreases from 65 Type 1 diabetic patients, partly from the Foulis collection, for the presence of enteroviral RNA by in situ hybridization and found enterovirus-positive islet cells in 4 patients, but not in control pancreases [60]. Putative enterovirus receptor molecules were expressed in β-cells, although it was unclear whether the detected virus was in islets containing functional β-cells.
A somewhat controversial study by Dotta and coworkers complemented this series of direct experimental approaches in pancreas tissue from 6 recent-onset patients [61]. VP1 staining was found in β-cells from 3 out of 6 patients, and virus extracted from one of these cases was sequenced and determined to be a Coxsackie B4 virus (CVB4). The CVB4 presence was found to affect glucose-stimulated insulin secretion upon infection of isolated human islets. However, an array of arguments may limit the significance of the findings. First and foremost, the 3 patients in which VP1 staining was observed displayed no signs of β-cell loss—only functional impairment to a variable degree—and had an unusual, predominantly NK (natural killer) cell-mediated islet infiltration without T cell reactivity to common islet antigens. The 3 pancreases devoid of VP1, in contrast, did manifest the more traditional T cell-dominated insulitis pattern accompanied by β-cell decline. Secondly, one of these 3 pancreases was a transplanted organ from a patient under an immunosuppressive regimen. This raised concerns over the relevance of viral infection to T1D etiology. Finally, some virologists have opposed the hypothesis that the isolated Coxsackie strain represented a laboratory contamination and not the isolation of a 'modern' CVB4 strain [62].
A recent case report looked at pancreatic tissue from an ICA autoantibody-positive, non-diabetic child. Whereas the β-cells were intact and free of insulitis, enterovirus was detected by immunohistochemistry selectively in the islets but not by in situ hybridization [63]. This anecdotal finding may have caught the disease process in its very early stages, with the virus already in place but still in the absence of an immune response.
New data indicate that the pancreas may not be the only site of infection. Oikarinen et al. analyzed 12 Finnish type 1 diabetic patients for the presence of enterovirus in small intestinal biopsies using in situ hybridization and immunohistochemistry. Enterovirus was detected in half of the type 1 diabetic patients but in none of the control subjects. The authors put forward the assumption that the gut, which retained normal features in all study subjects, could provide a viral reservoir that stands in close anatomical connection to the pancreas. Alternatively, it is argued that the gut could also be a site of immune stimulation for lymphocytes, which could subsequently home to the pancreas.
Despite the ambiguities related to sensitivity and specificity, immunohistochemical detection or in situ hybridization on co-stained pancreas sections are arguably the most straightforward methods to unveil the presence of viral antigens at a cellular level in human samples. Detecting viral sequences in whole pancreas lysate, e.g. by PCR, mystifies the relation to the target cell in T1D, because it may point to exocrine infections unrelated to T1D. Provided that autoimmune conditions are preexistent in individuals genetically at-risk of developing T1D, pancreatic non-β-cell-specific infections could merely act as the final culminating step pushing the remaining β-cells over the brink [64]. As such, the frequent detection of virus pattern in recently diagnosed patients may be regarded as a testament to this final insult, rather than a lingering chronic infection as suggested by Foulis et al. More elaborate studies in healthy pre-diabetic individuals are required to settle this debate. Whatever the case may be, it is unlikely that a certain viral strain will turn out to be the sole environmental trigger that leads to linear β-cell loss, as in all studies a significant fraction of cases was found to be free of viral markers. As a final consideration, eliminating viral infections completely may not be advisable, as our group recently provided functional evidence for a protective influence of viral infections in the context of T1D [65].
The NOD mouse: use with caution
We have already mentioned the NOD model's profound deviation from human T1D pathogenesis in the previous sections, and many investigators are now well aware of these discrepancies. Hence we will limit our discussion to the major histopathological differences that make the NOD mouse unique.
NOD mice are rodents, a fact that differentiates them from humans with regard to their basic islet architecture [66-70]. In humans, α, β, δ, and PP cells show random distribution throughout the islet and in many islets, β-cells represent the outermost layer of cells. In contrast, in murine islets, β-cells form the core, and α, δ, and pancreatic polypeptide producing (PP) cells form the outer layer. In human islets, the cellular constitution, i.e. the ratios of the various endocrine cell types, varies considerably, while in rodent islets frequencies remain reasonably constant. Both species have their islets surrounded by a continuous basal membrane with very little direct exocrine to endocrine cell-cell contact, which in the NOD was suggested to act as a protective barrier against destructive insulitis [71, 72].
