

Abstract
Biocompatible mesoporous silica nanoparticles, containing the fluorescence dye fluorescein isothiocyanate (FITC), provide a promising system to deliver hydrophobic anticancer drugs to cancer cells. In this study, we investigated the mechanism of uptake of fluorescent mesoporous silica nanoparticles (FMSN) by cancer cells. Incubation with FMSN at different temperatures showed that the uptake was higher at 37°C than at 4°C. Metabolic inhibitors impeded uptake of FMSN into cells. The inhibition of FMSN uptake by nocodazole treatment suggests that microtubule functions are required. We also report utilization of mesoporous silica nanoparticles to deliver a hydrophobic anticancer drug paclitaxel to PANC-1 cancer cells and to induce inhibition of proliferation. Mesoporous silica nanoparticles may provide a valuable vehicle to deliver hydrophobic anticancer drugs to human cancer cells.
Keywords: mesoporous silica nanoparticle, endocytosis, hydrophobic drugs, drug delivery, cancer
Introduction
The last decade saw increasing interest in using nanoparticles for biomedical applications, including disease treatment, diagnosis, tracking, and drug delivery. Research into rational delivery and targeting of therapeutic and diagnostic agents is at the forefront of cancer research [1, 2]. Nanoparticles offer great potential to deliver therapeutic agents into targeted organs or cells and are actively developed for application in cancer therapy. Among nanoparticles in development, polymeric micelles, polymeric and ceramic nanoparticles, viral-derived capsid nanoparticles, liposomes, albumin nanoparticles and silica nanoparticles have been widely studied. One application of such nanocarriers is to encapsulate, covalently attach, or adsorb therapeutic agents to the nanoparticles to overcome drug solubility problems. This is of importance, as large portions of new drug candidates emerging from high-throughput drug screening initiatives are water insoluble [3]. Another application is to use nanoparticles for targeting to specific cells and organs within the body. This is accomplished by functionalizing their surface with antibody or appropriate ligands [4].
Mesoporous silica nanoparticles possess several attractive features such as large surface areas and porous interiors that can be used to store various molecules [5]. In addition, their pore size and environment as well as their size and shape can be easily modified, making them a suitable choice as intracellular drug carriers [6]. In our previous study [7], we successfully incorporated a representative hydrophobic anticancer drug camptothecin (CPT) into the pores of fluorescent mesoporous silica nanoparticles (FMSN) and delivered CPT into a variety of human cancer cells to induce cell death, demonstrating that mesoporous silica nanoparticles are promising in overcoming the insolubility problem of anticancer drugs.
In this paper, we first addressed the mechanism of uptake of FMSN into cancer cells. Different incubation temperatures and various chemicals were used to gain insight into the uptake mechanism. We also examined whether a hydrophobic anticancer drug paclitaxel can be delivered into cancer cells by utilizing FMSN as a drug nanocarrier. Here, we report that FMSN can be taken up by human cancer cells through energy-dependent endocytosis mechanism and that FMSN can be used as a delivery vehicle for paclitaxel.
Experimental
Synthesis of Nanoparticles
All chemicals used for the synthesis of the nanoparticles were purchased from Sigma-Aldrich and used without further purification. In a round-bottom flask, 12 μL aminopropyltriethoxysilane (APTS) was added to a solution of 5.5 mg fluorescein isothiocyanate (FITC) and 3 mL absolute ethanol and stirred under inert atmosphere. In another container, 0.5 g cetyltrimethylammonium bromide (CTAB) was dissolved in a solution of 240 mL distilled water and 1.75 mL sodium hydroxide (2 M) and stirred vigorously at 80°C. The solution of FITC-APTS was stirred for 2 h before adding 2.5 mL tetraethylorthosilicate (TEOS). Once the temperature of the CTAB solution had stabilized, the ethanol solution containing TEOS and FITC-APTS was added. After 15 min, 0.63 mL of 3-trihydroxysilylpropyl methylphosphonate was added to the mixture. Two hours later, the solution was cooled to room temperature, and the particles were filtered and washed with methanol. The particles were dried at room temperature overnight. To remove the surfactants from the pores of the particles, 850 mg of particles were dispersed in a solution of 90 mL methanol and 5 mL hydrochloric acid (12.1 M) and refluxed for 24 h. The particles were then filtered, washed thoroughly with methanol, and dried at room temperature.
