

Abstract
Objectives
While some eruption disorders occur as part of a medical syndrome, primary failure of eruption (PFE) – defined as a localized failure of secondary tooth eruption -exists without systemic involvement. Recent studies support that heredity may play an important role in the pathogenesis of PFE. The objective of our human genetic study is to investigate the genetic contribution to PFE.
Materials and Methods
Four candidate genes POSTN, RUNX2, AMELX, and AMBN) were investigated due to their relationship to tooth eruption or putative relationship to each other. Families and individuals were ascertained based on the clinical diagnosis of PFE. Pedigrees were constructed and analyzed by inspection to determine the mode of inheritance in 4 families. The candidate genes were directly sequenced for both unrelated affected individuals and unaffected individuals. A genome wide scan using 500 microsatellite markers followed by linkage analysis was carried out for one family.
Results
Pedigree analysis of families suggests an autosomal dominant inheritance pattern with complete penetrance and variable expressivity. Sequence analysis revealed 2 non-functional polymorphisms in the POSTN gene and no other sequence variations in the remaining candidate genes. Genotyping and linkage analysis of one family yielded a LOD score of 1.51 for markers D13S272; D15S118 and D17S831 on chromosomes 13, 15 and 17 respectively.
Conclusions
While LOD scores were not significant evidence of linkage, extension of current pedigrees and novel SNP chip technology holds great promise for identification of a causative locus for PFE.
Clinical Relevance
When the process of normal tooth eruption fails, it may result in a clinically guarded or hopeless prognosis. Our studies aim to understand the etiological basis of Primary Failure of Eruption (PFE) toward the development of future orthodontic or pharmocologic interventions that will successfully treat this problem.
Keywords: mutational analysis, eruption failure, linkage studies
Introduction
Cellular and molecular advances in osteoclastogenesis have increased our understanding of the mechanisms controlling tooth eruption. As a result of recent studies in mice (1), we know that normal tooth eruption is a temporally coordinated process in which the dental follicle interacts with both osteoclasts and osteoblasts. A disturbance of this process can occur secondary to a blocked eruption path (mechanical failure of eruption) or as a primary failure of the eruption apparatus itself (primary failure of eruption). Primary Failure of Eruption (PFE), first described by Proffit and Vig (2), is defined as a localized eruption failure of permanent teeth - even when subject to orthodontic force. Recent clinical characterization has revealed that several types of non-syndromic PFE exist including familial and non-familial forms affecting posterior segments either unilaterally or bilaterally (3). However, whether it is part of a medical syndrome or a sporadic event, eruption failure is likely a heritable trait and the elucidation of its genetic contribution will undoubtedly lead to improved diagnosis and treatment approaches.
Surgical studies in dogs provided initial evidence that the role of the dental follicle was central to the eruption process - if the follicle was removed eruption failed, but if left intact without a tooth, eruption occurred (4). Hence, the dental follicle plays an important role by initiating the coordinated process of alveolar bone resorption and apposition necessary to allow a tooth to move through bone. Our current understanding of the genetic and molecular control of tooth eruption has largely stemmed from studies in mice (5). For example, osteopetrotic mice (6,7,8) and RANKL knockout mice (9) have an eruption failure phenotype, but injection of CSF-1 into these animals at a developmental age prior to the onset of eruption induces eruption (10).
Advances in molecular biology have contributed to our understanding of the microenvironment surrounding tooth eruption. For instance, the complex relationship between receptor activator of nuclear factor kB ligand (RANK-L) and macrophage-colony stimulating factor (CSF) in the formation and activation of osteoclasts has been well- described (5) and makes them potential candidates genes for PFE. However, the search for candidate genes responsible for PFE still presents a unique challenge since this dental condition presents in the absence of any systemic disorders. Hence, the consideration of ‘tooth-specific’ genes, or those genes not implicated in systemic conditions, becomes a high-priority in evaluating candidate genes for PFE. For instance, gene products of Periostin (POSTN), Amelogenin (AMELX), and Ameloblastin (AMBN) play an important role in tooth developmental processes and provide interesting candidates for eruption failure for many reasons. AMELX, an enamel matrix protein, was shown to be a negative regulator of osteoclastogenesis inhibiting the expression of RANKL, M-CSF (11). One study revealed that although AMBN and AMELX are important for mineralization, both also share a role in the development and regeneration of the periodontal ligament (12) making either potential candidate genes for eruption disorders. Periostin, initially identified as osteoblast-specific factor-2, was renamed periostin to avoid its confusion with a transcription factor, Cbfa1 (13). Periostin null mice showed an eruption failure when compared to wild-type mice (14). Finally, we propose another potential candidate gene, RUNX2. Although previous studies confirm the role of RUNX2 (also referred to as CBFA1) in both dental and skeletal defects seen in cleidocranial dysplasia (CCD) - including abnormal somatic bone development, absence of clavicles, supernumerary teeth and eruption delay - one study extended the CCD phenotypic spectrum to include a dental phenotype with a radiologically normal skeleton (15). This evidence that an isolated dental phenotype (supernumerary teeth) can result from a RUNX2 mutation provides compelling rationale for choosing it as a candidate gene for eruption failure.