As female NOD mice age, pathogenesis is heralded by non-destructive peri-insulitis, firstly consisting of dendritic cells and macrophages, followed by T cells and B cells [73]. After this initial phase, the disease progresses towards a complete T cell-mediated β-cell destruction by 4-6 months of age. In this stepwise sequence of events CD8 T cells are thought to be essential to initiate islet infiltration and are one of the earliest cell types to be seen in the islets [74, 75]. By the end of the peri-insulitic phase, overwhelming numbers of T cells can be seen engulfing the islets, to such an extent that they appear to organize in tertiary lymphoid structures [76-78]. These were shown to exhibit features common to lymph nodes such as distinct T and B cell zones, and expression of key chemokines and adhesion molecules. Overall, it is clear that the pathology in NOD animals, although often designated as 'chronic' in nature, is in fact a rather acute and aggressive autoimmune disease as compared to the subtle, CD8 dominated insulitis distinguished in humans.
Viral infection as an underlying mechanism in the NOD mouse can of course be excluded, as the disease usually manifests spontaneously. However, a substantial body of knowledge shows that certain pancreotropic virus strains can significantly influence disease outcome. Work by Steven Tracy's group has elucidated many aspects of the complex interplay between coxsackie viruses and islets at various stages of disease progression in the NOD [79, 80]. In brief, infection of young NOD mice confers protection, which contrasts with the outcome in older, pre-diabetic mice where it accelerates disease. We believe these data offer a possible rationale explaining why such a high incidence of viral infection can be seen in recent-onset individuals, as viral infection at this point tips the balance towards clinically overt diabetes. This would also explain the long acknowledged seasonal onset of T1D and the high frequency of anti-viral antibodies in recent-onset patients, as these individuals can be postulated to have recently encountered a virus that triggered disease, rather than them being infected for years with a chronic pancreatropic virus. On the other hand, early infections might constitute an essential requirement for protection later in life, as corroborated by our recent results [65].
Comparing these considerations with what we know about human disease, it is obvious that we must ensure proper interpretation of pre-clinical data in the NOD model. When we take successful reversal of disease upon immunotherapy of recent-onset NOD mice as an example, we must consider that patients may not have an abundant reserve of insulin-deficient yet viable β-cells left to recover, or probably do not have equivalent flexibility pertaining to β-cell proliferation. Promising factors in this respect include the common phenomenon of the 'honeymoon phase' following diagnosis, possible offering a suitable window for such interventions.
Emerging diversity and the relapsing-remitting hypothesis
How should we reconcile this data and, more importantly, do they collectively support or favor one of the existing mechanistic models? We have recently proposed a hypothetical model for T1D that pictures the disease as being driven by the interplay between epitope spreading, β-cell proliferation and Treg control of auto-reactive T cell responses [53]. Figure 2 displays this model while adding the histopathological features outlined in this article to the timeline. We believe that this kind of relapsing-remitting, gradual course may account for at least some of the variability that can be seen in the cross-sectional studies we have discussed. It also explains why insulitis generally appears to be minimal, especially in the pre-diabetic stage. A complementary cause for the observed inter-patient variability could be the consideration that type 1 and type 2 diabetes are opposite ends of a continuous spectrum, and that the clinical manifestation depends on the velocity of loss and the factors that influence it [81]. Applied to our model, this would introduce a variable timeline between initiation of β-cell decay and onset, and rationalize the usually more aggressive course in children versus adult onset cases [82, 83].
Figure 2. Correlation of histopathology data with the relapsing-remitting hypothesis.
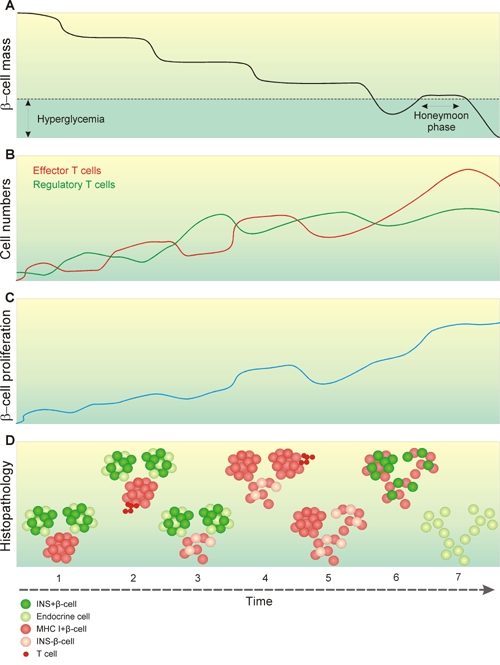