Materials Characterization
FMSN were analyzed using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). For the TEM analysis, a drop of diluted FMSN solution was deposited onto copper grid and dried at room temperature. The images were collected using JEOL JEM-1011 transmission electron microscopy (JEOL, Japan) operated at 80 kV with a Gatan digital camera (Gatan, USA). For the SEM analysis, a drop of diluted FMSN solution was deposited onto carbon tape, dried at 60°C, and sputtered with gold. The materials were analyzed using JEOL JSM-6700F Field Emission Scanning Electron Microscopy (JEOL, Japan).
Cell Culture
Human pancreatic cancer cell line, PANC-1, and hepatoma cell line, Hepa-1, obtained from the American Type Culture Collection were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (Sigma), 2% l-glutamine, 1% penicillin, and 1% streptomycin stock solutions, incubated at 37°C under a 5% CO2 95% air atmosphere. The media were changed every 3 days, and the cells were passaged by trypsinization before confluence.
Fluorescence and Confocal Microscopy
The fluorescence of FITC was used to monitor cellular uptake of FMSN. Cells were incubated with FMSN for 3 h at 37°C under a 5% CO2 95% air atmosphere in an eight-chamber Lab-Tek chamber slide system with covers (Nalge Nunc International), washed with phosphate-buffered saline (PBS), and examined with confocal microscopy (λex=470 nm; Carl Zeiss LSM 310 laser scanning confocal microscopy). To examine the distribution of FMSN within the cells, the same cells were then incubated with 6 μM of Acridine Orange (AO; Sigma), which specifically stains lysosomes/endosomes to red [8], for 1 h in DMEM without phenol red, washed with PBS, and examined by confocal microscopy again (λex=488 nm). The emission light was passed through a 560-nm short-pass dichroic mirror. The green emission was passed through a 530-nm bandpass filter, and the red emission was passed through a 600-nm longpass filter. Both emissions were simultaneously collected using two photomultiplier tubes.
Immunocytochemistry
Cells cultured in chamber slides were also labeled for the location of lysosomes according to standard immunocytochemistry procedures. Briefly, after treating with FMSN for 3 h, cells were fixed, permeabilized, and blocked before antibody labeling. To label LAMP1 (lysosome-associated membrane protein 1) [9], we used a rat anti-mouse monoclonal antibody (1D4B; Abcam, Cambridge, MA). Primary antibody was incubated with the cells for 1 h at room temperature, and then, a 1:500 dilution of secondary antibody (Alexa-594 goat anti-rat IgG, molecular probes, Eugene, OR) was incubated for 1 h at room temperature. Slides were mounted in Prolong antifade medium containing DAPI as counterstain (molecular probes), and then examined with confocal microscopy.
Cell Treatment and Confocal Microscopic Analysis
To determine whether the uptake of FMSN into cancer cells was energy-dependent, the cells were incubated with FMSN under different metabolic conditions. The uptake assay was performed in the presence of sodium azide, sucrose, bafilomycin A, nocodazole, or brefeldin A. Cells were seeded on eight-chamber Lab-Tek chamber slides for 24 h, then preincubated at 4°C with either 0.1% sodium azide, or sucrose (0.45 M), or bafilomycin A (0.05 μM), or nocodazole (2 μg/ml), or brefeldin A (5 μg/ml) for 10 min. The FMSN suspension in PBS was then added, and the cells were incubated for an additional 3 h at 37°C. The uptake was stopped by washing the cells three times with cold PBS, and then the cells were fixed with 4% paraformaldehyde in PBS for 1 h at room temperature and washed with PBS. The chambers of slides were removed, and each slide was mounted with fluorescence-free glycerol-based mounting medium (Fluoromount-G; Southern Biotechnology Associates, USA) by cover glasses. Cells were analyzed by confocal microscopy (Carl Zeiss LSM 310 laser scanning confocal microscopy).