The objective of our study is to define the genetic contribution of eruption failure in humans and to test the hypothesis that human PFE is due to one or more distinct mutations in key candidate genes. Our rationale for investigating the genetic etiology of the PFE is based on studies in mice that confirm the role of several candidate genes in eruption failure and studies in humans that show the segregation of this trait in families.
Materials and Methods
Diagnosis and inheritance of clinical eruption failure phenotype
Approval for this study was granted by the Biomedical Institutional Review Board at the University of North Carolina at Chapel Hill. Consent to participate (including a release for dental records) was obtained from every adult participant or from a parental guardian in the case of minors. Individuals and families were recruited based on the presence of the PFE. Through subsequent interviews, pedigrees (Fig. 1A–D) were extended for a total of 17 individuals from 4 families. There was also one individual evaluated who did not report affected relatives. Family A consisted of 12 individuals (9 were available for clinical and molecular studies); family B consisted of 13 members (3 were available for clinical and molecular studies); Family C consisted of 12 individuals (3 were available for clinical studies); and Family D consisted of 44 individuals according to the proband and historian (2 were available for clinical and molecular studies). The age range was from 5 – 72 years. A total of 18 individuals (representative of 4 families and 1 isolated case) were available for pre-treatment clinical photographs, panoramic and cephalometric radiographs following the initial clinical evaluation. Briefly, the clinical evaluation was carried out as follows 1) a positive diagnosis of the PFE was made for all available individuals based on the presence of at least one unerupted tooth following unsuccessful orthodontic treatment in the absence of any mechanical or secondary barrier; 2) skeletal relationships including vertical (overbite or open bite) and sagittal (Angle’s Class I, Class II or Class III molar) were made based on clinical exam and cephalometric radiographs; and 3) anomalies in growth and/or stature were determined to be absent or present.
Figure 1.

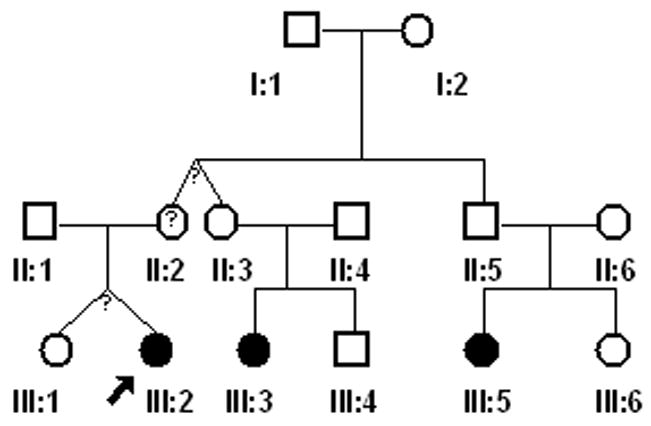
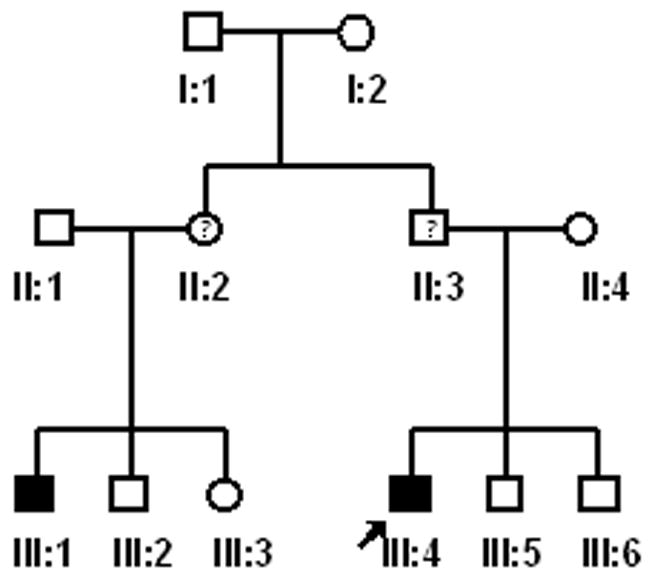
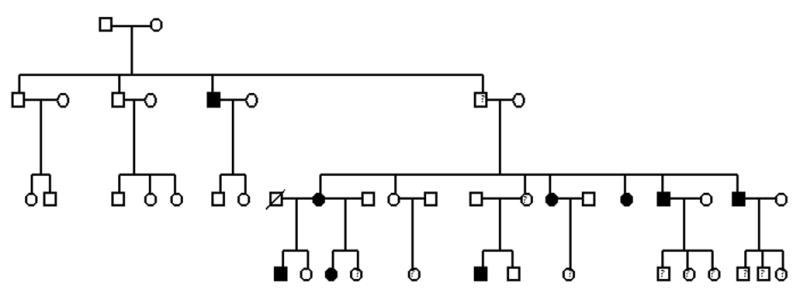
(A–D). Pedigrees of 4 families showing segregation of the PFE. Affection status is as follows: darkened circles or squares denote an affected individual; clear denotes unaffected; circles or squares with a question mark indicate affected status unknown. Squares indicate male; circles indicate female; diagonal line indicates deceased.
Sequencing and mutational analysis of candidate genes