A: We described the progression of diabetes development earlier as a stepwise, non-linear decline of β-cell mass over time [53], up to the point where patients lose 60-90% of their functional β-cells and become diabetic. A series of environmental triggers may cause these stages of β-cell decay, explaining why signs of viral infection can be found around the time of diagnosis, following the final step of degeneration. The honeymoon phase is a transient phase of normoglycemia after clinical manifestation, which may serve as a suitable window for therapeutic intervention. B: The stepwise nature of β-cell decay supposedly results from the interplay between auto-reactive T cells and regulatory T cells. Episodes of β-cell loss are caused by a disequilibrium giving auto-reactivity the upper hand. C: Although evidence of significant β-cell proliferations as a rescue mechanism has only been obtained in animal models, the slowly progressing, chronic nature of the prediabetic phase indirectly suggests that some form of β-cell survival takes place. D: Reconciling this model with the combined data from histological reports: 1. One of the earliest histological signs may be the upregulation of MHC Class I in response to an environmental agent. At this point only a minority of islets is affected in a confined pancreatic lobule, and all β-cells remain fully functional. Random sampling such as e.g. by biopsy easily misses out on these subtle events. 2. Some islets become infiltrated by auto-reactive T cells. 3. As regulatory T cells transiently regain supremacy, insulitis subsides leaving a minor fraction of β-cells either death or insulin-deficient. 4. As the prediabetic phase progresses, more islets become MHC Class I+ and insulitis spreads to more lobules. At this point just before clinical onset, the disease should be easily detectable in e.g. pre-screened multiple auto-antibody positive patients. 5. In recent-onset individuals, the majority of β-cell mass is lost, and what is seen histologically are remnants of a process that has been ongoing for years. 6. During the honeymoon phase, some β-cells regain function and support a transient period of normoglycemia. 7. In longstanding cases, the vast majority of islets are devoid of functional β-cells and inflammation, although there is evidence that at least a fraction of patients retain a minor β-cell mass. Together, this stepwise relapsing-remitting course may explain the variability that has been reported between patients at various stages of disease development.
Conclusions
In summary we can conclude that information on the human histopathology at various stages of T1D is currently rather limited. Whereas a multitude of functional data on the immunology and possible treatment of the disease have been derived from the NOD model, it is clear that the human disease presents itself in a much more subtle and variable manner. Correlating the available reports with our hypothetical model of T1D as a relapsing-remitting disease, we believe much of the variability seen in cross-sectional studies can be explained by the stepwise, gradual progression of autoimmunity. Collaborative projects such as nPOD should be able to challenge and expand our understanding of the histopathology by generating a new influx of patient material. In the end we believe that an enhanced insight into local events in the islets may not only be of fundamentally scientific importance, but will prove pivotal in efficient translation of future therapies beyond the NOD mouse model.
Conflict of interest statement: The authors declare that they have no competing conflict of interests.
Acknowledgments
We would like to thank Natalie Amirian and Carlo Randise-Hinchliff for excellent technical support in preparation of histological sections presented in Figure 1. We greatly acknowledge the people contributing to the Network for Pancreatic Organ Donors (nPOD) for procurement, sectioning and shipment of the samples.
References
- 1.Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes. 1965;14(10):619–633. doi: 10.2337/diab.14.10.619. [DOI] [PubMed] [Google Scholar]
- 2.Bottazzo GF, Florin-Christensen A, Doniach D. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet. 1974;2(7892):1279–1283. doi: 10.1016/s0140-6736(74)90140-8. [DOI] [PubMed] [Google Scholar]
- 3.MacCuish AC, Irvine WJ, Barnes EW, Duncan LJ. Antibodies to pancreatic islet cells in insulin-dependent diabetics with coexistent autoimmune disease. Lancet. 1974;2(7896):1529–1531. doi: 10.1016/s0140-6736(74)90281-5. [DOI] [PubMed] [Google Scholar]
- 4.Nerup J, Platz P, Andersen OO, Christy M, Lyngsoe J, Poulsen JE, Ryder LP, Nielsen LS, Thomsen M, Svejgaard A. HL-A antigens and diabetes mellitus. Lancet. 1974;2(7885):864–866. doi: 10.1016/s0140-6736(74)91201-x. [DOI] [PubMed] [Google Scholar]
- 5.Singal DP, Blajchman MA. Histocompatibility (HL-A) antigens, lymphocytotoxic antibodies and tissue antibodies in patients with diabetes mellitus. Diabetes. 1973;22:429–432. doi: 10.2337/diab.22.6.429. [DOI] [PubMed] [Google Scholar]
- 6.Foulis AK. Pancreatic pathology in type 1 diabetes in human. Novartis Found Symp. 2008;292:2–13. [PubMed] [Google Scholar]
- 7.Atkinson MA, Gianani R. The pancreas in human type 1 diabetes: providing new answers to age-old questions. Curr Opin Endocrinol Diabetes Obes. 2009;16:279–285. doi: 10.1097/MED.0b013e32832e06ba. [DOI] [PubMed] [Google Scholar]
- 8. http://www.jdrfnpod.org.