Loading Anticancer Drug in the Pores of FMSN
To load the paclitaxel drug molecules into the pores of the nanoparticles, 25 mg of FMSN were soaked in a solution containing 1 mg paclitaxel (Sigma-Aldrich, USA) and 2 mL DMSO. After 24 h, the mixture was centrifuged, and the supernatant was removed completely. The paclitaxel-loaded FMSN was dried under vacuum to remove trace DMSO. The paclitaxel-loaded FMSN were washed and sonicated with PBS two times, and the supernatants were collected for absorbance measurement. The concentration of paclitaxel in the supernatants that represents weakly adsorbed drug molecules was calculated by measuring the absorbance of paclitaxel at 273 nm. To measure the amount of paclitaxel that is stored inside FMSN, the paclitaxel-loaded FMSN were then resuspended in methanol and sonicated thoroughly to remove the paclitaxel from the pores of FMSN (paclitaxel has high solubility in methanol). After centrifugation, the supernatant was collected, and the process was repeated two more times to completely remove the drug from the pores of FMSN. The concentration of paclitaxel in the supernatant was calculated by measuring the absorbance of paclitaxel at 273 nm. The amount of paclitaxel adsorbed outside of FMSN was less than 5% of the amount that was stored inside FMSN.
Cell Growth Assay
Cells were cultured in 96-well plates (3,000 cells/well) with fresh DMEM at 37°C in a 5% CO2/95% air atmosphere for 24 h, then washed with PBS, and the medium was changed to a fresh medium along with paclitaxel in DMSO, paclitaxel-loaded FMSN in PBS, unloaded FMSN, or paclitaxel suspended in PBS at the indicated concentrations. The cells were washed with PBS, 24 h later, to remove paclitaxel and FMSN that were not taken up by the cells and continued incubating in DMEM for an additional 48 h. WST-8 solution (cell counting kit-8, Dojindo Molecular Technologies, Inc.), 10%, was then added to cells and incubated for another 2 h. The absorbance of each well was measured at 450 nm with a plate reader. As the absorbance is proportional to the number of viable cells in the medium, the viable cell number was determined using a prepared calibration curve.
Cell Morphology Analysis
Cells were seeded in eight-chamber Lab-Tek chamber for 24 h, washed with PBS, and then the medium was changed to a fresh medium along with paclitaxel in DMSO, paclitaxel-loaded FMSN in PBS, FMSN or paclitaxel suspended in PBS at the indicated concentrations. After 24 h, the medium was changed to fresh DMEM, and incubation was continued for an additional 24 h. The cells were then washed with PBS and analyzed by light microscopy.
Statistical Analysis
All results are expressed as means±SD. Statistical comparisons were made using Student's t test after analysis of variance. The results were considered to be significantly different at P value<0.05.
Results and Discussion
Characteristics and Cellular Uptake of FMSN
Mesoporous silica nanoparticles provide a promising vehicle to deliver anticancer drugs to cancer cells. The nanoparticles used in this study were less than 130 nm in diameter and contained pores that were around 2 nm in diameter. Approximately 750 pores are present per particle. Figure 1 shows electron microscopy analysis of the morphology of the nanoparticles and the hexagonal arrays of the pores. FITC dyes were covalently bonded within the pores of the nanoparticles to enable the monitoring of FMSN by using fluorescent microscopy. In addition, the surface of FMSN was modified with inert and hydrophilic phosphonate group to prevent aggregation caused by the interparticle hydrogen bonding interaction between the anionic silanol groups and the unreacted cationic amine groups. Preparation of FMSN is described in the Experimental section and in our previous publication [7].
Fig. 1.