Extraction and purification of DNA was carried out using buccal cell (PureGene kit, Gentra Systems, MN) or saliva using Oragene kits (DNA Genotek, CA) for 6 of 18 individuals analyzed above. Sequencing and mutational analysis was limited to selected individuals (probands and isolated cases) since related individuals share the same genotype. High-priority candidate genes were selected based on their relationship to tooth eruption or their putative relationship to each other. PCR amplification of four candidate genes (POSTN RUNX2, AMELX, and AMBN) was carried out for selected individuals (n=6) using primers as shown in Table I (POSTN) or as described previously for AMLEX (16) RUNX2 and AMBN (17). PCR was carried out using FailSafe PCR buffer (Epicentre Biotechnologies, Madison, WI) and under the following conditions: 10 min 95°C activation/premelt step, followed by 35 cycles of 30 s of 94°C melt, 30 s of 60°C anneal, and 3 min of 72°C extension. PCR products were purified using ExoSaplt (USB, Cleveland OH), then sequenced (Applied Biosystems, Foster City, CA) as recommended by the manufacturer. Sequencing was performed at the UNC Genome Analysis Facility.
Table 1.
Parametric and non-parametric linkage analyses reveal regions on Chromosomes 13q21; 15q11-q13 and 17p13.2.
| Locus* | Marker | LOD+ | ZLR# | parametric (p)/non-para (npl) |
|---|---|---|---|---|
| 13q21 | D13S272 | 1.51 | 2.63 | p |
| 15q11-q13 | D15S118 | 1.51 | 2.63 | npl |
| 17p13.2 | D17S831 | 1.51 | 2.63 | npl |
Locus refers to map position as identified by MARKER
LOD denotes logarithm (base 10) of odds
ZLR refers to the maximum likelihood ratio z-score
Genotyping and linkage analysis
Genotyping was carried using 500 microsatellite markers (deCODE Genetics, Reykjavik, Iceland) via an automated 8 cM genome-wide microsatellite repeat scan for 9 individuals in Family A (Fig. 1) that was judged adequate to show ‘suggestive’ evidence of linkage. Diagnosis was made based on the dichotomous affection status (ie presence or absence of PFE). Although for the purposes of future studies, patients were also diagnosed with specific types of PFE as described previously (3). Map distances were taken from the deCODE map based on human genome build 36 at NCBI. Parametric and non-parametric (model free) multipoint linkage analyses (18) were performed using Allegro software version 2.1 by Allegro Microsystems, Inc., Worcester, MA (19). Non-parametric linkage (NPL) analysis was run to account for the possibility of alternate modes of inheritance. NPL has been described as a powerful approach compared to the commonly used parametric methods and has been referred to as the method of choice for pedigree studies of complex traits (20). We assumed an autosomal dominant inheritance with incomplete penetrance (97%) for the parametric analysis and an affected allele frequency of 0.0001. Calculations using marker allele frequencies were based on the allele frequencies generated from an Icelandic population (Caucasian sample). Linkage calculations were made at 1cM intervals and LOD scores generated for both parametric and non-parametric analyses.