- 9.Foulis AK, Stewart JA. The pancreas in recent-onset type 1 (insulin-dependent) diabetes mellitus: insulin content of islets, insulitis and associated changes in the exocrine acinar tissue. Diabetologia. 1984;26(6):456–461. doi: 10.1007/BF00262221. [DOI] [PubMed] [Google Scholar]
- 10.Bottazzo GF, Dean BM, McNally JM, MacKay EH, Swift PG, Gamble DR. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N Engl J Med. 1985;313:353–360. doi: 10.1056/NEJM198508083130604. [DOI] [PubMed] [Google Scholar]
- 11.Foulis AK, Liddle CN, Farquharson MA, Richmond JA, Weir RS. The histopathology of the pancreas in type 1 (insulin-dependent) diabetes mellitus: a 25-year review of deaths in patients under 20 years of age in the United Kingdom. Diabetologia. 1986;29(5):267–274. doi: 10.1007/BF00452061. [DOI] [PubMed] [Google Scholar]
- 12.Foulis AK, McGill M, Farquharson MA. Insulitis in type 1 (insulin-dependent) diabetes mellitus in man - macrophages, lymphocytes, and interferon-gamma containing cells. J Pathol. 1991;165:97–103. doi: 10.1002/path.1711650203. [DOI] [PubMed] [Google Scholar]
- 13.Hanafusa T, Miyazaki A, Miyagawa J, Tamura S, Inada M, Yamada K, Shinji Y, Katsura H, Yamagata K, Itoh N. et al. Examination of islets in the pancreas biopsy specimens from newly diagnosed type 1 (insulin-dependent) diabetic patients. Diabetologia. 1990;33:105–111. doi: 10.1007/BF00401048. [DOI] [PubMed] [Google Scholar]
- 14.Itoh N, Hanafusa T, Miyazaki A, Miyagawa J, Yamagata K, Yamamoto K, Waguri M, Imagawa A, Tamura S, Inada M. et al. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest. 1993;92:2313–2322. doi: 10.1172/JCI116835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanninen A, Jalkanen S, Salmi M, Toikkanen S, Nikolakaros G, Simell O. Macrophages, T cell receptor usage, and endothelial cell activation in the pancreas at the onset of insulin-dependent diabetes mellitus. J Clin Invest. 1992;90:1901–1910. doi: 10.1172/JCI116067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Somoza N, Vargas F, Roura-Mir C, Vives-Pi M, Fernandez-Figueras MT, Ariza A, Gomis R, Bragado R, Marti M, Jaraquemada D. et al. Pancreas in recent onset insulin-dependent diabetes mellitus. Changes in HLA, adhesion molecules and autoantigens, restricted T cell receptor V beta usage, and cytokine profile. J Immunol. 1994;153:1360–1377. [PubMed] [Google Scholar]
- 17.Spencer J, Peakman M. Post-mortem analysis of islet pathology in type 1 diabetes illuminates the life and death of the beta cell. Clin Exp Immunol. 2009;155:125–127. doi: 10.1111/j.1365-2249.2008.03864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. 2009;155:173–181. doi: 10.1111/j.1365-2249.2008.03860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uno S, Imagawa A, Okita K, Sayama K, Moriwaki M, Iwahashi H, Yamagata K, Tamura S, Matsuzawa Y, Hanafusa T. et al. Macrophages and dendritic cells infiltrating islets with or without beta cells produce tumour necrosis factor-alpha in patients with recent-onset type 1 diabetes. Diabetologia. 2007;50:596–601. doi: 10.1007/s00125-006-0569-9. [DOI] [PubMed] [Google Scholar]
- 20.Imagawa A, Hanafusa T, Tamura S, Moriwaki M, Itoh N, Yamamoto K, Iwahashi H, Yamagata K, Waguri M, Nanmo T. et al. Pancreatic biopsy as a procedure for detecting in situ autoimmune phenomena in type 1 diabetes: close correlation between serological markers and histological evidence of cellular autoimmunity. Diabetes. 2001;50:1269–1273. doi: 10.2337/diabetes.50.6.1269. [DOI] [PubMed] [Google Scholar]
- 21.In't Veld P, Lievens D, De Grijse J, Ling Z, Van der Auwera B, Pipeleers-Marichal M, Gorus F, Pipeleers D. Screening for insulitis in adult autoantibody-positive organ donors. Diabetes. 2007;56:2400–2404. doi: 10.2337/db07-0416. [DOI] [PubMed] [Google Scholar]
- 22.Butler AE, Galasso R, Meier JJ, Basu R, Rizza RA, Butler PC. Modestly increased beta cell apoptosis but no increased beta cell replication in recent-onset type 1 diabetic patients who died of diabetic ketoacidosis. Diabetologia. 2007;50:2323–2331. doi: 10.1007/s00125-007-0794-x. [DOI] [PubMed] [Google Scholar]