Characterization of FMSN. a Transmission electron microscopy image and b Scanning electron microscopy image (SEM) of FMSN
Uptake of FMSN by cancer cells was observed using confocal microscopy. Cancer cells were incubated with FMSN and then washed with PBS to remove nanoparticles that were outside the cell. The fluorescence of the nanoparticles, which were derivatized with fluorescein, was monitored by confocal microscopy. Figure 2 shows intracellular location of FMSN in PANC-1 (A) and Hepa-1 cells (B). PANC-1 cells were treated with FMSN followed by staining with Acridine Orange. This dye specifically stains lysosomes red and the whole cell green (Fig. 2a, left panel). Location of FMSN was identified by their green fluorescence (Fig. 2a, right panel). As shown in Fig. 2a, the green fluorescence of FMSN overlapped with the red fluorescence of Acridine Orange, indicating that FMSN are taken into lysosomes shortly after they are taken up by cells. Similar results were observed in Hepa-1 cells as shown in Fig. 2b. In this case, we used an antibody against LAMP1 (lysosome associated membrane protein 1) to detect lysosomes. Red fluorescence from LAMP1 in Hepa-1 cells overlapped with the green fluorescence from FMSN, resulting in a yellow composite staining pattern as shown in Fig. 2b (fusion).
Fig. 2.
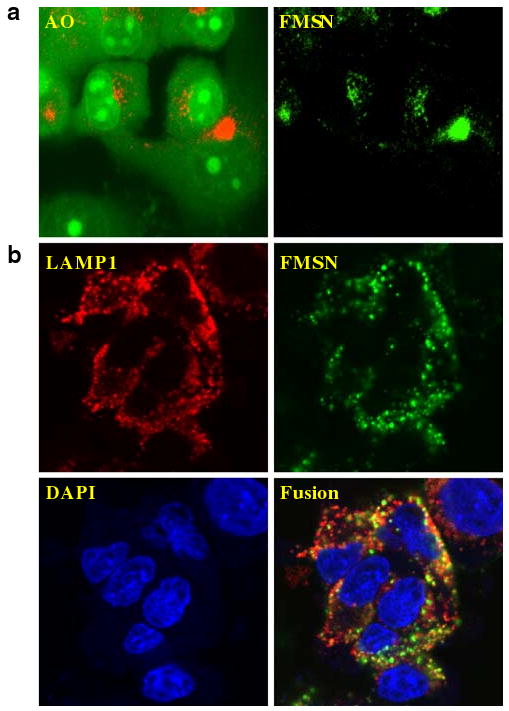
Uptake of FMSN by cancer cells. a PANC-1 cells stained with Acridine Orange (left) and the fluorescence of the nanoparticles within the same cell (right). The red fluorescence showed the location of lysosomes. b After incubation of Hepa-1 cells with FMSN for 3 h, location of FMSN was compared with that of LAMP1 that was determined by using anti-mouse monoclonal antibody (1D4B) and secondary antibody (Alexa-594 goat anti-rat IgG). Nuclei were stained with DAPI
Energy-Dependent Endocytosis of FMSN by Cells
A number of processes including phagocytosis and endocytosis could account for the uptake of nanoparticles into cells. To further characterize the mechanism of uptake of FMSN, the cells were first incubated with FMSN at 37°C or 4°C. As shown in Fig. 3, the lower temperature (4°C) significantly impeded cellular uptake of FMSN in PANC-1 cells, compared with the uptake at 37°C. This result raised the possibility that FMSN enter cells in an energy-dependent manner. To further investigate this point, effects of different metabolic inhibitors on the uptake of FMSN into cells were examined. The cells were incubated with several metabolic inhibitors, such as sodium azide [10], sucrose [11], and bafilomycin A [12], FMSN were added, and the cells were cultured. Uptake of FMSN was examined using confocal microscopy. As shown in Fig. 4, preincubation with different metabolic inhibitors, including sodium azide (which depletes intracellular ATP), sucrose (which suppresses coated pit function), and bafilomycin A (which inhibits the v-ATPase function), suppressed the uptake of FMSN into PANC-1 cells. Sodium azide is widely used as an inhibitor of cellular respiration, decreasing intracellular ATP concentration. Thus, its inhibition of the uptake of FMSN suggested that the uptake was an energy-dependent process. Clathrin-coated pits are the primary plasma membrane location [13] involved in the uptake of molecules through endocytosis. The hypertonic environment induced by sucrose, which disrupts coated-pit function, impeded FMSN uptake, suggesting that the uptake process involved clathrin-mediated endocytosis. The proton pump vacuolar ATPase (V-ATPase) energizes plasma membranes in mammalian cells [14]. Pretreatment with bafilomycin A which is a strong V-ATPase inhibitor completely suppressed the uptake of FMSN, indicating that the uptake occurred through a V-ATPase-dependent transport mechanism.