Results
Phenotypic characterization
The initial diagnosis of PFE was made based on the sole criteria of ‘failure to erupt’ following orthodontic forces. The PFE affection status was used for subsequent linkage studies, while additional phenotypic descriptions were made to fully characterize the craniofacial features that may be associated with the PFE phenotype. Individuals were also classified as PFE Type I, Type II, or combination as previously described (3). The cephalometric and clinical evaluation for 18 individuals included an examination of vertical and sagittal relationships and revealed that while more individuals also appeared Class III the sample size was not statistically sufficient enough to evaluate whether a correlation existed. Family A showed a co-segregation of the Class III and PFE traits.
Genotyping and linkage mapping
Pedigree analysis by visual inspection for 4 families suggests an autosomal dominant inheritance with incomplete penetrance. The following criteria were used to distinguish autosomal dominance from recessive patterns of inheritance: males and females are equally affected; the phenotype appears in every generation; and (e) unaffected individuals do not transmit the phenotype. In families B and C the second generation reported uncertainty about the eruption phenotype and were not available for analysis. Families A and D most resembled the autosomal dominant inheritance pattern.
Non-parametric linkage analysis revealed that 3 loci; 13q21, 15q11-q13 and 17p13.2 (Table 1) on 3 chromosomes were just under the threshold to be considered suggestive of linkage using the criteria of a LOD threshold of 1.86 proposed by Rao and Gu (21) for suggestive linkage in genome-wide linkage studies. We identified candidate genes of biologic interest for 3 loci, 13q21; 15q11-q13 and 17p13.2 using bioinformatic approaches (http://www.ncbi.nlm.nih.gov/projects/omim/Homology/). The candidate intervals described above included a total of 749 genes (Table 2), but we determined that 7 genes were of interest based on biological pathways or relationships to genes involved in tooth development. Several other genes in candidate regions were either previously undescribed or of unknown function. One of our goals was to try to exclude regions of key signaling molecules in tooth development to narrow our search for candidate genes; however, we were unable to exclude any regions (i.e., LOD = <-2.0) using our modest sample size (Family A).
Table 2.
Candidate intervals contain over 700 genes. Seven potential candidate genes were selected based on biologic relevance.
| Chromosome | Region | LOD | #of genes in region | Potential Candidates |
|---|---|---|---|---|
| 13 | D13S271-D13S272 | 1.51 | 84 | Proximal region to POSTN |
| 15 | D15S118-D15S998 | 1.51 | 258 |
PACE4 - convertase involved in the processing of TGFB1 FGF7 - growth factor specific for epithelial cells MYH12 - cellular proliferation or for the polarized movement IGF1R - anti-apoptotic agent |
| 17 | 17p11.2-ter | 1.50 | 407 |
MYH2 - drives diverse motile processes such as cellular locomotion MYH1 – similar to above |
Mutational analysis
Candidate gene analysis of POSTN, RUNX2, AMELX, and AMBN for both unrelated affected individuals and unaffected individuals did not reveal functional polymorphisms or disease-causing mutations. We did, however identify 2 polymorphisms in the intronic region of the POSTN gene for 1 affected non-familial individual with Type I PFE (Fig. 2). Using bioinformatic approaches, we determined that the intronic polymorphisms identified, c.1905+27G>A and c.2100+22G>A, 1) were not nonfunctional alterations, and 2) did not abolish or create splice sites of POSTN.
Figure 2.