- 23.Velthuis JH, Unger WW, van der Slik AR, Duinkerken G, Engelse M, Schaapherder AF, Ringers J, van Kooten C, de Koning EJ, Roep BO. Accumulation of autoreactive effector T cells and allo-specific regulatory T cells in the pancreas allograft of a type 1 diabetic recipient. Diabetologia. 2009;52:494–503. doi: 10.1007/s00125-008-1237-z. [DOI] [PubMed] [Google Scholar]
- 24.Coppieters K, von Herrath M. Taking a closer look at the pancreas. Diabetologia. 2008;51:2145–2147. doi: 10.1007/s00125-008-1181-y. [DOI] [PubMed] [Google Scholar]
- 25.Foulis AK, Farquharson MA, Hardman R. Aberrant expression of class II major histocompatibility complex molecules by B cells and hyperexpression of class I major histocompatibility complex molecules by insulin containing islets in type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1987;30:333–343. doi: 10.1007/BF00299027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imagawa A, Hanafusa T, Itoh N, Waguri M, Yamamoto K, Miyagawa J, Moriwaki M, Yamagata K, Iwahashi H, Sada M. et al. Immunological abnormalities in islets at diagnosis paralleled further deterioration of glycaemic control in patients with recent-onset Type I (insulin-dependent) diabetes mellitus. Diabetologia. 1999;42:574–578. doi: 10.1007/s001250051197. [DOI] [PubMed] [Google Scholar]
- 27.Foulis AK, Farquharson MA. Aberrant expression of HLA-DR antigens by insulin-containing beta-cells in recent-onset type I diabetes mellitus. Diabetes. 1986;35:1215–1224. doi: 10.2337/diab.35.11.1215. [DOI] [PubMed] [Google Scholar]
- 28.Foulis AK, Farquharson MA, Meager A. Immunoreactive alpha-interferon in insulin-secreting beta cells in type 1 diabetes mellitus. Lancet. 1987;2:1423–1427. doi: 10.1016/s0140-6736(87)91128-7. [DOI] [PubMed] [Google Scholar]
- 29.Huang X, Yuang J, Goddard A, Foulis A, James RF, Lernmark A, Pujol-Borrell R, Rabinovitch A, Somoza N, Stewart TA. Interferon expression in the pancreases of patients with type I diabetes. Diabetes. 1995;44:658–664. doi: 10.2337/diab.44.6.658. [DOI] [PubMed] [Google Scholar]
- 30.Devendra D, Eisenbarth GS. Interferon alpha - a potential link in the pathogenesis of viral-induced type 1 diabetes and autoimmunity. Clin Immunol. 2004;111:225–233. doi: 10.1016/j.clim.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 31.Imagawa A, Hanafusa T, Itoh N, Miyagawa J, Nakajima H, Namba M, Kuwajima M, Tamura S, Kawata S, Matsuzawa Y. et al. Islet-infiltrating t lymphocytes in insulin-dependent diabetic patients express CD80 (B7-1) and CD86 (B7-2) J Autoimmun. 1996;9:391–396. doi: 10.1006/jaut.1996.0053. [DOI] [PubMed] [Google Scholar]
- 32.Federici M, Hribal M, Perego L, Ranalli M, Caradonna Z, Perego C, Usellini L, Nano R, Bonini P, Bertuzzi F. et al. High glucose causes apoptosis in cultured human pancreatic islets of Langerhans: a potential role for regulation of specific Bcl family genes toward an apoptotic cell death program. Diabetes. 2001;50:1290–1301. doi: 10.2337/diabetes.50.6.1290. [DOI] [PubMed] [Google Scholar]
- 33.Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, Donath MY. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002;110:851–860. doi: 10.1172/JCI15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lupi R, Dotta F, Marselli L, Del Guerra S, Masini M, Santangelo C, Patane G, Boggi U, Piro S, Anello M. et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes. 2002;51:1437–1442. doi: 10.2337/diabetes.51.5.1437. [DOI] [PubMed] [Google Scholar]
- 35.Moriwaki M, Itoh N, Miyagawa J, Yamamoto K, Imagawa A, Yamagata K, Iwahashi H, Nakajima H, Namba M, Nagata S. et al. Fas and Fas ligand expression in inflamed islets in pancreas sections of patients with recent-onset Type I diabetes mellitus. Diabetologia. 1999;42:1332–1340. doi: 10.1007/s001250051446. [DOI] [PubMed] [Google Scholar]