Fig. 3.
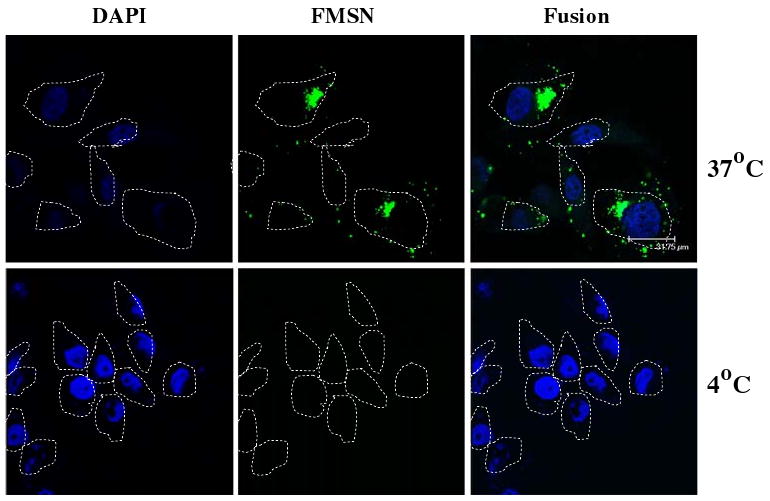
Confocal laser scanning microscopy images of FMSN taken up by cancer cells. PANC-1 cells were incubated at 37°C or 4°C for 30 min, and then the uptake was examined. Nuclei were stained with DAPI. Left panel: nuclei; middle panel: FMSN; right panel: merged image. Cell periphery is indicated by dotted lines
Fig. 4.
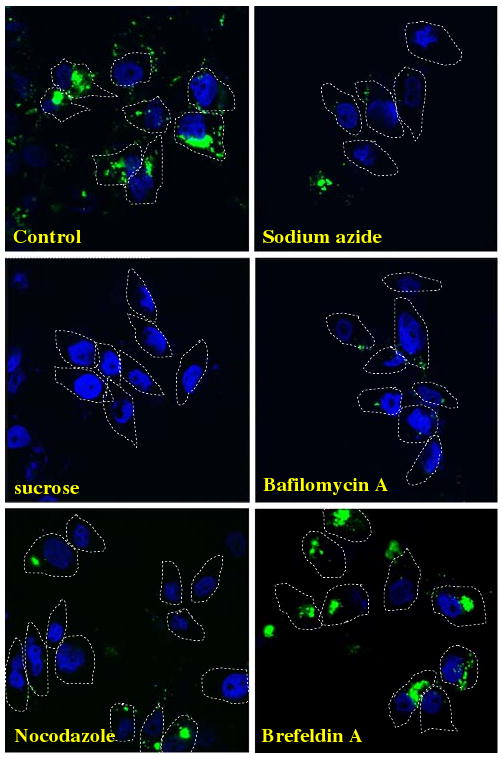
Confocal microscopy images of FMSN uptake in the presence of several metabolic inhibitors. PANC-1 cells were preincubated with metabolic inhibitors at the concentration indicated in the Experimental section and were then incubated with FMSN at 37°C for 1 h. Cells were fixed and observed with confocal microscopy. Nuclei were stained with DAPI
To investigate the effect of other endocytosis inhibitors, the microtubule depolymerizer nocodazole [15] or the plasma membrane polarity inhibitor brefeldin A [16] was preincubated with PANC-1 cells before the addition of FMSN. As shown in Fig. 4, while brefeldin A did not show any effect on FMSN uptake, nocodazole significantly disrupted the uptake, suggesting that a dynamic microtubule network, which is important for vesicular transport, is necessary for FMSN uptake.