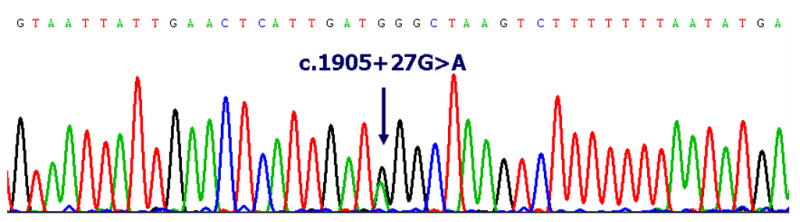
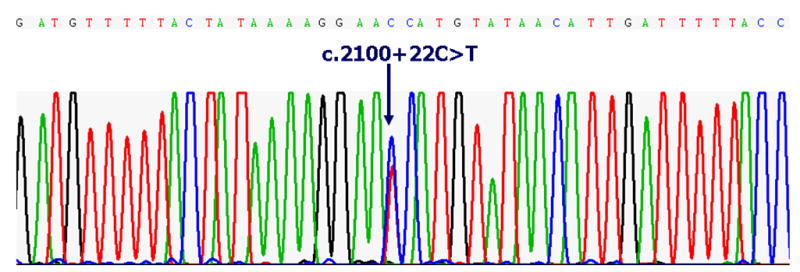
(A–B). Electrophlerograms of the POSTN sequence from a sporadic case (non-familial) of PFE. Pairwise alignment using bioinformatic tools indicate 2 non-functional SNPs (as labeled above) in the intronic region of POSTN.
Discussion
Our clinical and pedigree analysis findings support that theory that PFE is an inherited disorder. Our mutational analysis and linkage studies may indicate regions that are linked to the PFE trait. Specifically, we found that region 13q21 is in the vicinity of the POSTN locus at 13q11.1 where we identified two intronic polymorphisms. While the candidate gene analysis data was negative for the coding mutations in AMELX, AMBN, RUNX2 and POSTN, we found it informative and encouraging for future genetic studies to identify 2 polymorphisms that are proximal to a candidate region suggestive of linkage. While our linkage studies using the deCODE screen were not conclusive, the regions that we identified using linkage analysis are just under the threshold of ‘suggestive’ for linkage and provide potential regions for future studies with a larger and more informative sample size. However, the most optimal approach is to identify large informative families and expand our current pedigrees. This will provide the additional power needed to conclusively establish linkage to causative genes, and also enable us to further refine correlations of PFE to other features, such as Class III malocclusion.
An intriguing and critical issue to consider for future studies is the selection of high priority candidate genes for further interrogation. These questions and many others were addressed at the COAST conference where these proceedings were presented. In the absence of highly significant LOD scores, finding the most relevant genes to pursue is an indeed a complex task and to a large extent depends on our understanding of the biological event(s) creating the ‘eruptive force’ that is lacking in PFE. Our evaluation of mineralization genes, while not a logical first choice, may have great value. During the 2008 COAST conference Dr. Malcom Snead suggested that genes involved in mineralization may act in concert with those involved in osteoclastogenesis (RANKL, CSF1, Cfos, etc.). More specifically, his theory purports that during the eruption of a tooth the osteoclastic activity might be stimulated at the cessation of enamel formation, while enamel matrix gene expression is winding down. Further, the lack of amelogenin degradation proteins causes a local upregulation in RANKL and the osteolytic activity seen around the crown of the tooth. RANKL is suppressed by amelogenin as shown by the amelogenin knock out that suffers from increased osteoclastic and cemetoclastic activity (22). Hence the eruptive force is provided by immigration of stem cells into the site, which either aids the osteoclastic activity or enhances the ‘engine’ of eruption.
Researchers are rapidly working toward a complete understanding of the precise coordination of molecular and cellular events surrounding eruption. A combination of advances in genomics, proteomics and clinical sciences provide hope that future diagnostic and clinical regimes will offer pharmacogenetic strategies to definitively reverse an otherwise guarded or hopeless clinical prognosis.