- 36.Stassi G, De Maria R, Trucco G, Rudert W, Testi R, Galluzzo A, Giordano C, Trucco M. Nitric oxide primes pancreatic beta cells for Fas-mediated destruction in insulin-dependent diabetes mellitus. J Exp Med. 1997;186:1193–1200. doi: 10.1084/jem.186.8.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia. 2005;48:2221–2228. doi: 10.1007/s00125-005-1949-2. [DOI] [PubMed] [Google Scholar]
- 38.DeFranco S, Bonissoni S, Cerutti F, Bona G, Bottarel F, Cadario F, Brusco A, Loffredo G, Rabbone I, Corrias A. et al. Defective function of Fas in patients with type 1 diabetes associated with other autoimmune diseases. Diabetes. 2001;50:483–488. doi: 10.2337/diabetes.50.3.483. [DOI] [PubMed] [Google Scholar]
- 39.Giordano C, De Maria R, Stassi G, Todaro M, Richiusa P, Giordano M, Testi R, Galluzzo A. Defective expression of the apoptosis-inducing CD95 (Fas/APO-1) molecule on T and B cells in IDDM. Diabetologia. 1995;38:1449–1454. doi: 10.1007/BF00400606. [DOI] [PubMed] [Google Scholar]
- 40.Nakayama M, Nagata M, Yasuda H, Arisawa K, Kotani R, Yamada K, Chowdhury SA, Chakrabarty S, Jin ZZ, Yagita H. et al. Fas/Fas ligand interactions play an essential role in the initiation of murine autoimmune diabetes. Diabetes. 2002;51:1391–1397. doi: 10.2337/diabetes.51.5.1391. [DOI] [PubMed] [Google Scholar]
- 41.Chervonsky AV, Wang Y, Wong FS, Visintin I, Flavell RA, Janeway CA Jr, Matis LA. The role of Fas in autoimmune diabetes. Cell. 1997;89:17–24. doi: 10.1016/s0092-8674(00)80178-6. [DOI] [PubMed] [Google Scholar]
- 42.Savinov AY, Tcherepanov A, Green EA, Flavell RA, Chervonsky AV. Contribution of Fas to diabetes development. Proc Natl Acad Sci U S A. 2003;100:628–632. doi: 10.1073/pnas.0237359100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanlioglu AD, Dirice E, Elpek O, Korcum AF, Balci MK, Omer A, Griffith TS, Sanlioglu S. High levels of endogenous tumor necrosis factor-related apoptosis-inducing ligand expression correlate with increased cell death in human pancreas. Pancreas. 2008;36:385–393. doi: 10.1097/MPA.0b013e318158a4e5. [DOI] [PubMed] [Google Scholar]
- 44.Akirav E, Kushner JA, Herold KC. Beta-cell mass and type 1 diabetes: going, going, gone? Diabetes. 2008;57:2883–2888. doi: 10.2337/db07-1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meier JJ, Lin JC, Butler AE, Galasso R, Martinez DS, Butler PC. Direct evidence of attempted beta cell regeneration in an 89-year-old patient with recent-onset type 1 diabetes. Diabetologia. 2006;49:1838–1844. doi: 10.1007/s00125-006-0308-2. [DOI] [PubMed] [Google Scholar]
- 46.Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57:1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin-Pagola A, Sisino G, Allende G, Dominguez-Bendala J, Gianani R, Reijonen H, Nepom GT, Ricordi C, Ruiz P, Sageshima J. et al. Insulin protein and proliferation in ductal cells in the transplanted pancreas of patients with type 1 diabetes and recurrence of autoimmunity. Diabetologia. 2008;51:1803–1813. doi: 10.1007/s00125-008-1105-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campbell-Thompson M, Dixon LR, Wasserfall C, Monroe M, McGuigan JM, Schatz D, Crawford JM, Atkinson MA. Pancreatic adenocarcinoma patients with localised chronic severe pancreatitis show an increased number of single beta cells, without alterations in fractional insulin area. Diabetologia. 2009;52:262–270. doi: 10.1007/s00125-008-1200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sherry NA, Kushner JA, Glandt M, Kitamura T, Brillantes AM, Herold KC. Effects of autoimmunity and immune therapy on beta-cell turnover in type 1 diabetes. Diabetes. 2006;55:3238–3245. doi: 10.2337/db05-1034. [DOI] [PubMed] [Google Scholar]
- 50.Phillips JM, O'Reilly L, Bland C, Foulis AK, Cooke A. Patients with chronic pancreatitis have islet progenitor cells in their ducts, but reversal of overt diabetes in NOD mice by anti-CD3 shows no evidence for islet regeneration. Diabetes. 2007;56:634–640. doi: 10.2337/db06-0832. [DOI] [PubMed] [Google Scholar]