Significance of the energy-dependent endocytosis on the uptake of silica-based nanoparticles has been reported with silica nanoparticles that are different from the FMSN used in this study. Chung TH et al. [17] reported an actin-dependent endocytosis of mesoporous silica nanoparticles in 3T3-L1 cells. Using the organic fluorescent dye, rhodamine B, Kim et al. [18] showed that silica-overcoated magnetic nanoparticles were taken up by A549 cells through energy-dependent endocytosis. Gemeinhart et al. [19] reported that dense silica nanoparticles were internalized by an endosome-lysosomal route in Chinese hamster ovarian cells. Xing et al. [20] used rhodamine 6G isothiocyanate (RITC)-doped silica-coated nanoparticles to show an energy-dependent endocytic process by HeLa cells. Taken together, these studies demonstrated the importance of temperature and energy-dependent endocytosis in the uptake of FMSN into human cancer cells.
Delivery of Hydrophobic Anticancer Drug Paclitaxel to Cancer Cells by FMSN
Many anticancer drugs have poor water solubility, which hampers the ability of drugs to be administered via the intravenous route. Therefore, development of novel delivery systems for these drugs without resorting to the use of organic solvents is critical for cancer therapy. We have previously used FMSN to deliver a hydrophobic anticancer agent camptothecin into cancer cells to induce apoptosis [7]. To investigate whether this can be applied to other drugs, we have tested whether another hydrophobic drug paclitaxel can be delivered to cancer cells by FMSN.
By soaking FMSN in a concentrated paclitaxel/DMSO solution, the drug molecules can enter the pores. After the organic solvent was removed by centrifugation and high vacuum, the hydrophobic drugs would be retained inside the pores. Because delivering paclitaxel into cancer cells is expected to lead to growth inhibition and cell death [21], the cytotoxic effects of paclitaxel-loaded FMSN was examined. Growth inhibition of PANC-1 cells was observed with the cells treated with the suspension of paclitaxel-loaded FMSN in PBS. The extent of inhibition was comparable to that observed using paclitaxel dissolved in DMSO (Fig. 5a). In addition, we observed morphologic change of the cells treated with paclitaxel-loaded FMSN in PBS or paclitaxel dissolved in DMSO. Cells became round and smaller upon the treatment (Fig. 5b). These results are consistent with the effects of paclitaxel, which hyper-stabilizes microtubule structure and causes morphologic changes in addition to its growth inhibitory effects.
Fig. 5.
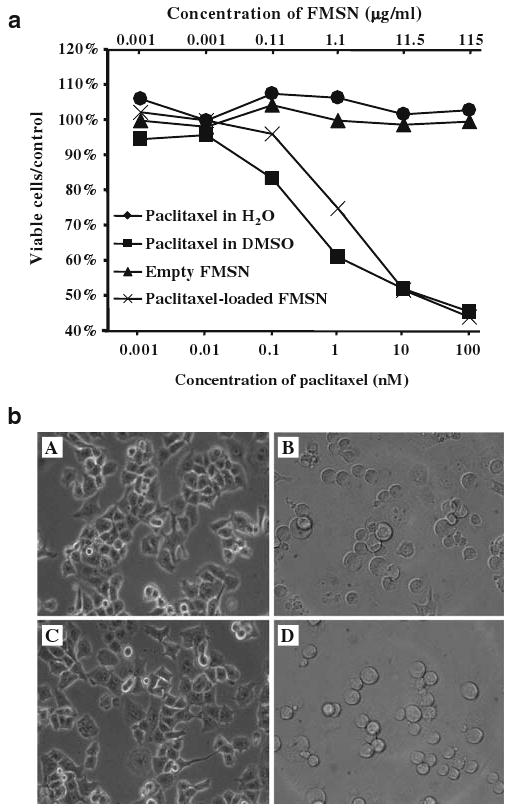
Cell growth inhibition assay. a PANC-1 cells were treated with empty FMSN in PBS (filled triangles), paclitaxel in DMSO (filled squares), paclitaxel in H2O (filled circles), or paclitaxel-loaded FMSN in PBS (ex mark), and the percentage of viable cells was compared to control wells without treatment (Y-axis). The concentration of FMSN is shown on top of each figure (μg/ml), while that of paclitaxel in DMSO, in PBS, or loaded in FMSN (μg/ml), is shown under each figure (nM). b Representative images of PANC-1 cells. PANC-1 cells were treated with A 1.15 μg/ml of empty FMSN, B 1 nM paclitaxel in DMSO, C 1 nM paclitaxel in H2O, or D 1.15 mg/ml of paclitaxel-loaded FMSN for 24 h, and then were observed with light microscopy
As shown in Fig. 5a, empty FMSN did not cause inhibition of proliferation of PANC-1 cells. Also, we did not observe any cytotoxic effects of FMSN with other pancreatic cancer cell lines Capan-1 and AsPc-1, a colon cancer cell line SW480 and a stomach cancer cell line MKN45 [7]. In addition, FMSN did not cause cytotoxic effects in a macrophage cell line RAW264.7 (data not shown), in contrast to other nanoparticles such as ZnO and cationic nanospheres that cause cytotoxic effects. These results suggest that the mesoporous silica nanoparticles themselves are not toxic to cultured mammalian cells. Other studies also point to biocompatibility of silica nanoparticles in human cells [22–24].