APPENDIX.
| Human Periostin Primers | |||
|---|---|---|---|
| Primer pair | Product size | Forward primer | Reverse Primer |
| 1 | 227 | GAGCTCTCCAAAGCCCACT | GCTGAGGAAAAAGAAAAATGG |
| 2 | 243 | TCAACCAATCTTTAGATACGAAAA | CAGGGGAAGGCATATACATCA |
| 3 | 228 | TCAACAAATCAAGTCAATAAGTGAAA | AGCCACCTCAACCCTTTCTT |
| 4 | 312 | ATGTCTTGGGAAACACAGCA | GTTAAAATGAAATAACAATAACAGCAA |
| 5 | 395 | CAAGAGAGTAGAAATAGGGTGATCG | GCCCTAGATTTGGGGGTAAA |
| 6 | 250 | GTCAATGGGTATCTCTGGTTTC | AGGAGAAGAAGAGGGATTTCA |
| 7 | 283 | TGTGTTGCAATCTTTTTCTGG | TTGGTAAGGACTAGCCTCTTTTT |
| 8 | 369 | GGGTCAAATGTTATTTCCCTTG | CTGCACATTCATTTTTCATGG |
| 9 | 261 | TGATGGAGATAATAGCAATGATGA | TGCAAACAATCATCATAAAGAAAAA |
| 10 | 278 | ACCGGGAGAAACTTCAATGT | TCATTGAAACAGCATTATTTGACA |
| 11 | 244 | GGGAAATTTCAAGCAAGGAA | CAAAAATATGCCAATAGCTCTCA |
| 12 | 250 | CATGCACTGTAGGTGAACATACCT | AAGTGAGATCCTGAAATGAAAGG |
| 13 | 678 | TGTTTCTTAGTCCTTATCAAAAAGTCA | CTGAGATCATGCCACTGCAC |
| 14 | 242 | AGTGGTGCCATGGTTTTGTT | TGAGATTGGCCATAGTTTGGA |
| 15 | 281 | AAAACCTTTGTTCAGGTTTTTATG | TGAGCATGCCATTGGTATAA |
| 16 | 238 | TTGAGTCACACCGAATGTTGA | TTCAAAATAATCACAGCGAAGG |
| 17 | 217 | TGGATGTATAGTCACAATATGAGGGTA | TTTGGGGATTAACTATAATTTGATCTT |
| 18 | 700 | CATGGCCCACAAGTGTAAAA | AAAATCTGCTTGCTCAGTCTTT |
| 19 | 238 | AACCATGGTCTCCTGTCTGA | GGCTAGCTTTTCTCTCTGATCG |
| 20 | 712 | TGAAAGTAAAAGATCAACTGCACA | ACAAGGCTCGGTCTTTTCAA |
Acknowledgments
We gratefully acknowledge the support of the family and dentists who participated in this study; the assistance of Christopher Corbin, Darnell Gregory and Melody Torain in the preparation of the data. This research was supported by the Southern Association of Orthodontists; American Association of Orthodontists Foundation; University of North Carolina at Chapel Hill Faculty Development Funds to SF-B; NIH grants 1K23RR17442 to (SAFB) and M01RR-00046.
References
- 1.Wise GE, King GJ. Mechanisms of tooth eruption and orthodontic tooth movement. J Dent Res. 2008;87:414–434. doi: 10.1177/154405910808700509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Proffit WR, Vig KWL. Primary failure of eruption: a possible cause of posterior open bite. Am J Orthod. 1981;80:173–190. doi: 10.1016/0002-9416(81)90217-7. [DOI] [PubMed] [Google Scholar]
- 3.Frazier-Bowers SA, Koehler KE, Ackerman JL, Proffit WR. Primary failure of eruption: further characterization of a rare eruption disorder. Am J Orthod Dentofacial Orthop. 2007;131:578. doi: 10.1016/j.ajodo.2006.09.038. [DOI] [PubMed] [Google Scholar]
- 4.Cahill DR, Marks SC., Jr Tooth eruption: evidence for the central role of the dental follicle. J Oral Path. 1980;9:189–200. doi: 10.1111/j.1600-0714.1980.tb00377.x. [DOI] [PubMed] [Google Scholar]
- 5.Wise GE, King GJ. Mechanisms of tooth eruption and orthodontic tooth movement. J Dent Res. 2008;87:414–434. doi: 10.1177/154405910808700509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Felix R, Cecchini MG, Hofstetter W, Elford PR, Stutzer A, Fleisch H. Impairment of macrophage colony-stimulating factor production and lack of resident bone marrow macrophages in the osteopetrotic op/op mouse. J Bone Miner Res. 1990;5:781–789. doi: 10.1002/jbmr.5650050716. [DOI] [PubMed] [Google Scholar]
- 7.Wiktor-Jedrzejczak W, Bartocci A, Ferrante AW, Jr, Ahmed-Ansari A, Sell KW, Pollard JW, et al. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc Natl Acad Sci USA. 1990;87:4828–4832. doi: 10.1073/pnas.87.12.4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]