- 51.Maedler K, Fontana A, Ris F, Sergeev P, Toso C, Oberholzer J, Lehmann R, Bachmann F, Tasinato A, Spinas GA. et al. FLIP switches Fas-mediated glucose signaling in human pancreatic beta cells from apoptosis to cell replication. Proc Natl Acad Sci U S A. 2002;99:8236–8241. doi: 10.1073/pnas.122686299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Menge BA, Tannapfel A, Belyaev O, Drescher R, Muller C, Uhl W, Schmidt WE, Meier JJ. Partial pancreatectomy in adult humans does not provoke beta-cell regeneration. Diabetes. 2008;57:142–149. doi: 10.2337/db07-1294. [DOI] [PubMed] [Google Scholar]
- 53.von Herrath M, Sanda S, Herold K. Type 1 diabetes as a relapsing-remitting disease? Nat Rev Immunol. 2007;7:988–994. doi: 10.1038/nri2192. [DOI] [PubMed] [Google Scholar]
- 54.Meier JJ, Ritzel RA, Maedler K, Gurlo T, Butler PC. Increased vulnerability of newly forming beta cells to cytokine-induced cell death. Diabetologia. 2006;49:83–89. doi: 10.1007/s00125-005-0069-3. [DOI] [PubMed] [Google Scholar]
- 55.Honeyman M. How robust is the evidence for viruses in the induction of type 1 diabetes? Curr Opin Immunol. 2005;17:616–623. doi: 10.1016/j.coi.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 56.Yoon JW, Austin M, Onodera T, Notkins AL. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N Engl J Med. 1979;300:1173–1179. doi: 10.1056/NEJM197905243002102. [DOI] [PubMed] [Google Scholar]
- 57.Foulis AK, Farquharson MA, Cameron SO, McGill M, Schonke H, Kandolf R. A search for the presence of the enteroviral capsid protein VP1 in pancreases of patients with type 1 (insulin-dependent) diabetes and pancreases and hearts of infants who died of coxsackieviral myocarditis. Diabetologia. 1990;33:290–298. doi: 10.1007/BF00403323. [DOI] [PubMed] [Google Scholar]
- 58.Foulis AK, McGill M, Farquharson MA, Hilton DA. A search for evidence of viral infection in pancreases of newly diagnosed patients with IDDM. Diabetologia. 1997;40:53–61. doi: 10.1007/s001250050642. [DOI] [PubMed] [Google Scholar]
- 59.Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia. 2009;52:1143–1151. doi: 10.1007/s00125-009-1276-0. [DOI] [PubMed] [Google Scholar]
- 60.Ylipaasto P, Klingel K, Lindberg AM, Otonkoski T, Kandolf R, Hovi T, Roivainen M. Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet beta cells. Diabetologia. 2004;47:225–239. doi: 10.1007/s00125-003-1297-z. [DOI] [PubMed] [Google Scholar]
- 61.Dotta F, Censini S, van Halteren AG, Marselli L, Masini M, Dionisi S, Mosca F, Boggi U, Muda AO, Prato SD. et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci U S A. 2007;104:5115–5120. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Drescher KM, Tracy SM. The CVB and etiology of type 1 diabetes. Curr Top Microbiol Immunol. 2008;323:259–274. doi: 10.1007/978-3-540-75546-3_12. [DOI] [PubMed] [Google Scholar]
- 63.Oikarinen M, Tauriainen S, Honkanen T, Vuori K, Karhunen P, Vasama-Nolvi C, Oikarinen S, Verbeke C, Blair GE, Rantala I. et al. Analysis of pancreas tissue in a child positive for islet cell antibodies. Diabetologia. 2008;51:1796–1802. doi: 10.1007/s00125-008-1107-8. [DOI] [PubMed] [Google Scholar]
- 64.von Herrath MG, Fujinami RS, Whitton JL. Microorganisms and autoimmunity: making the barren field fertile? Nat Rev Microbiol. 2003;1:151–157. doi: 10.1038/nrmicro754. [DOI] [PubMed] [Google Scholar]
- 65.Filippi CM, Estes EA, Oldham JE, von Herrath MG. Immunoregulatory mechanisms triggered by viral infections protect from type 1 diabetes in mice. J Clin Invest. 2009;119:1515–1523. doi: 10.1172/JCI38503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brissova M, Fowler MJ, Nicholson WE, Chu A, Hirshberg B, Harlan DM, Powers AC. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem. 2005;53:1087–1097. doi: 10.1369/jhc.5C6684.2005. [DOI] [PubMed] [Google Scholar]
- 67.Orci L, Baetens D, Rufener C, Amherdt M, Ravazzola M, Studer P, Malaisse-Lagae F, Unger RH. Hypertrophy and hyperplasia of somatostatin-containing D-cells in diabetes. Proc Natl Acad Sci U S A. 1976;73:1338–1342. doi: 10.1073/pnas.73.4.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baetens D, Malaisse-Lagae F, Perrelet A, Orci L. Endocrine pancreas: three-dimensional reconstruction shows two types of islets of langerhans. Science. 1979;206:1323–1325. doi: 10.1126/science.390711. [DOI] [PubMed] [Google Scholar]