Together with our previous study demonstrating the successful loading of camptothecin and delivery into pancreatic cancer cells, FMSN have now been shown to serve as a delivery vehicle for two representative hydrophobic anticancer drugs, camptothecin and paclitaxel. It will be interesting to investigate whether this method is applicable to other anticancer drugs. Because the low water solubility of anticancer drugs is frequently encountered in the development of effective therapy for cancer, our studies suggest that the mesoporous silica nanoparticles offer a solution to this problem in drug development.
Conclusion
In this study, we have shown that fluorescent mesoporous nanoparticles (FMSN) are efficiently taken up by human cancer cells and that this cellular uptake requires energy use and temperature-dependent endocytosis. Work is ongoing to further define the endocytosis pathway utilized by the nanoparticles. We have also shown that a hydrophobic anticancer drug paclitaxel can be stored in FMSN and delivered to human cancer cells resulting in the inhibition of proliferation of these cells. These results point to the useful feature of FMSN as a valuable vehicle for anticancer drugs. We are currently attaching nanovalves to FMSN so that the stored anticancer drugs can be released in a controlled manner. We believe the utilization of FMSN will provide a promising avenue for the development of an effective delivery system for cancer therapy.
Acknowledgments
This work is supported by US NIH grants CA32737 (F.T.) and ES015498 (A.N.), NSF grant DMR0346601 (J.Z.) and a grant from Edna E. and Susan E. Riley foundation. Monty Liong and Michael Kovochich are supported by the UC Lead Campus for Nanotoxicology Training and Research, funded by UC TSR&TP.
Contributor Information
Jie Lu, Department of Microbiology, Immunology and Molecular Genetics, Jonsson Comprehensive Cancer Center, Molecular Biology Institute, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA, California NanoSystems Institute, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA.
Monty Liong, Department of Chemistry and Biochemistry, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA, California NanoSystems Institute, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA.
Sean Sherman, Department of Microbiology, Immunology and Molecular Genetics, Jonsson Comprehensive Cancer Center, Molecular Biology Institute, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA, California NanoSystems Institute, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA.
Tian Xia, Department of Medicine, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA, California NanoSystems Institute, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA.
Michael Kovochich, Department of Medicine, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA, California NanoSystems Institute, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA.
Andre E. Nel, Department of Medicine, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA, California NanoSystems Institute, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA
Jeffrey I. Zink, Department of Chemistry and Biochemistry, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA, zink@chem.ucla.edu, California NanoSystems Institute, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA
Fuyuhiko Tamanoi, Department of Microbiology, Immunology and Molecular Genetics, Jonsson Comprehensive Cancer Center, Molecular Biology Institute, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA, California NanoSystems Institute, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA, Department of Microbiology, Immunology and Molecular Genetics, California NanoSystems Institute, JCCC, University of California Los Angeles, 405 Hilgard Avenue, Los Angeles, CA 90095, USA, fuyut@microbio.ucla.edu.