- 9.Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development, and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 10.Iizuka T, Cielinski M, Aukerman SL, Marks SC., Jr The effects of colony-stimulating factor-1 on tooth eruption in the toothless (osteopetrotic) rat in relation to the critical periods for bone resorption during tooth eruption. Arch Oral Biol. 1992;37:629–636. doi: 10.1016/0003-9969(92)90125-r. [DOI] [PubMed] [Google Scholar]
- 11.Nishiguchi M, Yuasa K, Saito K, Fukumoto E, Yamada A, Hasegawa T, et al. Amelogenin is a negative regulator of osteoclastogenesis via downregulation of RANKL, M-CSF and fibronectin expression in osteoblasts. Arch Oral Biol. 2007;52:237–243. doi: 10.1016/j.archoralbio.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 12.Zeichner-David M, Chen LS, Hsu Z, Reyna J, Caton J, Bringas P. Amelogenin and ameloblastin show growth-factor like activity in periodontal ligament cells. Eur J Oral Sci. 2006;114(Suppl 1):244–53. doi: 10.1111/j.1600-0722.2006.00322.x. discussion 254–6, 381–2. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki H, Amizuka N, Kii I, Kawano Y, Nozawa-Inoue K, Suzuki A, et al. Immunohistochemical localization of periostin in tooth and its surrounding tissues in mouse mandibles during development. Anat Rec A Discov Mol Cell Evol Biol. 2004;281:1264–1275. doi: 10.1002/ar.a.20080. [DOI] [PubMed] [Google Scholar]
- 14.Kii I, Amizuka N, Minqi L, Kitajima S, Saga Y, Kudo A. Periostin is an extracellular matrix protein required for eruption of incisors in mice. Biochem Biophys Res Commun. 2006;342(3):766–72. doi: 10.1016/j.bbrc.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 15.Quack I, Vonderstrass B, Stock M, Aylsworth AS, Becker A, Brueton L, et al. Mutation analysis of core binding factor A1 in patients with cleidocranial dysplasia. Am J Hum Genet. 1999;65:1268–1278. doi: 10.1086/302622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hart S, Hart T, Gibson C, Wright JT. Mutational analysis of X-linked amelogenesis imperfecta in multiple families. Arch Oral Biol. 2000;45:79–86. doi: 10.1016/s0003-9969(99)00106-5. [DOI] [PubMed] [Google Scholar]
- 17.Mardh CK, Backman B, Simmons D, Golovleva I, Gu TT, Holmgren G, MacDougall M, Forsman-Semb K. Human ameloblastin gene: genomic organization and mutation analysis in amelogenesis imperfecta patients. Eur J Oral Sci. 2001;109:8–13. doi: 10.1034/j.1600-0722.2001.00979.x. [DOI] [PubMed] [Google Scholar]
- 18.Whittemore AS, Halpern J, Ahsan H. Covariate adjustment in family-based association studies. Genet Epidemiol. 2005;28:244–255. doi: 10.1002/gepi.20055. [DOI] [PubMed] [Google Scholar]
- 19.Gudbjartsson DF, Jonasson K, Frigge ML, Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat Genet. 2000;25:12 – 13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 20.Kruglyak L. Thresholds and sample sizes. Nat Genet. 1996;14:132–133. doi: 10.1038/ng1096-132. [DOI] [PubMed] [Google Scholar]
- 21.Rao DC, Gu C. False positives and false negatives in genome scans. Adv Genet. 2001;42:487–498. doi: 10.1016/s0065-2660(01)42038-4. [DOI] [PubMed] [Google Scholar]
- 22.Hatakeyama J, Philp D, Hatakeyama Y, Haruyama N, Shum L, Aragon MA, et al. Amelogenin-mediated Regulation of Osteoclastogenesis, and Periodontal Cell Proliferation and Migration. J Dent Res. 2006;85:144–149. doi: 10.1177/154405910608500206. [DOI] [PubMed] [Google Scholar]