- 69.Brelje TC, Scharp DW, Sorenson RL. Three-dimensional imaging of intact isolated islets of Langerhans with confocal microscopy. Diabetes. 1989;38:808–814. doi: 10.2337/diab.38.6.808. [DOI] [PubMed] [Google Scholar]
- 70.Cirulli V, Baetens D, Rutishauser U, Halban PA, Orci L, Rouiller DG. Expression of neural cell adhesion molecule (N-CAM) in rat islets and its role in islet cell type segregation. J Cell Sci. 1994;107(Pt 6):1429–1436. doi: 10.1242/jcs.107.6.1429. [DOI] [PubMed] [Google Scholar]
- 71.Wang RN, Paraskevas S, Rosenberg L. Characterization of integrin expression in islets isolated from hamster, canine, porcine, and human pancreas. J Histochem Cytochem. 1999;47:499–506. doi: 10.1177/002215549904700408. [DOI] [PubMed] [Google Scholar]
- 72.Irving-Rodgers HF, Ziolkowski AF, Parish CR, Sado Y, Ninomiya Y, Simeonovic CJ, Rodgers RJ. Molecular composition of the peri-islet basement membrane in NOD mice: a barrier against destructive insulitis. Diabetologia. 2008;51:1680–1688. doi: 10.1007/s00125-008-1085-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jansen A, Homo-Delarche F, Hooijkaas H, Leenen PJ, Dardenne M, Drexhage HA. Immunohistochemical characterization of monocytes-macrophages and dendritic cells involved in the initiation of the insulitis and beta-cell destruction in NOD mice. Diabetes. 1994;43:667–675. doi: 10.2337/diab.43.5.667. [DOI] [PubMed] [Google Scholar]
- 74.Jarpe AJ, Hickman MR, Anderson JT, Winter WE, Peck AB. Flow cytometric enumeration of mononuclear cell populations infiltrating the islets of Langerhans in prediabetic NOD mice: development of a model of autoimmune insulitis for type I diabetes. Reg Immunol. 1990;3:305–317. [PubMed] [Google Scholar]
- 75.Wang B, Gonzalez A, Benoist C, Mathis D. The role of CD8+ T cells in the initiation of insulin-dependent diabetes mellitus. Eur J Immunol. 1996;26:1762–1769. doi: 10.1002/eji.1830260815. [DOI] [PubMed] [Google Scholar]
- 76.Kendall PL, Yu G, Woodward EJ, Thomas JW. Tertiary lymphoid structures in the pancreas promote selection of B lymphocytes in autoimmune diabetes. J Immunol. 2007;178:5643–5651. doi: 10.4049/jimmunol.178.9.5643. [DOI] [PubMed] [Google Scholar]
- 77.Lee Y, Chin RK, Christiansen P, Sun Y, Tumanov AV, Wang J, Chervonsky AV, Fu YX. Recruitment and activation of naive T cells in the islets by lymphotoxin beta receptor-dependent tertiary lymphoid structure. Immunity. 2006;25:499–509. doi: 10.1016/j.immuni.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 78.Ludewig B, Odermatt B, Landmann S, Hengartner H, Zinkernagel RM. Dendritic cells induce autoimmune diabetes and maintain disease via de novo formation of local lymphoid tissue. J Exp Med. 1998;188:1493–1501. doi: 10.1084/jem.188.8.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Drescher KM, Kono K, Bopegamage S, Carson SD, Tracy S. Coxsackievirus B3 infection and type 1 diabetes development in NOD mice: insulitis determines susceptibility of pancreatic islets to virus infection. Virology. 2004;329:381–394. doi: 10.1016/j.virol.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 80.Tracy S, Drescher KM, Chapman NM, Kim KS, Carson SD, Pirruccello S, Lane PH, Romero JR, Leser JS. Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J Virol. 2002;76:12097–12111. doi: 10.1128/JVI.76.23.12097-12111.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wilkin TJ. Changing perspectives in diabetes: their impact on its classification. Diabetologia. 2007;50:1587–1592. doi: 10.1007/s00125-007-0665-5. [DOI] [PubMed] [Google Scholar]
- 82.Pipeleers D, In't Veld P, Pipeleers-Marichal M, Gorus F. The beta cell population in type 1 diabetes. Novartis Found Symp. 2008;292:19–24. doi: 10.1002/9780470697405.ch3. [DOI] [PubMed] [Google Scholar]
- 83.Pipeleers D, Ling Z. Pancreatic beta cells in insulin-dependent diabetes. Diabetes Metab Rev. 1992;8:209–227. doi: 10.1002/dmr.5610080303. [DOI] [PubMed] [Google Scholar]