References
- 1.Reynolds AR, Moein MS, Hodivala-Dilke K. Trends Mol Med. 2003;9:2–4. doi: 10.1016/s1471-4914(02)00004-7. [DOI] [PubMed] [Google Scholar]
- 2.Van Vlerken LE, Amiji MM. Expert Opin Drug Deliv. 2006;3:205–16. doi: 10.1517/17425247.3.2.205. [DOI] [PubMed] [Google Scholar]
- 3.Ebbesen M, Jensen TG. J Biomed Biotechnol. 2006;2006:51516. doi: 10.1155/JBB/2006/51516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brannon-Peppas L, Blanchette JO. Adv Drug Deliv Rev. 2004;56:1649–59. doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 5.Kresge CT, Leonowicz ME, Roth WJ, Vartuli JC, Beck JS. Nature. 1992;359:710–2. [Google Scholar]
- 6.Han YJ, Stucky GD, Butler A. J Am Chem Soc. 1999;121:9897–8. [Google Scholar]
- 7.Lu J, Liong M, Zink JI, Tamanoi F. Small. 2007;3:1341–6. doi: 10.1002/smll.200700005. [DOI] [PubMed] [Google Scholar]
- 8.Canonico PG, Bird JWC. J Cell Biol. 1969;43:367–71. doi: 10.1083/jcb.43.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Cha Y, Yuksel K, Gracy R, August J. J Biol Chem. 1988;263:8754–8. [PubMed] [Google Scholar]
- 10.Lichstein HC, Soule MH. J Bacteriol. 1944;47:231–8. doi: 10.1128/jb.47.3.231-238.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heuser JE, Anderson RG. J Cell Biol. 1989;108:389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wessing A, Bertram G, Zierold K. J Comp Physiol. 1993;163:452. doi: 10.1007/BF00346929. [DOI] [PubMed] [Google Scholar]
- 13.Pearse BM, Crowther RA. Annu Rev Biophys Biophys Chem. 1987;16:49–68. doi: 10.1146/annurev.bb.16.060187.000405. [DOI] [PubMed] [Google Scholar]
- 14.Harvey WR, Maddrell SHP, Telfer WH, Wieczorek H. Amer Zool. 1998;38:426–41. [Google Scholar]
- 15.Samson F, Donoso JA, Heller-Bettinger I, Watson D, Himes RH. J Pharmacol Exp Ther. 1979;208:411–7. [PubMed] [Google Scholar]
- 16.Roma MG, Milkiewicz P, Elias E, Coleman R. Hepatology. 2000;32:1342–56. doi: 10.1053/jhep.2000.20519. [DOI] [PubMed] [Google Scholar]
- 17.Chung TH, Wu SH, Yao M, Lu CW, Lin YS, Hung Y, et al. Biomaterials. 2007;28:2959–66. doi: 10.1016/j.biomaterials.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Kim JS, Yoon TJ, Yu KN, Noh MS, Woo M, Kim BG, et al. J Vet Sci. 2006;7:321–6. doi: 10.4142/jvs.2006.7.4.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gemeinhart RA, Luo D, Saltzman WM. Biotechnol Prog. 2005;21:532–7. doi: 10.1021/bp049648w. [DOI] [PubMed] [Google Scholar]
- 20.Xing X, He X, Peng J, Wang K, Tan W. J Nanosci Nanotechnol. 2005;5:1688–93. doi: 10.1166/jnn.2005.199. [DOI] [PubMed] [Google Scholar]
- 21.Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. J Am Chem Soc. 1971;93:2325–7. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 22.Slowing I, Trewyn BG, Lin VS. J Am Chem Soc. 2006;128:14792–3. doi: 10.1021/ja0645943. [DOI] [PubMed] [Google Scholar]
- 23.Luo D, Saltzman WM. Nat Biotechnol. 2000;18:893–5. doi: 10.1038/78523. [DOI] [PubMed] [Google Scholar]
- 24.Jin Y, Kannan S, Wu M, Zhao JX. Chem Res Toxicol. 2007;20:1126–33. doi: 10.1021/tx7001959. [DOI] [PubMed] [Google Scholar]